HIF1?/TET1 Pathway Mediates Hypoxia-Induced Adipocytokine Promoter Hypomethylation in Human Adipocytes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Culture of Primary Subcutaneous Pre-Adipocytes
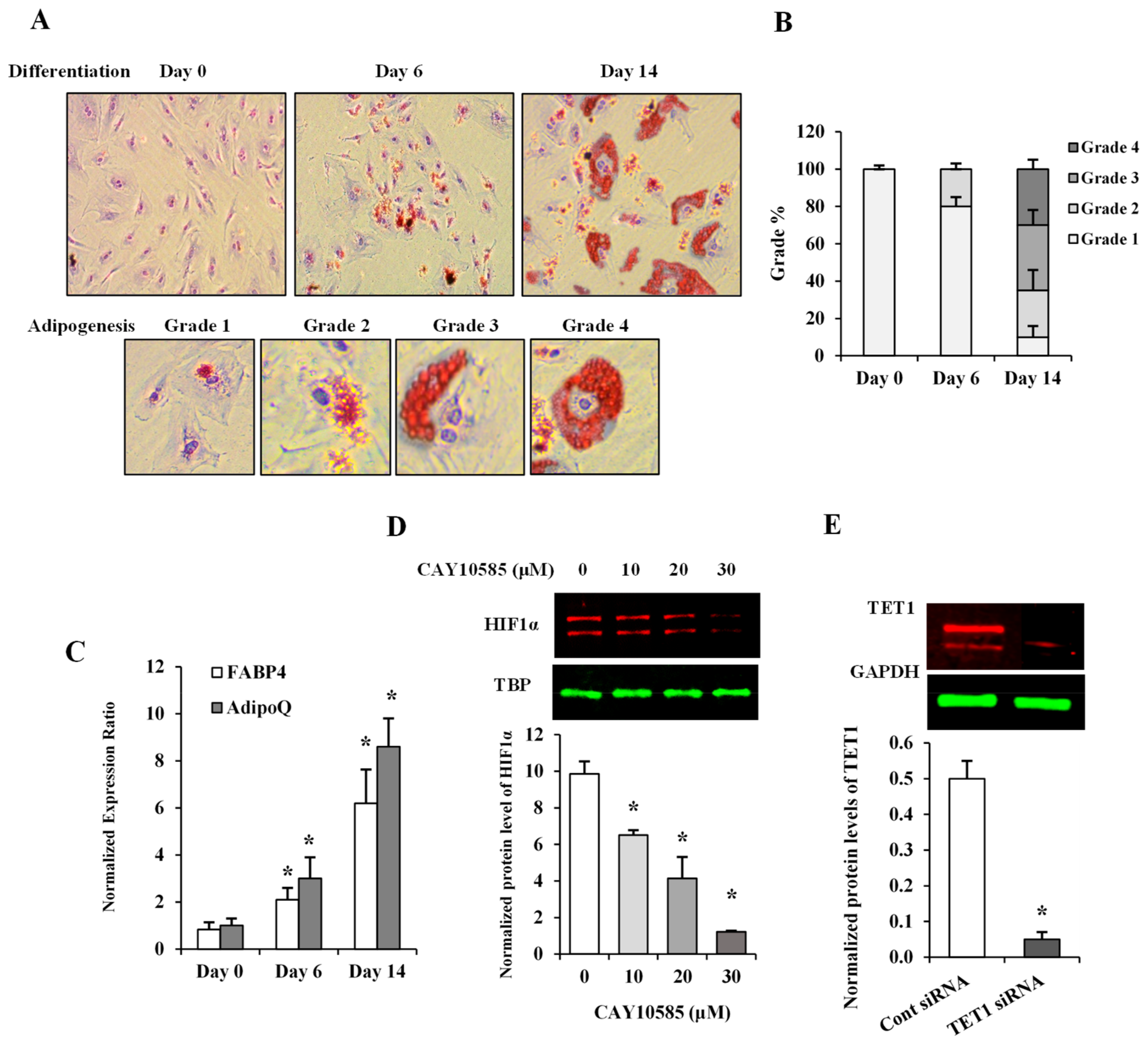
2.3. Oil Red O Staining
2.4. Induction of Hypoxia in Differentiated Adipocytes
2.5. TET1 Gene Silencing in Mature Adipocytes
2.6. Real-Time Polymerase Chain Reaction (PCR)
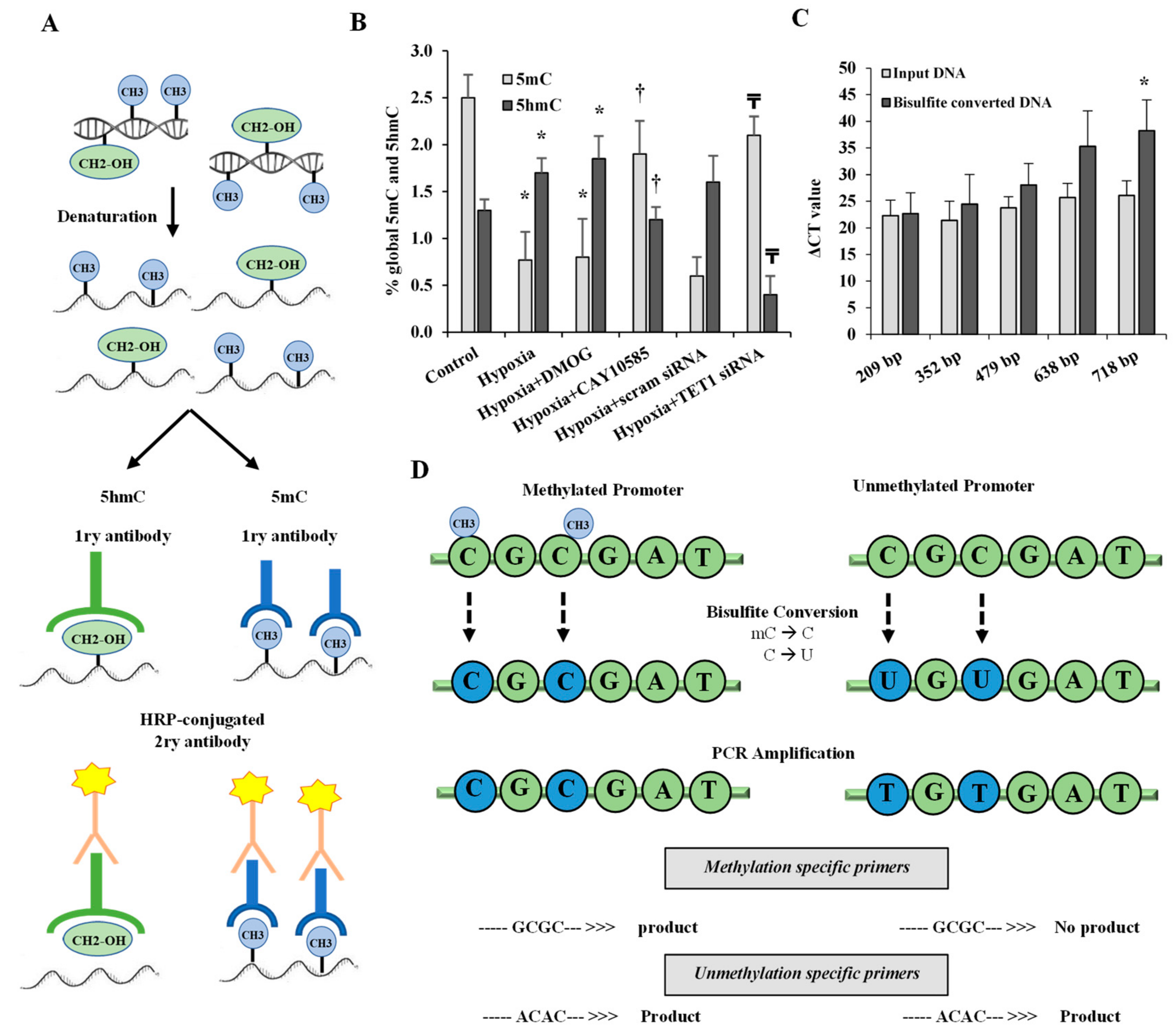
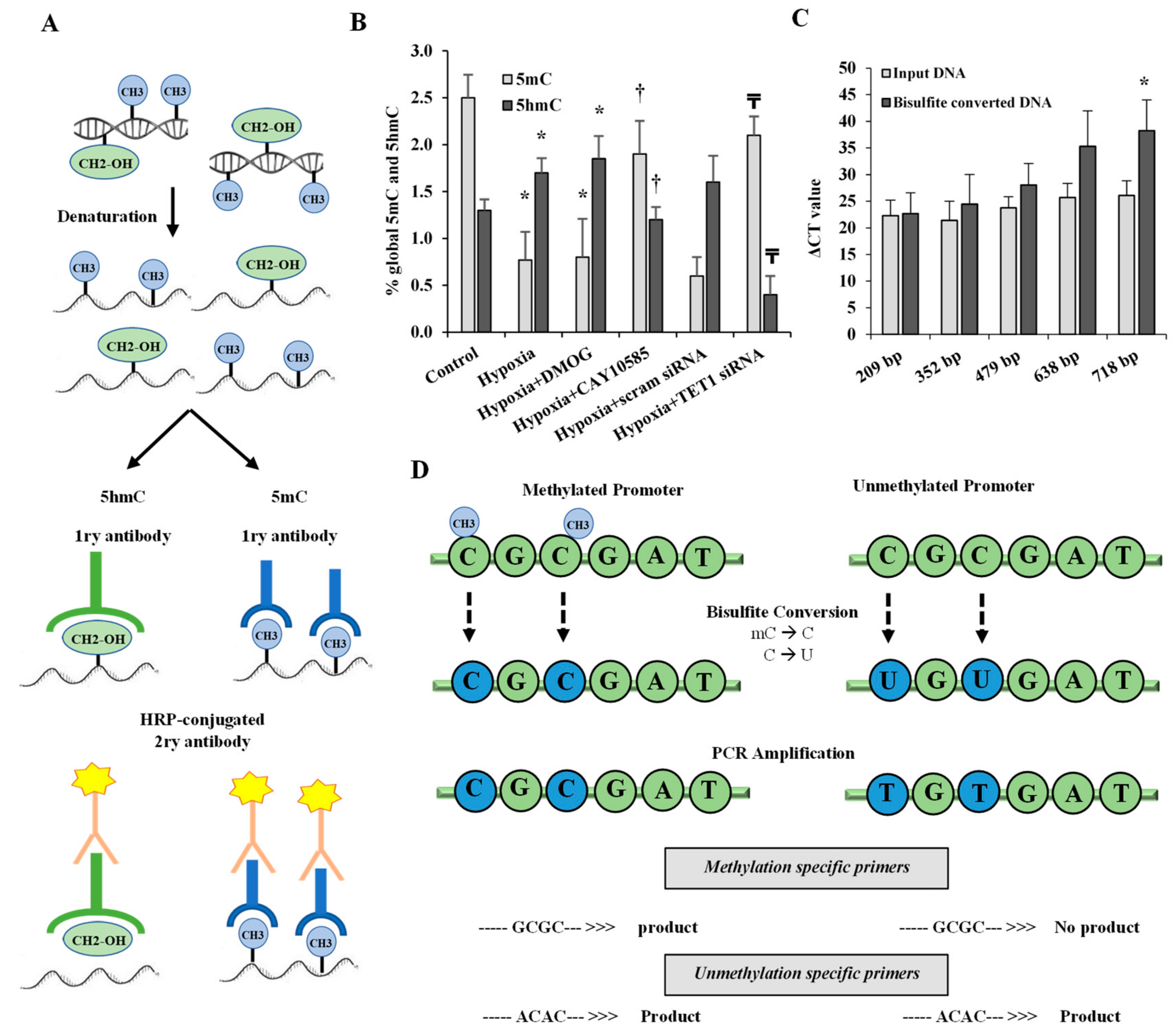
2.7. Global DNA Methylation and Hydroxymethylation Analysis
2.8. DNA Bisulfite Modification and Methylation-Specific PCR
2.9. Western Blotting
2.10. Measurement of DNMT Activity
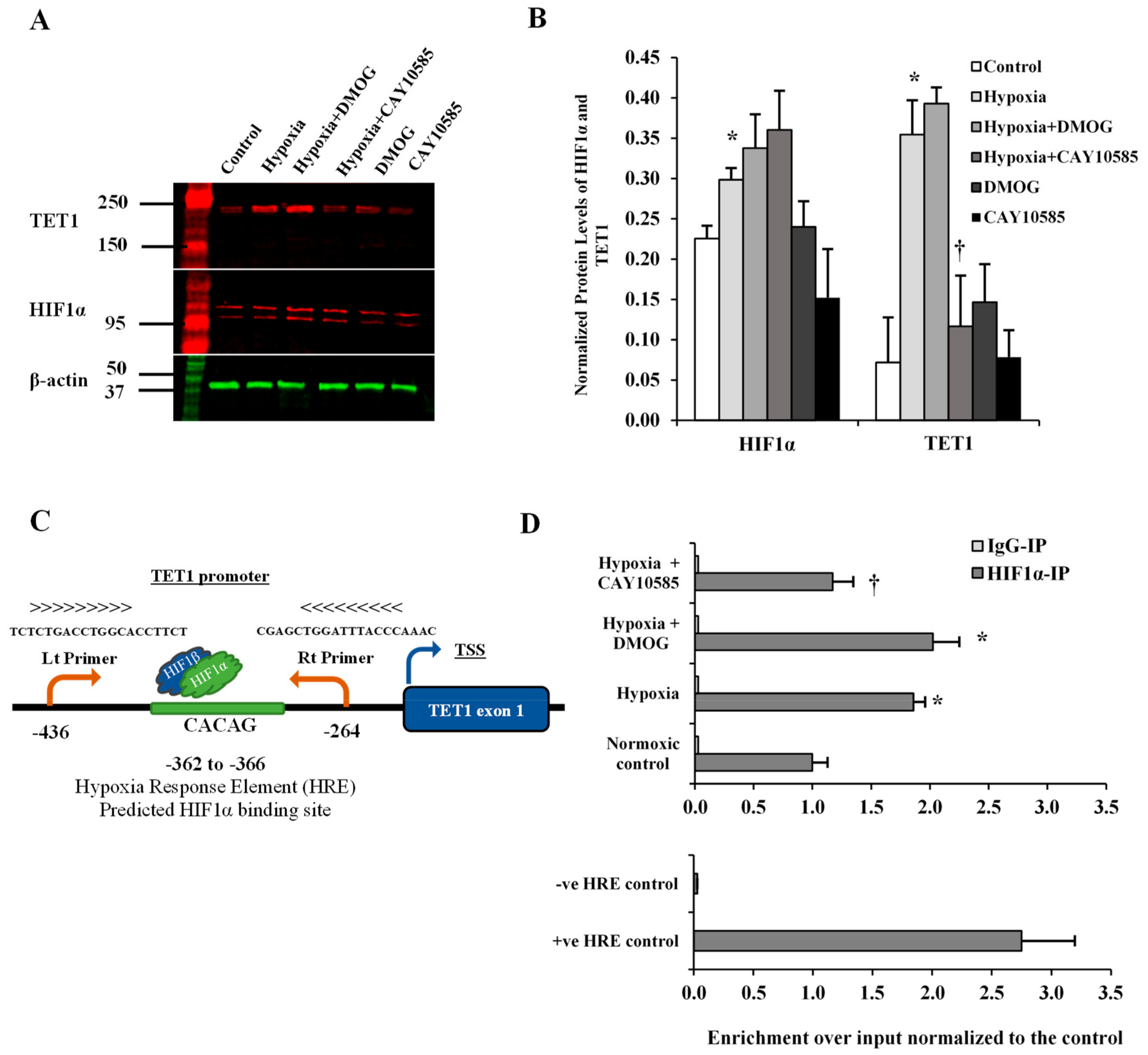
2.11. Chromatin Immunoprecipitation Assay (ChIP)
2.12. Statistical Analysis
3. Results
3.1. Description of Experimental Models
3.2. Effect of Hypoxia on TET1 Expression in Differentiated Adipocytes
3.3. Effect of Hypoxia and HIF1α/TET1 Pathway on Global DNA Methylation and Hydroxymethylation in Differentiated Adipocytes
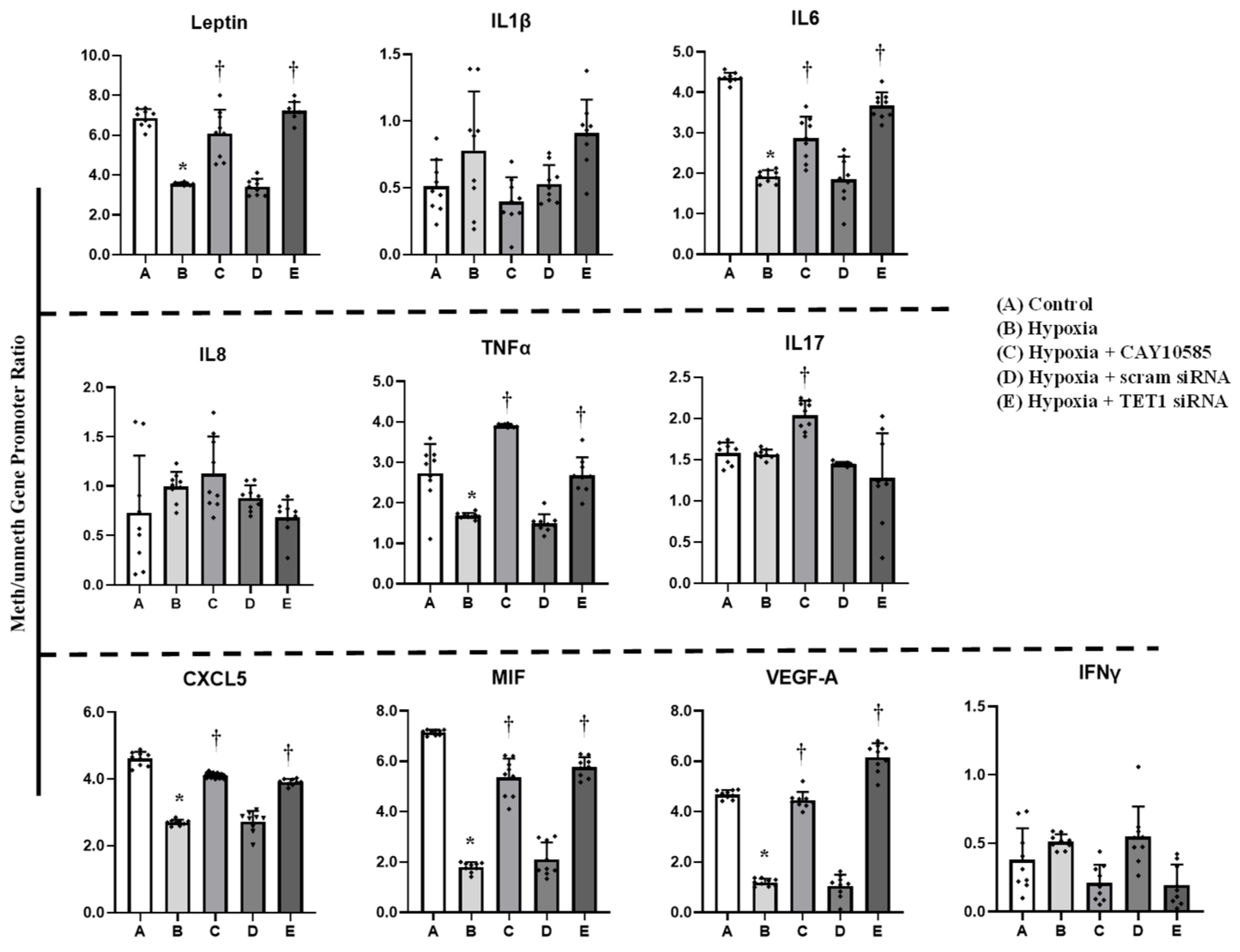
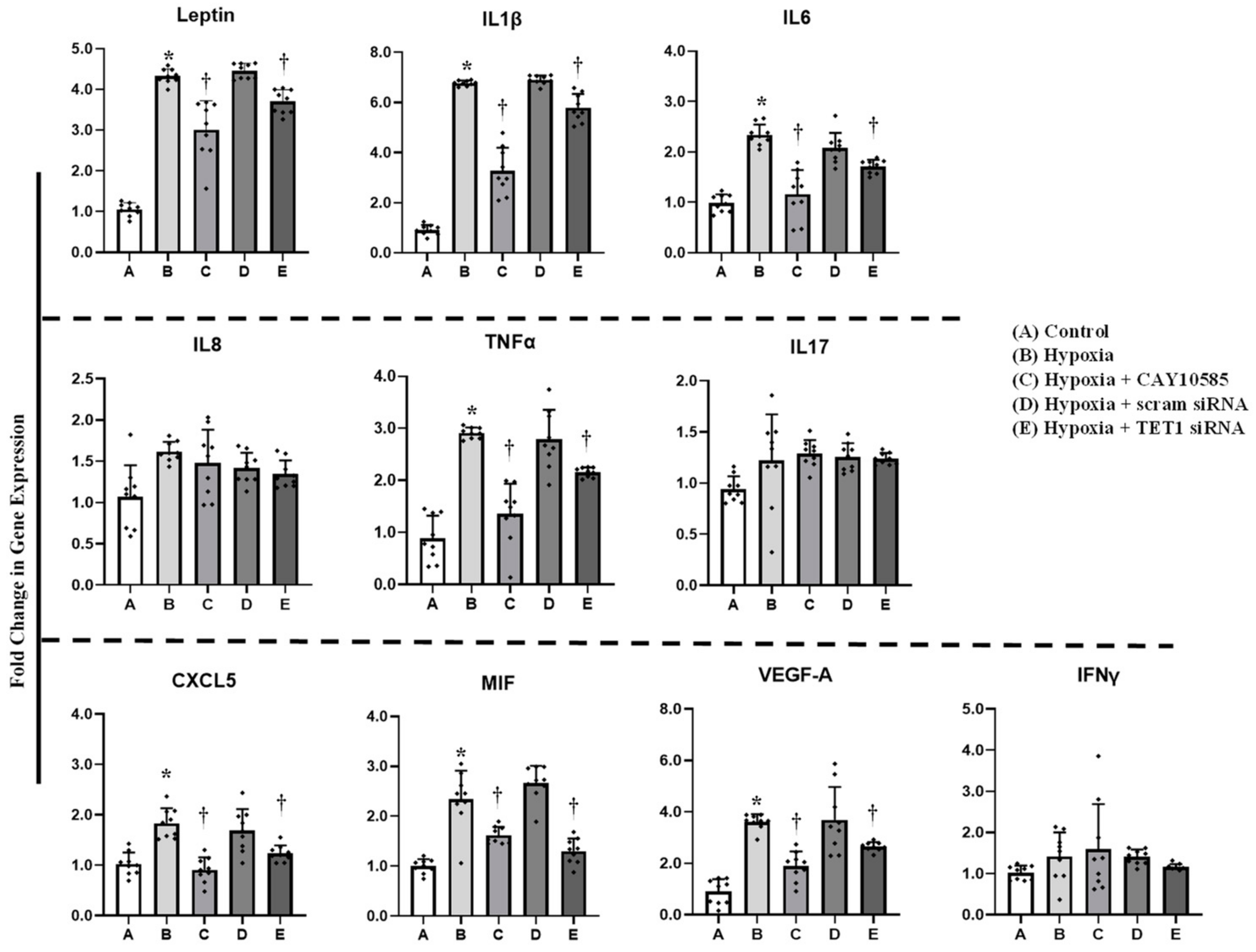
3.4. Effect of Hypoxia and HIF1α/TET1 Pathway on Adipocytokine Gene Expression and Promoter Methylation in Differentiated Adipocytes
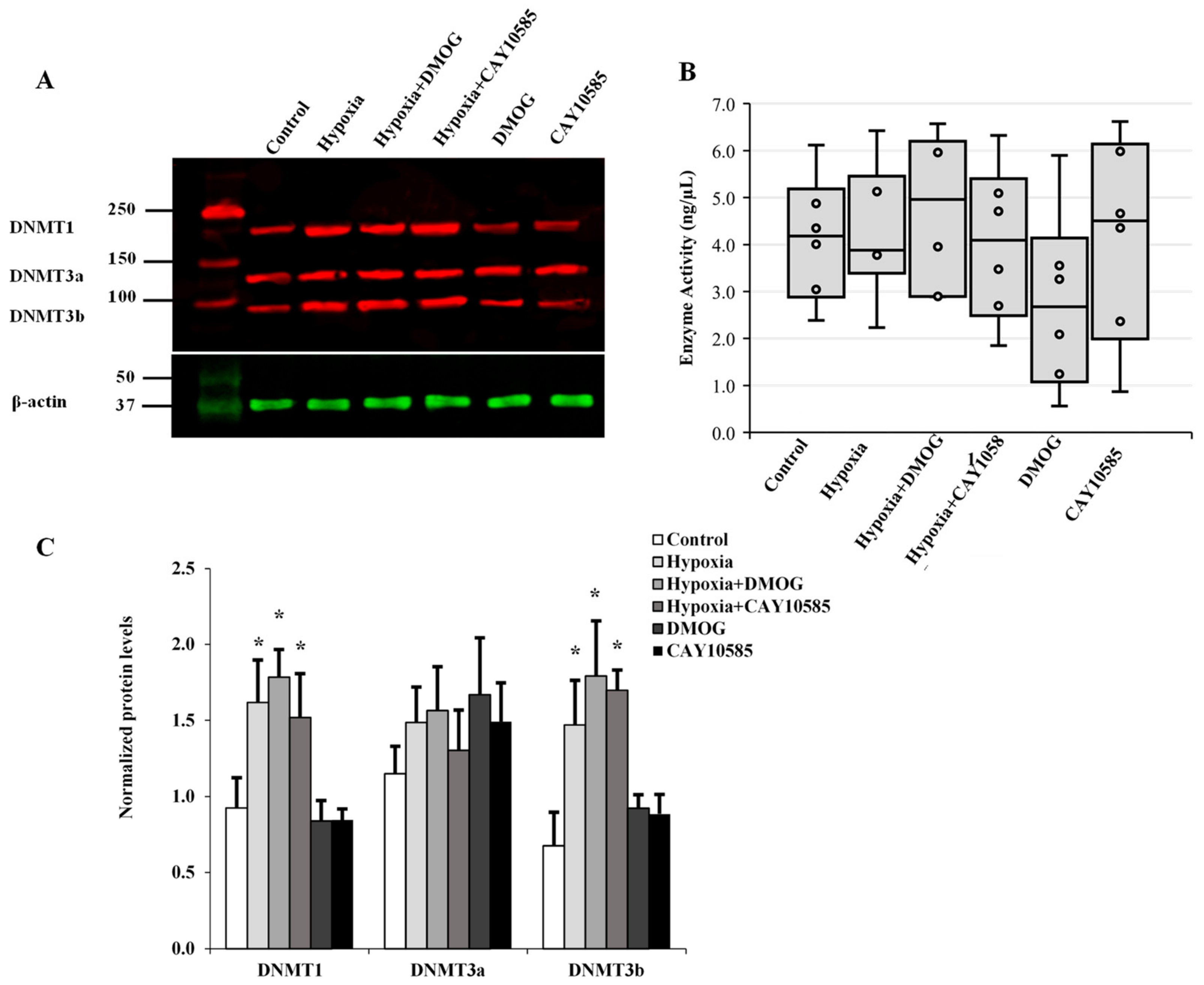
3.5. Evaluation of the Role of DNMT in Hypoxia-Induced Adipocytokine Hypomethylation
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Institute of Medicine. The Current State of Obesity Solutions in the United States: Workshop Summary; The National Academies Press: Washington, DC, USA, 2014; p. 94. [Google Scholar]
- Khaodhiar, L.; McCowen, K.C.; Blackburn, G.L. Obesity and its comorbid conditions. Clin. Cornerstone 1999, 2, 17–31. [Google Scholar] [CrossRef]
- Lian, X.; Gollasch, M. A Clinical Perspective: Contribution of Dysfunctional Perivascular Adipose Tissue (PVAT) to Cardiovascular Risk. Curr. Hypertens Rep. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Epigenetic changes by DNA methylation in chronic and intermittent hypoxia. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1096–L1100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, Y.; Jiang, S.; Liu, Y.; Huang, L.; Zhang, T.; Lu, G.; Gong, K.; Ji, X.; Shao, G. The effect of hypoxia preconditioning on DNA methyltransferase and PP1gamma in hippocampus of hypoxia preconditioned mice. High. Alt. Med. Biol. 2014, 15, 483–490. [Google Scholar] [CrossRef]
- Hu, X.Q.; Chen, M.; Dasgupta, C.; Xiao, D.; Huang, X.; Yang, S.; Zhang, L. Chronic hypoxia upregulates DNA methyltransferase and represses large conductance Ca2+-activated K+ channel function in ovine uterine arteries. Biol. Reprod. 2017, 96, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA demethylation through the activation of HIF-1alpha and transcriptional upregulation of MAT2A in hepatoma cells. Mol. Cancer 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Alivand, M.R.; Soheili, Z.S.; Pornour, M.; Solali, S.; Sabouni, F. Novel Epigenetic Controlling of Hypoxia Pathway Related to Overexpression and Promoter Hypomethylation of TET1 and TET2 in RPE Cells. J. Cell Biochem. 2017, 118, 3193–3204. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- English, A.; Jones, E.A.; Corscadden, D.; Henshaw, K.; Chapman, T.; Emery, P.; McGonagle, D. A comparative assessment of cartilage and joint fat pad as a potential source of cells for autologous therapy development in knee osteoarthritis. Rheumatology 2007, 46, 1676–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Zoll, S.; Sur, S.; van den Boom, D. A new method for accurate assessment of DNA quality after bisulfite treatment. Nucleic Acids Res. 2007, 35, e29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.L.; Shen, G.M.; Liu, X.L.; Wang, F.; Zhao, Y.Z.; Zhang, J.W. Hypoxia-inducible factor 1-mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions. J. Cell Mol. Med. 2012, 16, 1889–1899. [Google Scholar] [CrossRef]
- Neubauer, M.; Fischbach, C.; Bauer-Kreisel, P.; Lieb, E.; Hacker, M.; Tessmar, J.; Schulz, M.B.; Goepferich, A.; Blunk, T. Basic fibroblast growth factor enhances PPARgamma ligand-induced adipogenesis of mesenchymal stem cells. FEBS Lett. 2004, 577, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Kraus, W.L. Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Befani, C.; Liakos, P. Hypoxia upregulates integrin gene expression in microvascular endothelial cells and promotes their migration and capillary-like tube formation. Cell Biol. Int. 2017, 41, 769–778. [Google Scholar] [CrossRef]
- Lee, K.; Lee, J.H.; Boovanahalli, S.K.; Jin, Y.; Lee, M.; Jin, X.; Kim, J.H.; Hong, Y.S.; Lee, J.J. (Aryloxyacetylamino)benzoic acid analogues: A new class of hypoxia-inducible factor-1 inhibitors. J. Med. Chem. 2007, 50, 1675–1684. [Google Scholar] [CrossRef]
- Mariani, C.J.; Vasanthakumar, A.; Madzo, J.; Yesilkanal, A.; Bhagat, T.; Yu, Y.; Bhattacharyya, S.; Wenger, R.H.; Cohn, S.L.; Nanduri, J.; et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014, 7, 1343–1352. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Sun, W.; Yang, Z.; Guo, J.; Liu, H.; Liang, J. Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol. Lett. 2017, 14, 6457–6462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.; Park, J.H.; Weigel, C.; Liesenfeld, D.B.; Weichenhan, D.; Plass, C.; Seo, D.G.; Lindroth, A.M.; Park, Y.J. TET-mediated hydroxymethylcytosine at the Ppargamma locus is required for initiation of adipogenic differentiation. Int. J. Obes. (Lond) 2017, 41, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Landecho, M.F.; Tuero, C.; Valenti, V.; Bilbao, I.; de la Higuera, M.; Fruhbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef] [Green Version]
- Bullo, M.; Casas-Agustench, P.; Amigo-Correig, P.; Aranceta, J.; Salas-Salvado, J. Inflammation, obesity and comorbidities: The role of diet. Public Health Nutr. 2007, 10, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Unamuno, X.; Gomez-Ambrosi, J.; Rodriguez, A.; Becerril, S.; Fruhbeck, G.; Catalan, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur J. Clin Invest. 2018, 48, e12997. [Google Scholar] [CrossRef] [Green Version]
- Fujisaka, S.; Usui, I.; Ikutani, M.; Aminuddin, A.; Takikawa, A.; Tsuneyama, K.; Mahmood, A.; Goda, N.; Nagai, Y.; Takatsu, K.; et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1alpha-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 2013, 56, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Yun, Z. The Hypoxia-Inducible Factor Pathway in Adipocytes: The Role of HIF-2 in Adipose Inflammation and Hypertrophic Cardiomyopathy. Front. Endocrinol. (Lausanne) 2015, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabon, B.; Nagele, A.; Reddy, D.; Eagon, C.; Fleshman, J.W.; Sessler, D.I.; Kurz, A. Obesity decreases perioperative tissue oxygenation. Anesthesiology 2004, 100, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Mo, Y.; Ebenezer, D.; Bhattacharyya, S.; Liu, H.; Sundaravel, S.; Giricz, O.; Wontakal, S.; Cartier, J.; Caces, B.; et al. High resolution methylome analysis reveals widespread functional hypomethylation during adult human erythropoiesis. J. Biol. Chem. 2013, 288, 8805–8814. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wang, K.; Li, T.; Chen, J.; Xie, D.; Chang, X.; Yao, J.; Wu, J.; Zhou, Q.; Jia, Y.; et al. Hypoxia-induced TET1 facilitates trophoblast cell migration and invasion through HIF1alpha signaling pathway. Sci. Rep. 2017, 7, 8077. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.Z.; Chen, S.F.; Nieh, S.; Benner, C.; Ger, L.P.; Jan, C.I.; Ma, L.; Chen, C.H.; Hishida, T.; Chang, H.T.; et al. Hypoxia Drives Breast Tumor Malignancy through a TET-TNFalpha-p38-MAPK Signaling Axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef] [Green Version]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.P.; Chen, H.F.; Chen, S.Y.; Cheng, W.C.; Wang, H.W.; Shen, Z.J.; Song, C.; Teng, S.C.; He, C.; Wu, K.J. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome. Biol. 2014, 15, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiol. (Bethesda) 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharm. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh, D.Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Grosfeld, A.; Andre, J.; Hauguel-De Mouzon, S.; Berra, E.; Pouyssegur, J.; Guerre-Millo, M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J. Biol. Chem. 2002, 277, 42953–42957. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.J.; Chung, H.S.; Lee, B.R.; Kim, S.J.; Yoo, S.J.; Hong, S.H.; Kim, H.M. Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem. Biophys Res. Commun. 2003, 306, 805–811. [Google Scholar] [CrossRef]
- Budda, S.A.; Girton, A.; Henderson, J.G.; Zenewicz, L.A. Transcription Factor HIF-1alpha Controls Expression of the Cytokine IL-22 in CD4 T Cells. J. Immunol 2016, 197, 2646–2652. [Google Scholar] [CrossRef]
- Sun, F.; Abreu-Rodriguez, I.; Ye, S.; Gay, S.; Distler, O.; Neidhart, M.; Karouzakis, E. TET1 is an important transcriptional activator of TNFalpha expression in macrophages. PLoS ONE 2019, 14, e0218551. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Sinha, I.; Gao, H.; Brodin, D.; Thorell, A.; Ryden, M.; Andersson, D.P.; Henriksson, J.; Perfilyev, A.; Ling, C.; et al. The fat cell epigenetic signature in post-obese women is characterized by global hypomethylation and differential DNA methylation of adipogenesis genes. Int J. Obes. (Lond) 2015, 39, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Kvaloy, K.; Page, C.M.; Holmen, T.L. Epigenome-wide methylation differences in a group of lean and obese women—A HUNT Study. Sci. Rep. 2018, 8, 16330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, J.; Oliver, V.F.; Wang, G.; Zhu, H.; Zack, D.J.; Merbs, S.L.; Qian, J. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genom. 2015, 16, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018. [Google Scholar] [CrossRef] [Green Version]
- Van der Wijst, M.G.; Venkiteswaran, M.; Chen, H.; Xu, G.L.; Plosch, T.; Rots, M.G. Local chromatin microenvironment determines DNMT activity: From DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 2015, 10, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, M.; Hermann, A.; Pradhan, S.; Jeltsch, A. The activity of the murine DNA methyltransferase Dnmt1 is controlled by interaction of the catalytic domain with the N-terminal part of the enzyme leading to an allosteric activation of the enzyme after binding to methylated DNA. J. Mol Biol 2001, 309, 1189–1199. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. Allosteric control of mammalian DNA methyltransferases—A new regulatory paradigm. Nucleic Acids Res. 2016, 44, 8556–8575. [Google Scholar] [CrossRef] [Green Version]
- Dhe-Paganon, S.; Syeda, F.; Park, L. DNA methyl transferase 1: Regulatory mechanisms and implications in health and disease. Int. J. Biochem. Mol. Biol. 2011, 2, 58–66. [Google Scholar]
- Kint, S.; De Spiegelaere, W.; De Kesel, J.; Vandekerckhove, L.; Van Criekinge, W. Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR. PLoS ONE 2018, 13, e0199091. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | RefSeq Accession Number | Primer | Amplicon Size | Tm | Primer Sequence (5′-3′) Forward |
|---|---|---|---|---|---|
| leptin | NM_000230.3 | Fw | 150 | 60.0 | GGCTTTGGCCCTATCTTTTC |
| Rv | 60.0 | CCAAACCGGTGACTTTCTGT | |||
| IL1β | NM_000576.3 | Fw | 205 | 60.0 | GGGCCTCAAGGAAAAGAATC |
| Rv | 60.0 | TTCTGCTTGAGAGGTGCTGA | |||
| IL6 | NM_000600.5 | Fw | 180 | 59.8 | AGGAGACTTGCCTGGTGAAA |
| Rv | 59.8 | CAGGGGTGGTTATTGCATCT | |||
| IL8 | NM_000584.4 | Fw | 227 | 60.0 | TAGCAAAATTGAGGCCAAGG |
| Rv | 60.0 | AAACCAAGGCACAGTGGAAC | |||
| IL17 | NM_002190.3 | Fw | 216 | 60.0 | CCCCAGTTGATTGGAAGAAA |
| Rv | 60.0 | GAGGACCTTTTGGGATTGGT | |||
| CXCL5 | NM_002994.5 | Fw | 218 | 60.0 | GACGGTGGAAACAAGGAAAA |
| Rv | 59.9 | GCTTAAGCGGCAAACATAGG | |||
| MIF | NM_002415.2 | Fw | 234 | 59.6 | AGAACCGCTCCTACAGCAAG |
| Rv | 59.8 | ATTTCTCCCCACCAGAAGGT | |||
| VEGF | NM_001025366.3 | Fw | 186 | 60.0 | CCCACTGAGGAGTCCAACAT |
| Rv | 60.0 | TTTCTTGCGCTTTCGTTTTT | |||
| TNF-α | NM_000594.4 | Fw | 173 | 60.1 | TCCTTCAGACACCCTCAACC |
| Rv | 60.0 | AGGCCCCAGTTTGAATTCTT | |||
| IFNɣ | NM_000619.3 | Fw | 198 | 59.6 | TCCCATGGGTTGTGTGTTTA |
| Rv | 59.7 | AAGCACCAGGCATGAAATCT | |||
| TET1 | NM_030625.3 | Fw | 173 | 59.8 | AGGTCCAGGGCCAAATAACT |
| Rv | 59.9 | AGAAGGTGCCAGGTCAGAGA | |||
| β-actin | NG_007992.1 | Fw | 209 | 59.9 | AGAAAATCTGGCACCACACC |
| Rv | 59.9 | AACGGCAGAAGAGAGAACCA | |||
| β-actin | NG_007992.1 | Fw | 352 | 60.0 | AAACTGGAACGGTGAAGGTG |
| Rv | 59.9 | CTCAAGTTGGGGGACAAAAA | |||
| β-actin | NG_007992.1 | Fw | 479 | 59.9 | AAGATGACCCAGGTGAGTGG |
| Rv | 59.9 | GGGGTGTTGAAGGTCTCAAA | |||
| β-actin | NG_007992.1 | Fw | 638 | 59.9 | CTCTTCCAGCCTTCCTTCCT |
| Rv | 60.0 | AAAGCCATGCCAATCTCATC | |||
| β-actin | NG_007992.1 | Fw | 718 | 59.0 | CTCTTCCAGCCTTCCTTCCT |
| Rv | 60.0 | CACCTTCACCGTTCCAGTTT | |||
| FABP4 | NM_001442.3 | Fw | 181 | 60.0 | TACTGGGCCAGGAATTTGAC |
| Rv | 60.0 | GTGGAAGTGACGCCTTTCAT | |||
| AdipoQ | NM_001177800.2 | Fw | 173 | 60.0 | CCTAAGGGAGACATCGGTGA |
| Rv | 60.0 | GTAAAGCGAATGGGCATGTT | |||
| PDK1 (+ve) | NC_000002.12 | Fw | 100 | 62.0 | CGCCCTGTCCTTGAGCC |
| Rv | 62.9 | CGGTATGGAGCGTCCCCT | |||
| PDK1 (-ve) | NC_000002.12 | Fw | 486 | 56.5 | CTGATAAGACTACACTGGACG3 |
| Rv | 56.6 | CCCAAGATACTACACTACCATC |
| Gene Name | RefSeq Accession Number | Primer | Amplicon Size | Tm | GC% | Primer Sequence (5′-3′) Forward |
|---|---|---|---|---|---|---|
| leptin | NG_007450.1 | Fw M | 111 | 59.6 | 72.73 | TAGGATTAACGAGGGCGTAGTC |
| Rv M | 58.7 | 62.50 | AACCCCTTAAAAAAATACTTCGAA | |||
| Fw U | 113 | 57.4 | 72.00 | TTAGGATTAATGAGGGTGTAGTTGT | ||
| Rv U | 59.0 | 64.00 | CAACCCCTTAAAAAAATACTTCAAA | |||
| IL1β | NG_008851.1 | Fw M | 146 | 59.3 | 52.00 | TAATTTTAATATTTTGGGAGGTCGA |
| Rv M | 58.4 | 68.00 | CAACTAAAACTACAAACACCTACCG | |||
| Fw U | 144 | 57.4 | 52.00 | TAATTTTAATATTTTGGGAGGTTGA | ||
| Rv U | 57.0 | 72.00 | ACTAAAACTACAAACACCTACCACC | |||
| IL6 | NG_011640 | Fw M | 223 | 59.8 | 65.22 | GACGGATTATAGTGTACGGTTGC |
| Rv M | 59.2 | 52.17 | ATAAAATCATCCATTCTTCACCG | |||
| Fw U | 223 | 58.6 | 68.00 | GATGGATTATAGTGTATGGTTGTGG | ||
| Rv U | 59.1 | 48.00 | ATAAAATCATCCATTCTTCACCAAT | |||
| IL8 | NG_029889.1 | Fw M | 146 | 59.1 | 60.00 | TTTTTGAGTAGTTGGGATTATAGGC |
| Rv M | 59.1 | 69.57 | ACACTTTAAAAATCCGAAACGAA | |||
| Fw U | 149 | 57.9 | 61.54 | TTTTTTGAGTAGTTGGGATTATAGGT | ||
| Rv U | 58.7 | 68.00 | CAACACTTTAAAAATCCAAAACAAA | |||
| IL17 | NG_033021.1 | Fw M | 138 | 59.1 | 54.55 | TTTTTATGATTTTATTGGGGGC |
| Rv M | 55.3 | 44.00 | ATAAACAAAATATAACGCTATCGTC | |||
| Fw U | 142 | 59.3 | 48.00 | ATTTTTTTATGATTTTATTGGGGGT | ||
| Rv U | 50.5 | 46.15 | AATAAACAAAATATAACACTATCATC | |||
| CXCL5 | NC_000004.12 | Fw M | 186 | 56.9 | 56.00 | TAATTTTCGTTTTTTTAATTTTCGT |
| Rv M | 58.8 | 76.00 | GCTAACGATAAACCCTAACTACGTC | |||
| Fw U | 188 | 54.5 | 57.69 | TTAATTTTTGTTTTTTTAATTTTTGT | ||
| Rv U | 54.8 | 76.92 | CACTAACAATAAACCCTAACTACATC | |||
| MIF | NG_012099.1 | Fw M | 136 | 56.7 | 76.00 | GGTTTATCGTCGTATTTTTATTTTC |
| Rv M | 57.0 | 60.87 | CAACCTATTCTCCACTTAACGAC | |||
| Fw U | 137 | 54.9 | 76.92 | GTTTATTGTTGTATTTTTATTTTTGG | ||
| Rv U | 59.8 | 60.00 | TCCAACCTATTCTCCACTTAACAAC | |||
| VEGF | NG_008732.1 | Fw M | 236 | 58.8 | 66.67 | TAGTTAGAGTCGGGGTGTGTAGAC |
| Rv M | 59.1 | 76.19 | GAAAAACCGAACAAAAACGAA | |||
| Fw U | 241 | 58.7 | 68.00 | TAGTTAGAGTTGGGGTGTGTAGATG | ||
| Rv U | 59.0 | 70.83 | AAAACAAAAAACCAAACAAAAACA | |||
| TNF-α | NG_007462.1 | Fw M | 151 | 59.3 | 56.00 | GAGTATTGAAAGTATGATTCGGGAC |
| Rv M | 59.1 | 68.00 | CAACAAACAAAAAAACGTAATAACG | |||
| Fw U | 147 | 57.5 | 56.00 | GTATTGAAAGTATGATTTGGGATGT | ||
| Rv U | 55.7 | 72.00 | ACAAACAAAAAAACATAATAACACC | |||
| IFNγ | NG_015840.1 | Fw M | 194 | 55.0 | 46.15 | TTTTGATTAATATAGTGAAATTTCGT |
| Rv M | 58.6 | 68.00 | TCACCCAAACTAAAATACAATAACG | |||
| Fw U | 189 | 50.1 | 45.83 | TTGATTAATATAGTGAAATTTTGT | ||
| Rv U | 55.3 | 70.83 | CCCAAACTAAAATACAATAACACA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.M.; Phillips, S.A.; Mahmoud, A.M. HIF1?/TET1 Pathway Mediates Hypoxia-Induced Adipocytokine Promoter Hypomethylation in Human Adipocytes. Cells 2020, 9, 134. https://doi.org/10.3390/cells9010134
Ali MM, Phillips SA, Mahmoud AM. HIF1?/TET1 Pathway Mediates Hypoxia-Induced Adipocytokine Promoter Hypomethylation in Human Adipocytes. Cells. 2020; 9(1):134. https://doi.org/10.3390/cells9010134
Chicago/Turabian StyleAli, Mohamed M., Shane A. Phillips, and Abeer M. Mahmoud. 2020. "HIF1?/TET1 Pathway Mediates Hypoxia-Induced Adipocytokine Promoter Hypomethylation in Human Adipocytes" Cells 9, no. 1: 134. https://doi.org/10.3390/cells9010134
APA StyleAli, M. M., Phillips, S. A., & Mahmoud, A. M. (2020). HIF1?/TET1 Pathway Mediates Hypoxia-Induced Adipocytokine Promoter Hypomethylation in Human Adipocytes. Cells, 9(1), 134. https://doi.org/10.3390/cells9010134