Polyhexamethylene Guanidine Phosphate Damages Tight Junctions and the F-Actin Architecture by Activating Calpain-1 via the P2RX7/Ca2+ Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Immunofluorescence
2.5. Determination of Intracellular Ca2+ Level
2.6. Measurement of Calpain Activity
2.7. ATP Assay
2.8. Western Blotting
2.9. Statistical Analysis
3. Results
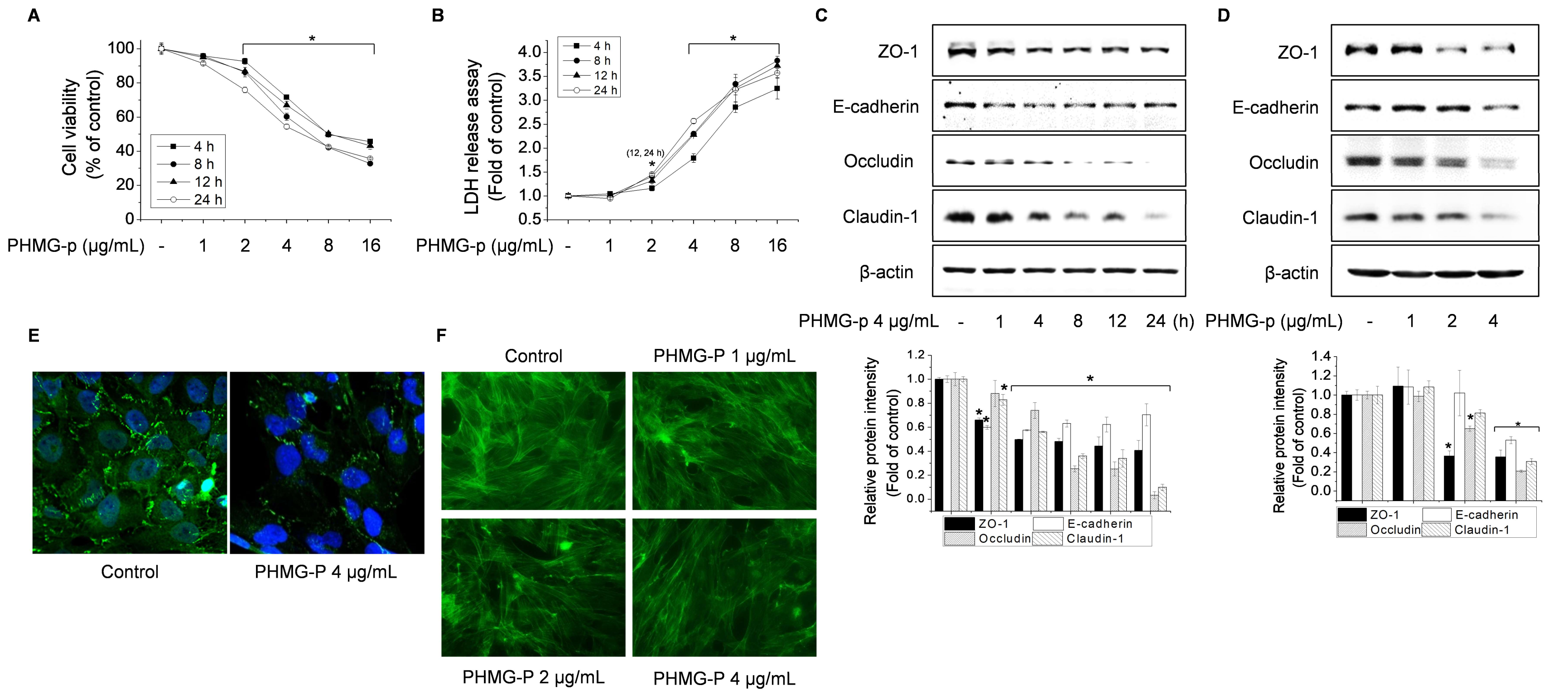
3.1. Effect of PHMG-p on TJ Proteins and the F-Actin Architecture in BEAS-2B Cells
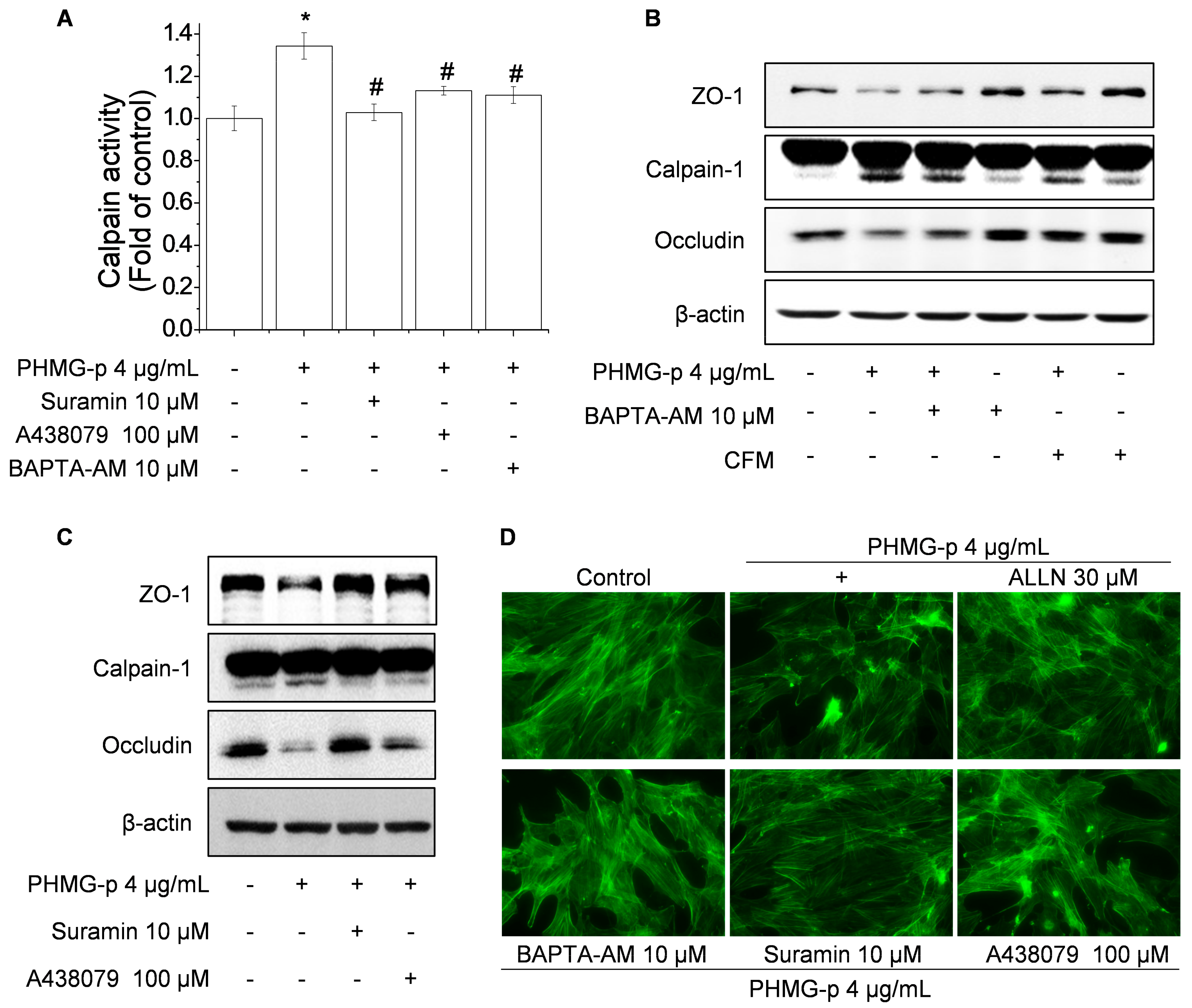
3.2. Role of Calpain in Impairment of TJ Proteins and F-Actin Architecture by PHMG-p
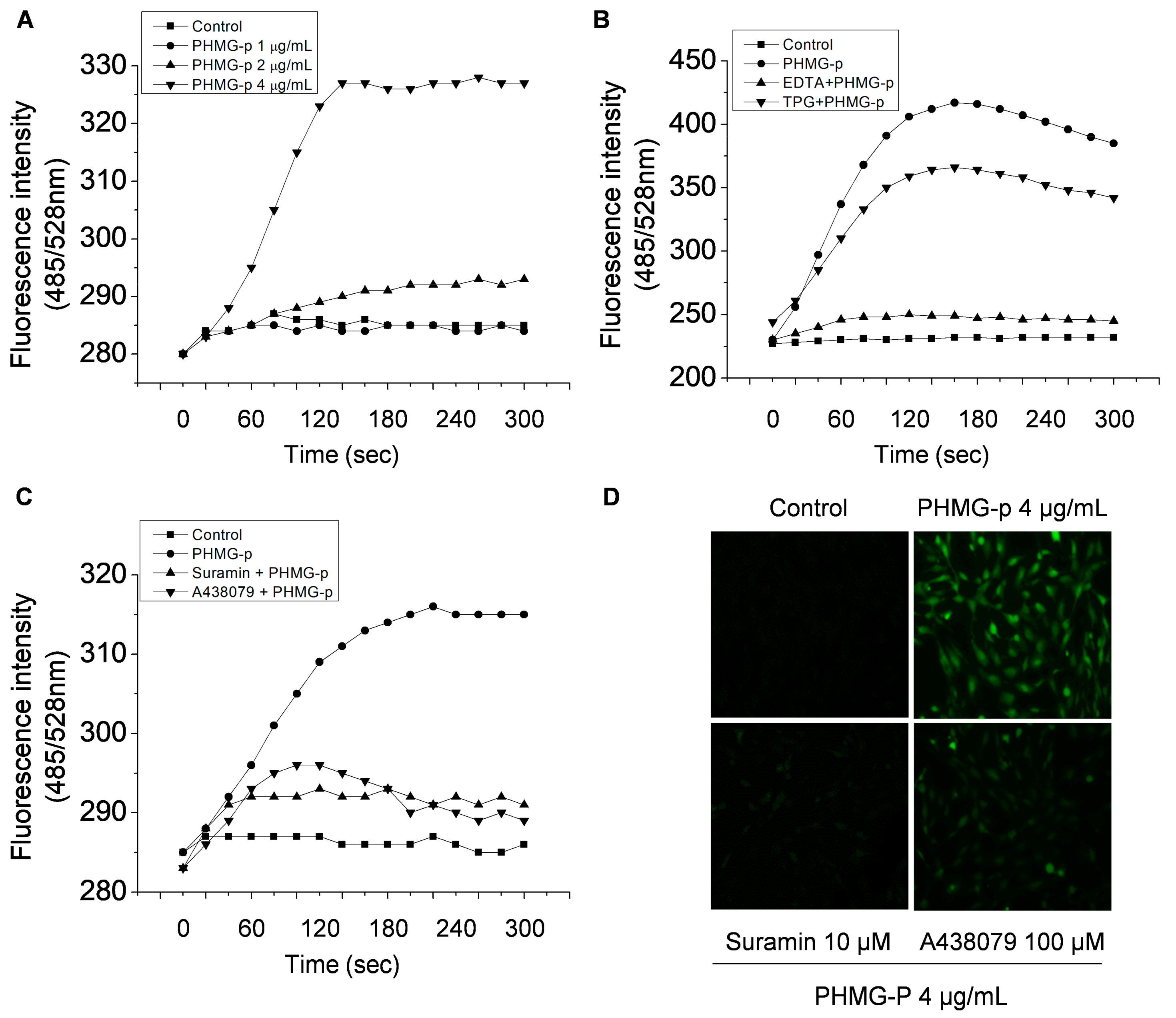
3.3. PHMG-p Induces Intracellular Ca2+ Influx via P2RX7
3.4. PHMG-p Induces Calpain-1 Activity and Calpain-1-Dependent Epithelial Barrier Dysfunction via P2RX7/Ca2+
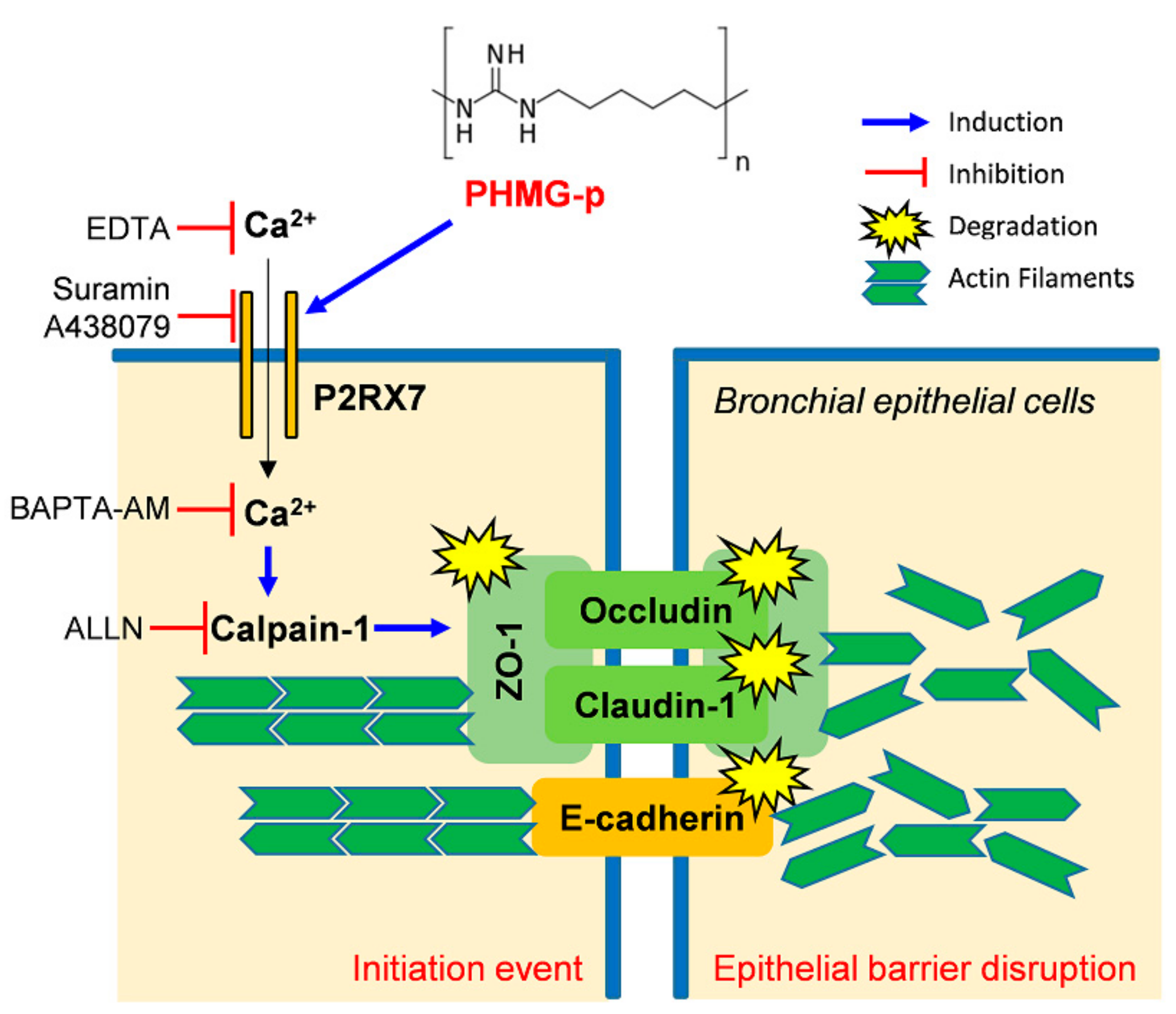
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AJs | adherens junctions |
| DMEM/F-12 | Dulbecco’s modified Eagle’s medium/Ham’s F-12 Nutrient Mixture |
| eATP | extracellular ATP |
| LDH | lactate dehydrogenase |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PHMG-p | polyhexamethylene guanidine phosphate |
| P2RX7 | P2X purinoreceptor 7 |
| TJs | tight junctions |
| ZO | zonula occludens |
References
- Wittekindt, O.H. Tight junctions in pulmonary epithelia during lung inflammation. Pflug. Arch. 2017, 469, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cromwell, O.; Hamid, Q.; Corrigan, C.J.; Barkans, J.; Meng, Q.; Collins, P.D.; Kay, A.B. Expression and generation of interleukin-8, IL-6 and granulocyte-macrophage colony-stimulating factor by bronchial epithelial cells and enhancement by IL-1 beta and tumour necrosis factor-alpha. Immunology 1992, 77, 330–337. [Google Scholar] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.F.; Weiland, J.E.; Pacht, E.R.; Gadek, J.E.; Davis, W.B. Protein permeability in the adult respiratory distress syndrome loss of size selectivity of the alveolar epithelium. J. Clin. Investig. 1986, 78, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional strands in tight junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokutta, S.; Weis, W.I. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu. Rev. Cell Dev. Biol. 2007, 23, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane proteins of tight junctions. Biochim. Biophys. Acta. 2008, 1778, 588–600. [Google Scholar] [CrossRef] [Green Version]
- Madara, J.L.; Moore, R.; Carlson, S. Alteration of intestinal tight junction structure and permeability by cytoskeletal contraction. Am. J. Physiol. 1987, 253, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I. Actin motors that drive formation and disassembly of epithelial apical junctions. Front. Biosci. 2008, 13, 6662–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Hwang, G.W.; Naganuma, A.; Chung, K.H. Adverse health effects of humidifier disinfectants in Korea: Lung toxicity of polyhexamethylene guanidine phosphate. J. Toxicol. Sci. 2016, 41, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Park, S.J.; Kim, S.; Lee, K.; Chang, J. Lung fibroblasts may play an important role in clearing apoptotic bodies of bronchial epithelial cells generated by exposure to PHMG-P-containing solution. Toxicol. Lett. 2018, 286, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jung, K.J.; Yoon, S.J.; Lee, K.; Kim, B. Polyhexamethyleneguanidine phosphate induces cytotoxicity through disruption of membrane integrity. Toxicology 2019, 414, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Misawa, Y.; Miyamoto, H.; Makino, M.; Nagai, K.; Shiraishi, T.; Nakagawa, Y.; Yamato, S.; Tachikawa, E.; Zenda, H. A comparative study of characteristics of current-type and conventional-type cationic bactericides. Biol. Pharm. Bull. 2001, 24, 1093–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oule, M.K.; Azinwi, R.; Bernier, A.M.; Kablan, T.; Maupertuis, A.M.; Mauler, S.; Nevry, R.K.; Dembele, K.; Forbes, L.; Diop, L. Polyhexamethylene guanidine hydrochloride-based disinfectant: A novel tool to fight meticillin-resistant Staphylococcus aureus and nosocomial infections. J. Med. Microbiol. 2008, 57, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Oule, M.K.; Quinn, K.; Dickman, M.; Bernier, A.M.; Rondeau, S.; DeMoissac, D.B.; Boisvert, A.; Diop, L. Akwaton, polyhexamethylene-guanidine hydrochloride-based sporicidal disinfectant: A novel tool to fight bacterial spores and nosocomial infections. J. Med. Microbiol. 2012, 61, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Broxton, P.; Woodcock, P.M.; Heatley, F.; Gilbert, P. Interaction of some polyhexamethylene biguanides and membrane phospholipids in Escherichia coli. J. Appl. Bacteriol. 1984, 57, 115–124. [Google Scholar] [CrossRef]
- Ikeda, T.; Ledwith, A.; Bamford, C.H.; Hann, R.A. Interaction of a polymeric biguanide biocide with phospholipid membranes. Biochim. Biophys. Acta 1984, 769, 57–66. [Google Scholar] [CrossRef]
- Ikeda, T.; Tazuke, S.; Watanabe, M. Interaction of biologically active molecules with phospholipid membranes. I. Fluorescence depolarization studies on the effect of polymeric biocide bearing biguanide groups in the main chain. Biochim. Biophys. Acta 1983, 735, 380–386. [Google Scholar] [CrossRef]
- Carmona-Ibeiro, A.M.; de Melo Carrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef] [Green Version]
- Sumitomo, T.; Nakata, M.; Higashino, M.; Jin, Y.; Terao, Y.; Fujinaga, Y.; Kawabata, S. Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J. Biol. Chem. 2011, 286, 2750–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part Fibre Toxicol. 2012, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A. New insights of P2X7 receptor signaling pathway in alveolar functions. J. Biomed. Sci. 2013, 20, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Jacobson, K.N.; McDermott, K.M.; Reddy, S.P.; Cress, A.E.; Tang, H.; Dudek, S.M.; Black, S.M.; Garcia, J.G.; Makino, A.; et al. Capsaicin-induced Ca2+ signaling is enhanced via upregulated TRPV1 channels in pulmonary artery smooth muscle cells from patients with idiopathic PAH. Am. J. Physiol. Cell Physiol. 2016, 312, 309–325. [Google Scholar] [CrossRef]
- Takenouchi, T.; Suzuki, S.; Shinkai, H.; Tsukimoto, M.; Sato, M.; Uenishi, H.; Kitani, H. Extracellular ATP does not induce P2X7 receptor-dependent responses in cultured renal- and liver-derived swine macrophages. Results Immunol. 2014, 4, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Schlingmann, B.; Molina, S.A.; Koval, M. Claudins: Gatekeepers of lung epithelial function. Semin. Cell Dev. Biol. 2015, 42, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Lee, K.; Park, C.W.; Song, J.A.; Shin, D.Y.; Park, Y.J.; Chung, K.H. Polyhexamethylene guanidine phosphate aerosol particles induce pulmonary inflammatory and fibrotic responses. Arch. Toxicol. 2016, 90, 617–632. [Google Scholar] [CrossRef]
- Nava, P.; Kamekura, R.; Nusrat, A. Cleavage of transmembrane junction proteins and their role in regulating epithelial homeostasis. Tissue Barriers 2013, 1, e24783. [Google Scholar] [CrossRef] [Green Version]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef] [Green Version]
- Heijink, I.H.; Brandenburg, S.M.; Postma, D.S.; van Oosterhout, A.J. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur. Respir. J. 2012, 39, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Novak, I. Purinergic signalling in the pancreas in health and disease. J. Endocrinol. 2012, 213, 123–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, D.; Natale, M.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. The role of P2X7 receptors in tissue fibrosis: A brief review. Purinergic Signal. 2015, 11, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Zhao, K.; Zhang, X.; Zhang, J.; Xu, B. ATP Induces Disruption of Tight Junction Proteins via IL-1 Beta-Dependent MMP-9 Activation of Human Blood-Brain Barrier In Vitro. Neural Plast. 2016, 2016, 8928530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesslau, K.P.; Stein, A.; Kasper, M.; Barth, K. P2X7 Receptor Indirectly Regulates the JAM-A Protein Content via Modulation of GSK-3β. Int. J. Mol. Sci. 2019, 20, 2298. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Kim, M.S.; Kim, S.H.; Joen, D.; Lee, K. Protective Effects of Nintedanib against Polyhexamethylene Guanidine Phosphate-Induced Lung Fibrosis in Mice. Molecules 2018, 23, 1974. [Google Scholar] [CrossRef] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Ferrari, D.; Pizzirani, C.; Gulinelli, S.; Callegari, G.; Chiozzi, P.; Idzko, M.; Panther, E.; Di Virgilio, F. Modulation of P2X7 receptor functions by polymyxin B: Crucial role of the hydrophobic tail of the antibiotic molecule. Br. J. Pharmacol. 2007, 150, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Elssner, A.; Duncan, M.; Gavrilin, M.; Wewers, M.D. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J. Immunol. 2004, 172, 4987–4994. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of microglia by amyloid {beta} requires P2X7 receptor expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef] [Green Version]
- Niemi, K.; Teirilä, L.; Lappalainen, J.; Rajamäki, K.; Baumann, M.H.; Öörni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef] [Green Version]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, S.W.; Lee, G.H.; Pham, H.T.; Choi, J.H.; Jeong, H.G. Polyhexamethylene Guanidine Phosphate Damages Tight Junctions and the F-Actin Architecture by Activating Calpain-1 via the P2RX7/Ca2+ Signaling Pathway. Cells 2020, 9, 59. https://doi.org/10.3390/cells9010059
Jin SW, Lee GH, Pham HT, Choi JH, Jeong HG. Polyhexamethylene Guanidine Phosphate Damages Tight Junctions and the F-Actin Architecture by Activating Calpain-1 via the P2RX7/Ca2+ Signaling Pathway. Cells. 2020; 9(1):59. https://doi.org/10.3390/cells9010059
Chicago/Turabian StyleJin, Sun Woo, Gi Ho Lee, Hoa Thi Pham, Jae Ho Choi, and Hye Gwang Jeong. 2020. "Polyhexamethylene Guanidine Phosphate Damages Tight Junctions and the F-Actin Architecture by Activating Calpain-1 via the P2RX7/Ca2+ Signaling Pathway" Cells 9, no. 1: 59. https://doi.org/10.3390/cells9010059
APA StyleJin, S. W., Lee, G. H., Pham, H. T., Choi, J. H., & Jeong, H. G. (2020). Polyhexamethylene Guanidine Phosphate Damages Tight Junctions and the F-Actin Architecture by Activating Calpain-1 via the P2RX7/Ca2+ Signaling Pathway. Cells, 9(1), 59. https://doi.org/10.3390/cells9010059