Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
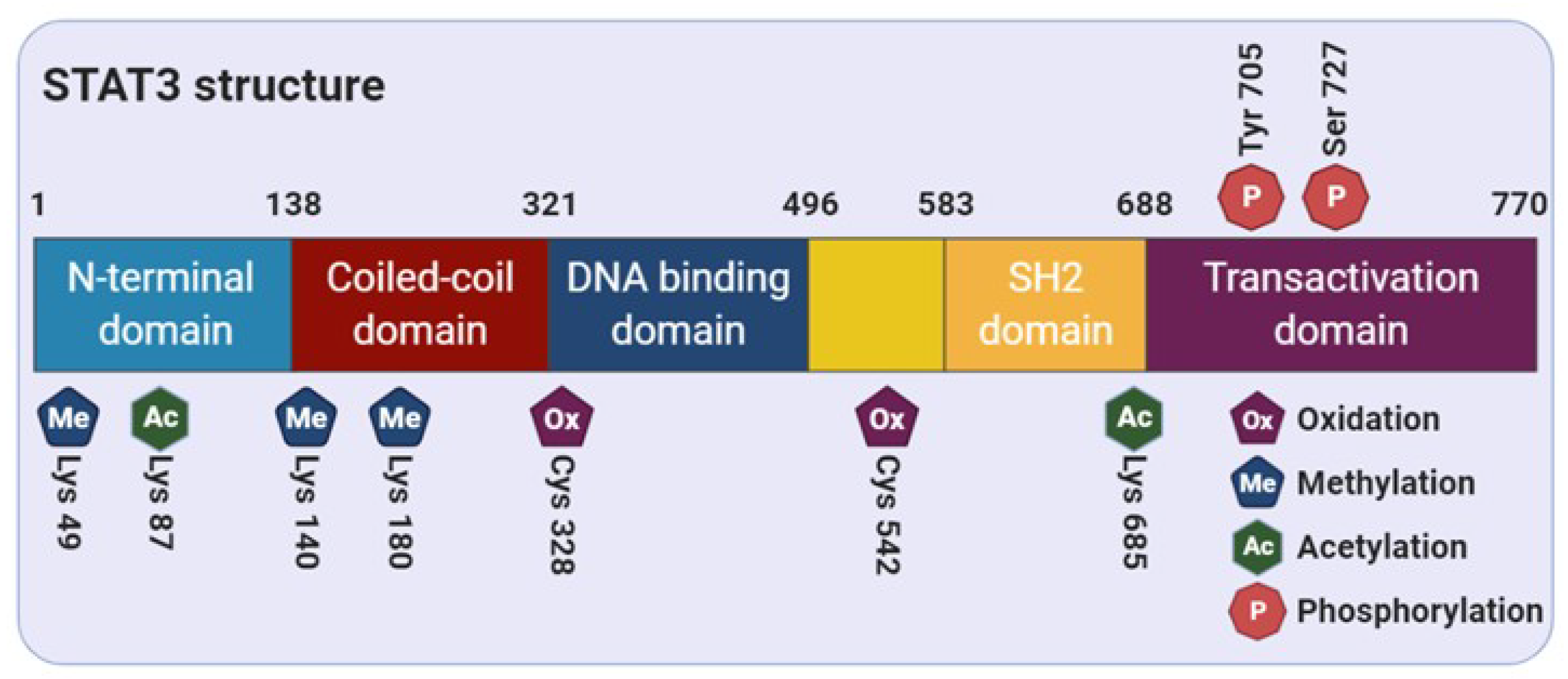
2. Structure of STAT3
3. Regulation of STAT3 Activity through Post-Translational Modifications
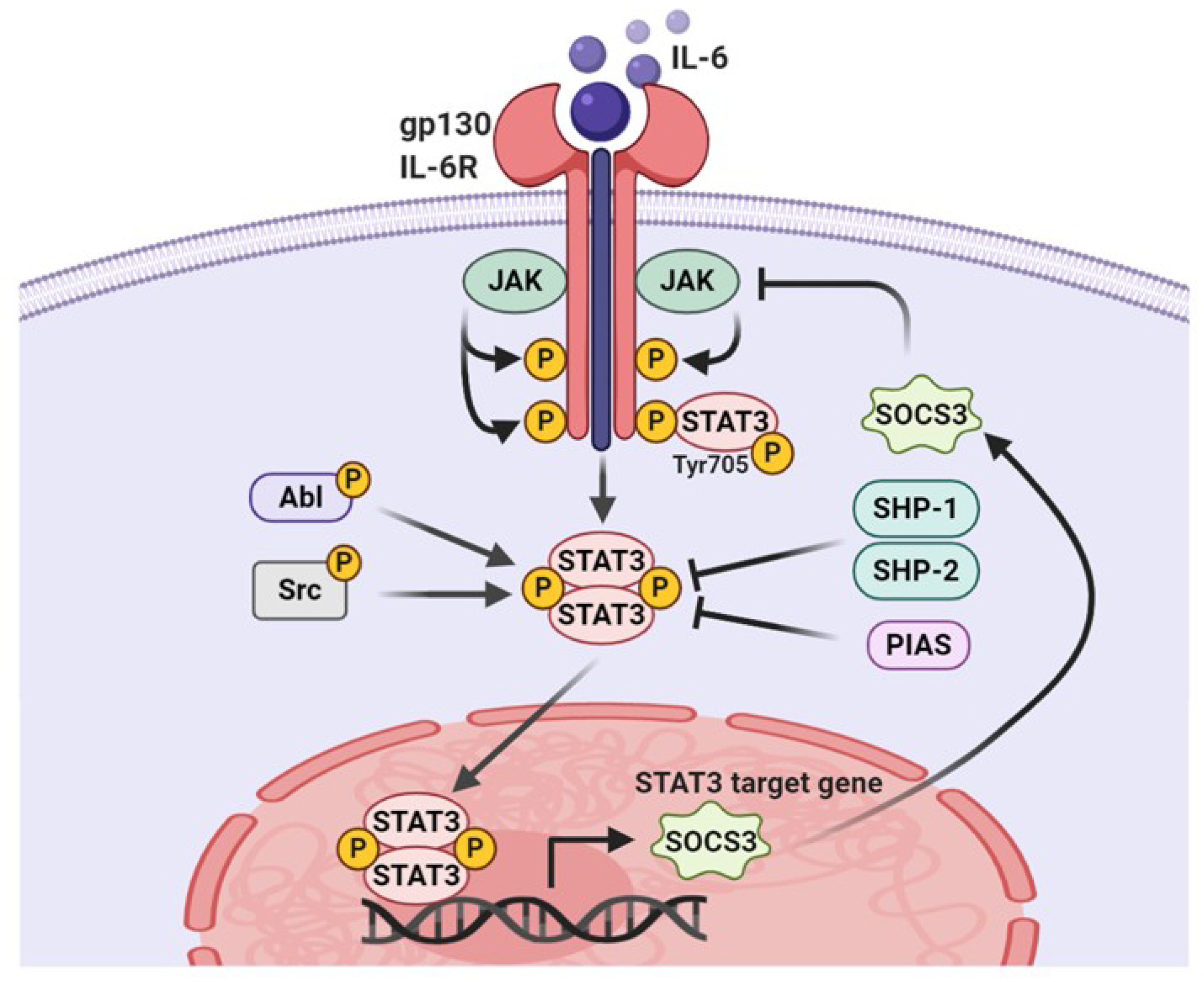
3.1. Tyrosine Phosphorylation
3.2. Serine Phosphorylation
3.3. Acetylation
3.4. Methylation
3.5. S-Glutathionylation
4. Redox Balance of STAT3 in Cellular Survival
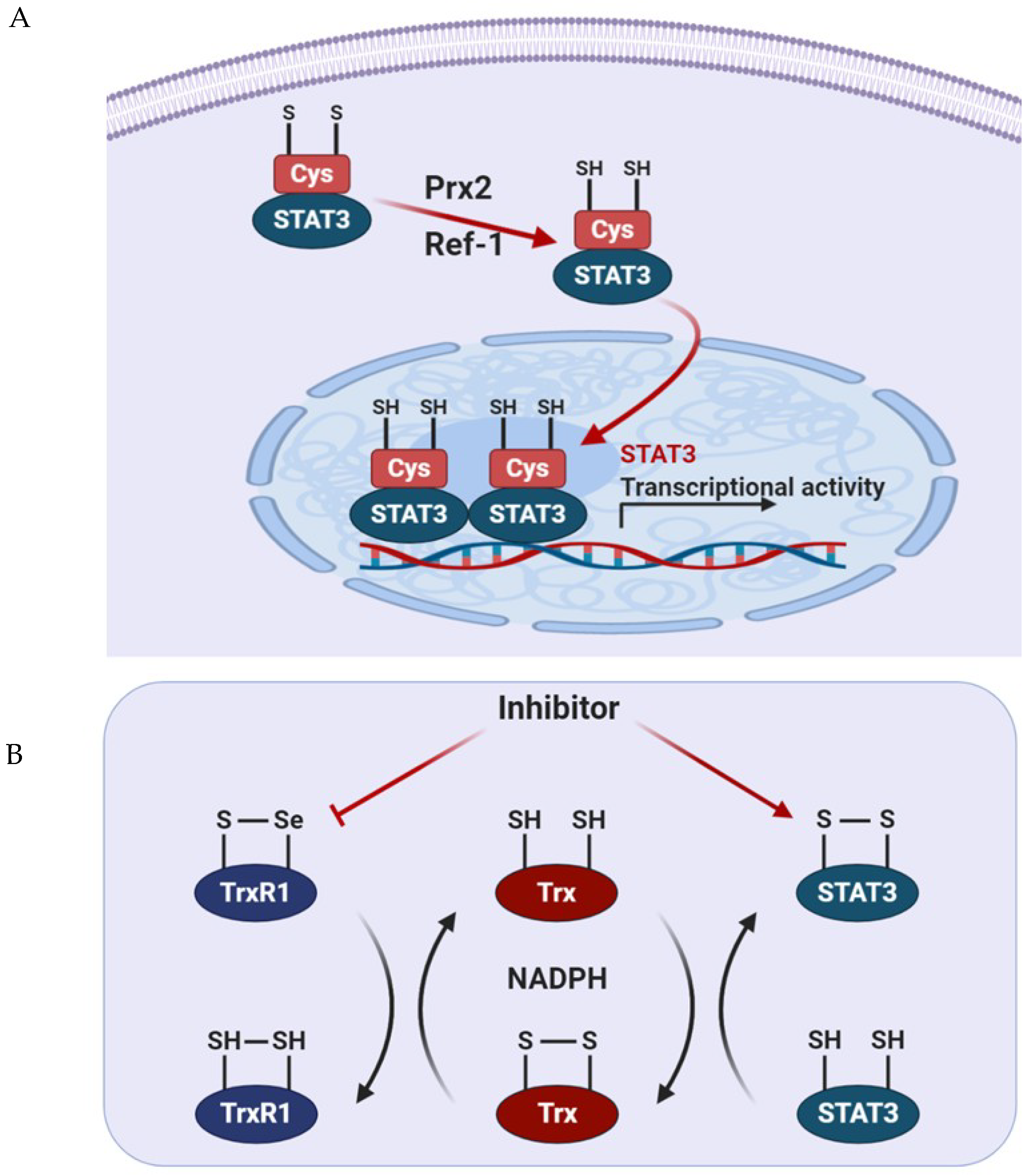
5. ROS Regulatory Molecules Involved in STAT3 Activity
5.1. Prx2
5.2. Trx1
5.3. Apurinic/Apyrimidinic Endonuclease 1/Redox Factor 1 (APE1/Ref-1)
6. Regulation of Metabolic Signaling Pathway through STAT3 in Cancer
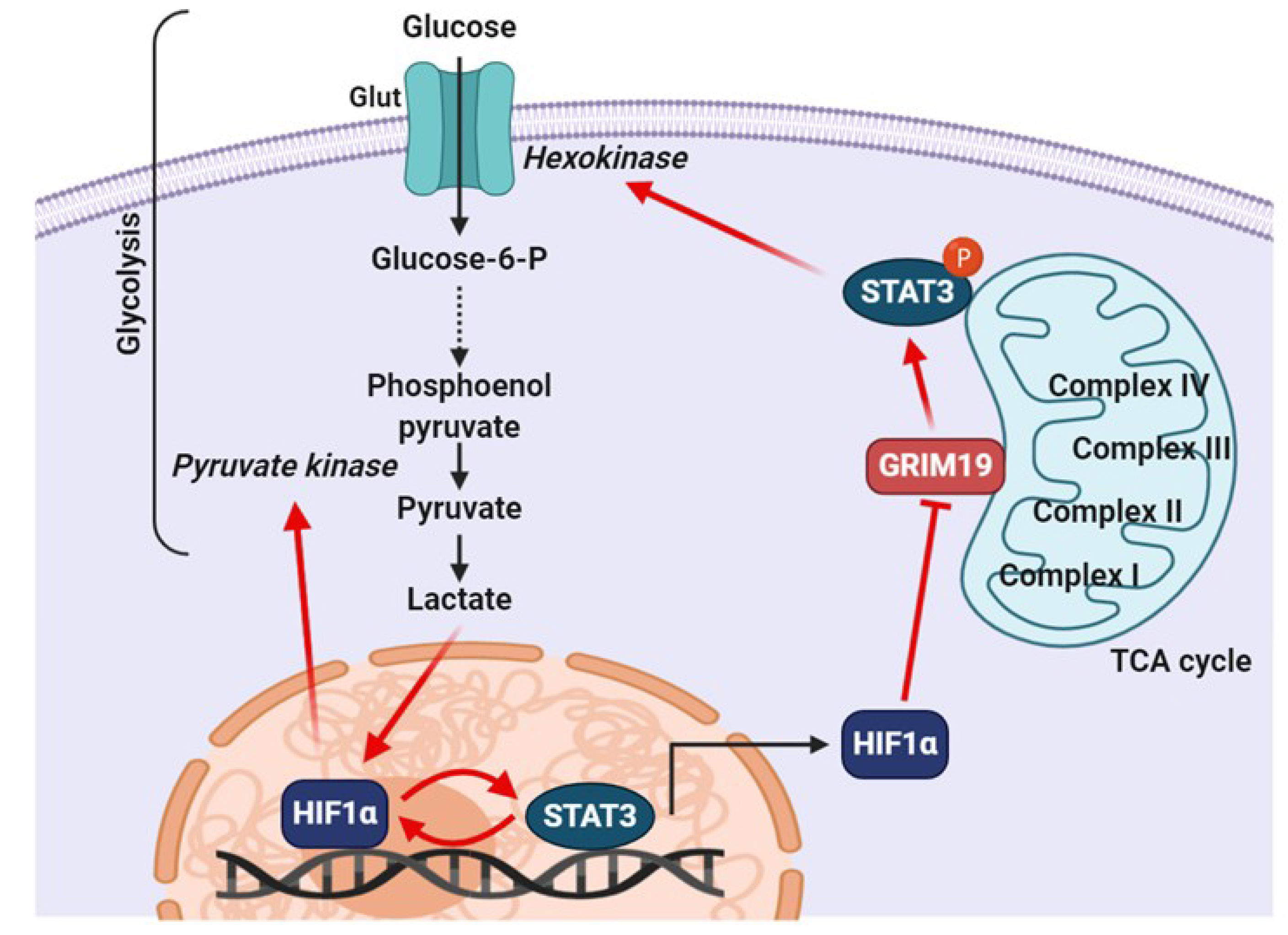
6.1. Relationship between STAT3 and HIF-1α under Hypoxic Conditions
6.2. Role of Mitochondrial STAT3 in Cancer Metabolism
6.3. Role of Tyrosine Phosphorylated STAT3 in Cancer Metabolism
6.4. Role of STAT3 Acetylation in Cancer Metabolism
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Song, T.L.; Nairismagi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Yi, E.H.; Ye, S.K. Signal transducer and activator of transcription 3 as a therapeutic target for cancer and the tumor microenvironment. Arch. Pharm. Res. 2016, 39, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Pal, S.K.; Reckamp, K.; Figlin, R.A.; Yu, H. STAT3: A target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 2011, 344, 41–59. [Google Scholar] [CrossRef]
- Barger, J.F.; Plas, D.R. Balancing biosynthesis and bioenergetics: Metabolic programs in oncogenesis. Endocr. Relat. Cancer 2010, 17, R287–R304. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef]
- Peppicelli, S.; Bianchini, F.; Calorini, L. Dynamic scenario of metabolic pathway adaptation in tumors and therapeutic approach. Oncoscience 2015, 2, 225–232. [Google Scholar] [CrossRef]
- Lamonte, G.; Tang, X.; Chen, J.L.; Wu, J.; Ding, C.K.; Keenan, M.M.; Sangokoya, C.; Kung, H.N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef]
- Smolkova, K.; Plecita-Hlavata, L.; Bellance, N.; Benard, G.; Rossignol, R.; Jezek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2011, 43, 950–968. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Chae, I.G.; Song, N.Y.; Kim, D.H.; Lee, M.Y.; Park, J.M.; Chun, K.S. Thymoquinone induces apoptosis of human renal carcinoma Caki-1 cells by inhibiting JAK2/STAT3 through pro-oxidant effect. Food Chem. Toxicol. 2020, 139, 111253. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, J.E.; Chae, I.G.; Park, G.; Lee, S.; Chun, K.S. Isoliquiritigenin inhibits the proliferation of human renal carcinoma Caki cells through the ROS-mediated regulation of the Jak2/STAT3 pathway. Oncol. Rep. 2017, 38, 575–583. [Google Scholar] [CrossRef]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Ren, Z.; Mao, X.; Mertens, C.; Krishnaraj, R.; Qin, J.; Mandal, P.K.; Romanowski, M.J.; McMurray, J.S.; Chen, X. Crystal structure of unphosphorylated STAT3 core fragment. Biochem. Biophys. Res. Commun. 2008, 374, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Y.L.; Hoey, T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science 1996, 273, 794–797. [Google Scholar] [CrossRef]
- Vogt, M.; Domoszlai, T.; Kleshchanok, D.; Lehmann, S.; Schmitt, A.; Poli, V.; Richtering, W.; Muller-Newen, G. The role of the N-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J. Cell Sci. 2011, 124, 900–909. [Google Scholar] [CrossRef]
- Pranada, A.L.; Metz, S.; Herrmann, A.; Heinrich, P.C.; Muller-Newen, G. Real time analysis of STAT3 nucleocytoplasmic shuttling. J. Biol. Chem. 2004, 279, 15114–15123. [Google Scholar] [CrossRef]
- Zhang, X.; Yue, P.; Page, B.D.; Li, T.; Zhao, W.; Namanja, A.T.; Paladino, D.; Zhao, J.; Chen, Y.; Gunning, P.T.; et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. USA 2012, 109, 9623–9628. [Google Scholar] [CrossRef]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Corzano, R.; Rashid, R.; Lin, J.; Senthil, M.; Hedvat, M.; Schroeder, A.; Mao, A.; Herrmann, A.; Yim, J.; et al. Alkylation of cysteine 468 in Stat3 defines a novel site for therapeutic development. ACS Chem. Biol. 2011, 6, 432–443. [Google Scholar] [CrossRef]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Hitosugi, T.; Chen, J. Post-translational modifications and the Warburg effect. Oncogene 2014, 33, 4279–4285. [Google Scholar] [CrossRef]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Linossi, E.M.; Nicholson, S.E. Kinase inhibition, competitive binding and proteasomal degradation: Resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol. Rev. 2015, 266, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzman, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Gough, D.J.; Koetz, L.; Levy, D.E. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Yan, H.; Ye, S.; Tong, C.; Ying, Q.L. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells 2014, 32, 1149–1160. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Huo, X.; Agoston, A.T.; Zhang, X.; Theiss, A.L.; Cheng, E.; Zhang, Q.; Zaika, A.; Pham, T.H.; Wang, D.H.; et al. Mitochondrial STAT3 contributes to transformation of Barrett’s epithelial cells that express oncogenic Ras in a p53-independent fashion. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G146–G161. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 Undergoes Acetylation-dependent Mitochondrial Translocation to Regulate Pyruvate Metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.C.; Hsieh, C.W.; Lin, Y.C.; Wung, B.S. The glutaredoxin/glutathione system modulates NF-kappaB activity by glutathionylation of p65 in cinnamaldehyde-treated endothelial cells. Toxicol. Sci. 2010, 116, 151–163. [Google Scholar] [CrossRef]
- Kurdi, M.; Booz, G.W. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets JAK1 activation by generating ROS. J. Cell. Physiol. 2007, 212, 424–431. [Google Scholar] [CrossRef]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef]
- Li, L.; Cheung, S.H.; Evans, E.L.; Shaw, P.E. Modulation of gene expression and tumor cell growth by redox modification of STAT3. Cancer Res. 2010, 70, 8222–8232. [Google Scholar] [CrossRef]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, W.; Li, D.; Zhan, Q. Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J. Biol. Chem. 2013, 288, 6552–6560. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Choi, M.J.; Jeong, K.J.; Kim, J.J.; Hwang, M.H.; Shin, S.C.; Park, C.G.; Lee, H.Y. A ROS/STAT3/HIF-1alpha signaling cascade mediates EGF-induced TWIST1 expression and prostate cancer cell invasion. Prostate 2014, 74, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Bak, Y.; Park, Y.H.; Jang, G.B.; Nam, J.S.; Yoo, J.E.; Park, Y.N.; Bak, I.S.; Kim, J.M.; Yoon, D.Y.; et al. Peroxiredoxin II Is Essential for Maintaining Stemness by Redox Regulation in Liver Cancer Cells. Stem Cells 2016, 34, 1188–1197. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010, 2, 823–842. [Google Scholar] [CrossRef]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.J.; Abdul-Hay, M.; Gough, D.J.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell. Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef]
- Carlsson, R.; Ozen, I.; Barbariga, M.; Gaceb, A.; Roth, M.; Paul, G. STAT3 precedes HIF1alpha transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS ONE 2018, 13, e0194146. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Arner, E.S. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Sachweh, M.C.; Stafford, W.C.; Drummond, C.J.; McCarthy, A.R.; Higgins, M.; Campbell, J.; Brodin, B.; Arner, E.S.; Lain, S. Redox effects and cytotoxic profiles of MJ25 and auranofin towards malignant melanoma cells. Oncotarget 2015, 6, 16488–16506. [Google Scholar] [CrossRef] [PubMed]
- Busker, S.; Qian, W.; Haraldsson, M.; Espinosa, B.; Johansson, L.; Attarha, S.; Kolosenko, I.; Liu, J.; Dagnell, M.; Grander, D.; et al. Irreversible TrxR1 inhibitors block STAT3 activity and induce cancer cell death. Sci. Adv. 2020, 6, eaax7945. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.L.; He, F.; Ye, J.Z.; Wu, H.N.; Zhang, J.Y.; Liu, Z.H.; Li, Y.Q.; Luo, X.L.; Lin, Y.; Liang, R. APE1 overexpression is associated with poor survival in patients with solid tumors: A meta-analysis. Oncotarget 2017, 8, 59720–59728. [Google Scholar] [CrossRef]
- Flaherty, D.M.; Monick, M.M.; Hunninghake, G.W. AP endonucleases and the many functions of Ref-1. Am. J. Respir. Cell Mol. Biol. 2001, 25, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L., 2nd; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M.; Suzuki, S.; Irani, K. Redox factor-1/APE suppresses oxidative stress by inhibiting the rac1 GTPase. FASEB J. 2002, 16, 889–890. [Google Scholar] [CrossRef]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Gray, M.J.; Zhang, J.; Ellis, L.M.; Semenza, G.L.; Evans, D.B.; Watowich, S.S.; Gallick, G.E. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 2005, 24, 3110–3120. [Google Scholar] [CrossRef]
- Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1815. [Google Scholar] [CrossRef]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1alpha: The Warburg’s vicious circle. JAKSTAT 2012, 1, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Jung, J.E.; Lee, H.G.; Cho, I.H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef]
- Guo, F.J.; Shao, Y.P.; Wang, Y.P.; Jin, Y.M.; Liu, S.S.; Wang, Q.Y. MIR-92 stimulates VEGF by inhibiting von Hippel-Lindau gene product in epithelial ovarian cancer. J. Biol. Regul. Homeost. Agents 2017, 31, 615–624. [Google Scholar]
- Gough, D.J.; Marie, I.J.; Lobry, C.; Aifantis, I.; Levy, D.E. STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood 2014, 124, 2252–2261. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask STAT3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef]
- Huang, G.; Chen, Y.; Lu, H.; Cao, X. Coupling mitochondrial respiratory chain to cell death: An essential role of mitochondrial complex I in the interferon-beta and retinoic acid-induced cancer cell death. Cell Death Differ. 2007, 14, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Li, S.; Hsu, P.; Qu, C.K. Induction of a tumor-associated activating mutation in protein tyrosine phosphatase Ptpn11 (Shp2) enhances mitochondrial metabolism, leading to oxidative stress and senescence. J. Biol. Chem. 2013, 288, 25727–25738. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Morciano, G.; Caroccia, N.; Ghetti, E.; Orecchia, V.; Viavattene, D.; Giorgi, C.; Pinton, P.; Poli, V. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death Differ. 2019, 26, 932–942. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, P.K.; Bollrath, J.; Pallangyo, C.K.; Matsutani, T.; Canli, O.; De Oliveira, T.; Diamanti, M.A.; Muller, N.; Gamrekelashvili, J.; Putoczki, T.; et al. Mitophagy in Intestinal Epithelial Cells Triggers Adaptive Immunity during Tumorigenesis. Cell 2018, 174, 88–101.e16. [Google Scholar] [CrossRef]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef]
- Zheng, R.; Chen, G.; Li, X.; Wei, X.; Liu, C.; Derwahl, M. Effect of IL-6 on proliferation of human thyroid anaplastic cancer stem cells. Int. J. Clin. Exp. Pathol. 2019, 12, 3992–4001. [Google Scholar] [PubMed]
- Zhang, X.Y.; Li, M.; Sun, K.; Chen, X.J.; Meng, J.; Wu, L.; Zhang, P.; Tong, X.; Jiang, W.W. Decreased expression of GRIM-19 by DNA hypermethylation promotes aerobic glycolysis and cell proliferation in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 101–115. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef]
- Jain, S.; Wei, J.; Mitrani, L.R.; Bishopric, N.H. Auto-acetylation stabilizes p300 in cardiac myocytes during acute oxidative stress, promoting STAT3 accumulation and cell survival. Breast Cancer Res. Treat. 2012, 135, 103–114. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Soto-Cruz, I. Energy Metabolism in Cancer: The Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers (Basel) 2020, 12, 1224. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, K.-S.; Jang, J.-H.; Kim, D.-H. Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer. Cells 2020, 9, 2202. https://doi.org/10.3390/cells9102202
Chun K-S, Jang J-H, Kim D-H. Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer. Cells. 2020; 9(10):2202. https://doi.org/10.3390/cells9102202
Chicago/Turabian StyleChun, Kyung-Soo, Jeong-Hoon Jang, and Do-Hee Kim. 2020. "Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer" Cells 9, no. 10: 2202. https://doi.org/10.3390/cells9102202
APA StyleChun, K.-S., Jang, J.-H., & Kim, D.-H. (2020). Perspectives Regarding the Intersections between STAT3 and Oxidative Metabolism in Cancer. Cells, 9(10), 2202. https://doi.org/10.3390/cells9102202