HDAC6 Regulates Radiosensitivity of Non-Small Cell Lung Cancer by Promoting Degradation of Chk1


_Zhang.png)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies, Chemicals, and Reagents
2.2. Establishment of HDAC6 Knockout Cell Lines
2.3. Cell Culture
2.4. Generation of H1299 and A549 Inducible HDAC6 Knockdown Cells
2.5. Generation of A549, H157, and H1975 HDAC6 Knockout Cells
2.6. Irradiation
2.7. Trypan Blue Exclusion
2.8. Colony Formation Assay
2.9. Constructs and Transfection
2.10. GST Pull-Down Assay
2.11. Generation of Chk1 Inducible Knockdown Cells
2.12. RT-PCR
2.13. In Vivo Ubiquitination Assay
2.14. In Vitro Ubiquitination Assay
2.15. Animals
3. Results
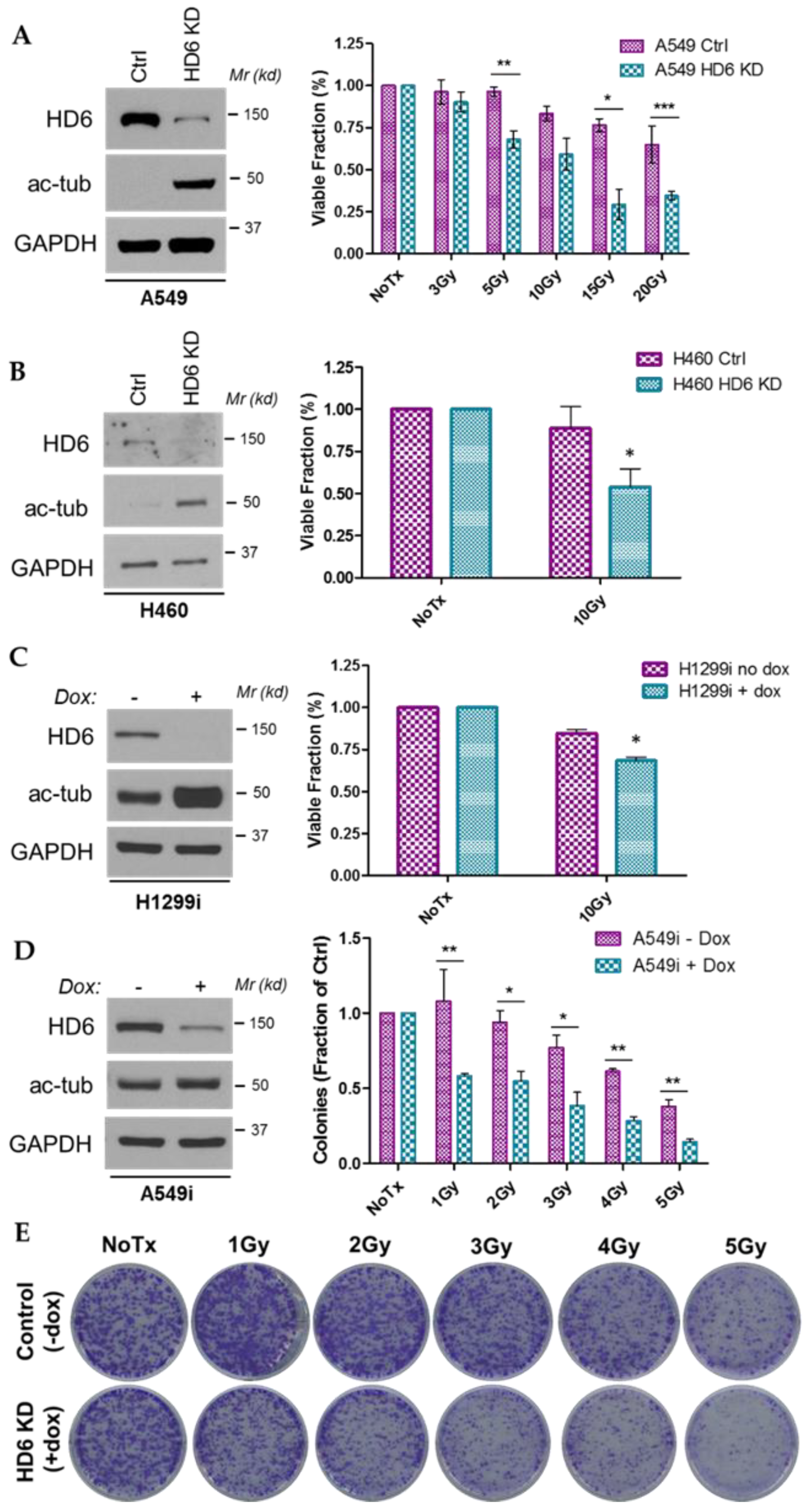
3.1. HDAC6 Depletion Sensitizes Several NSCLC Cell Lines to IR
3.2. HDAC6 Knockdown A549 Cells Exhibit Prolonged G2 Arrest upon IR
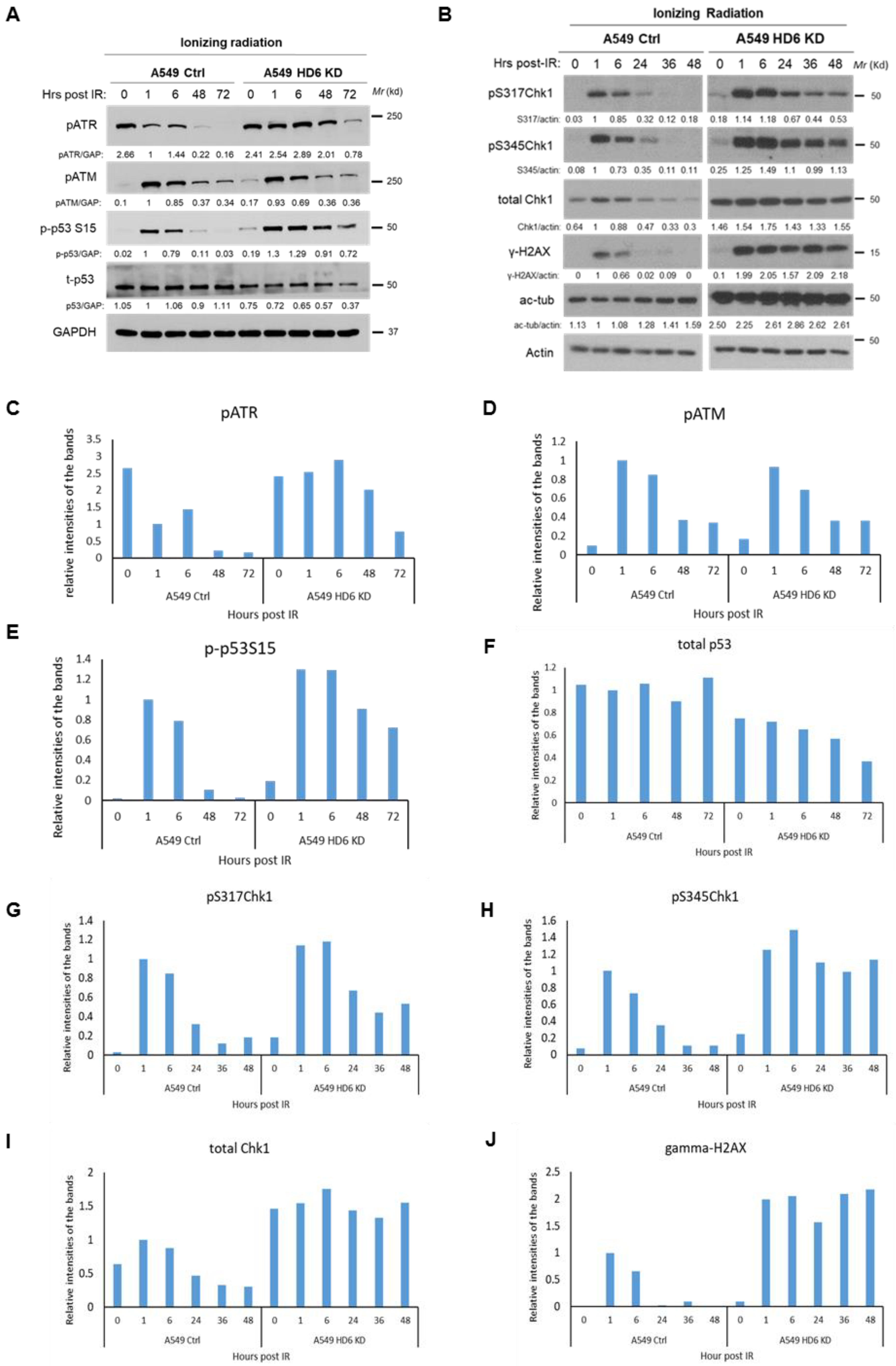
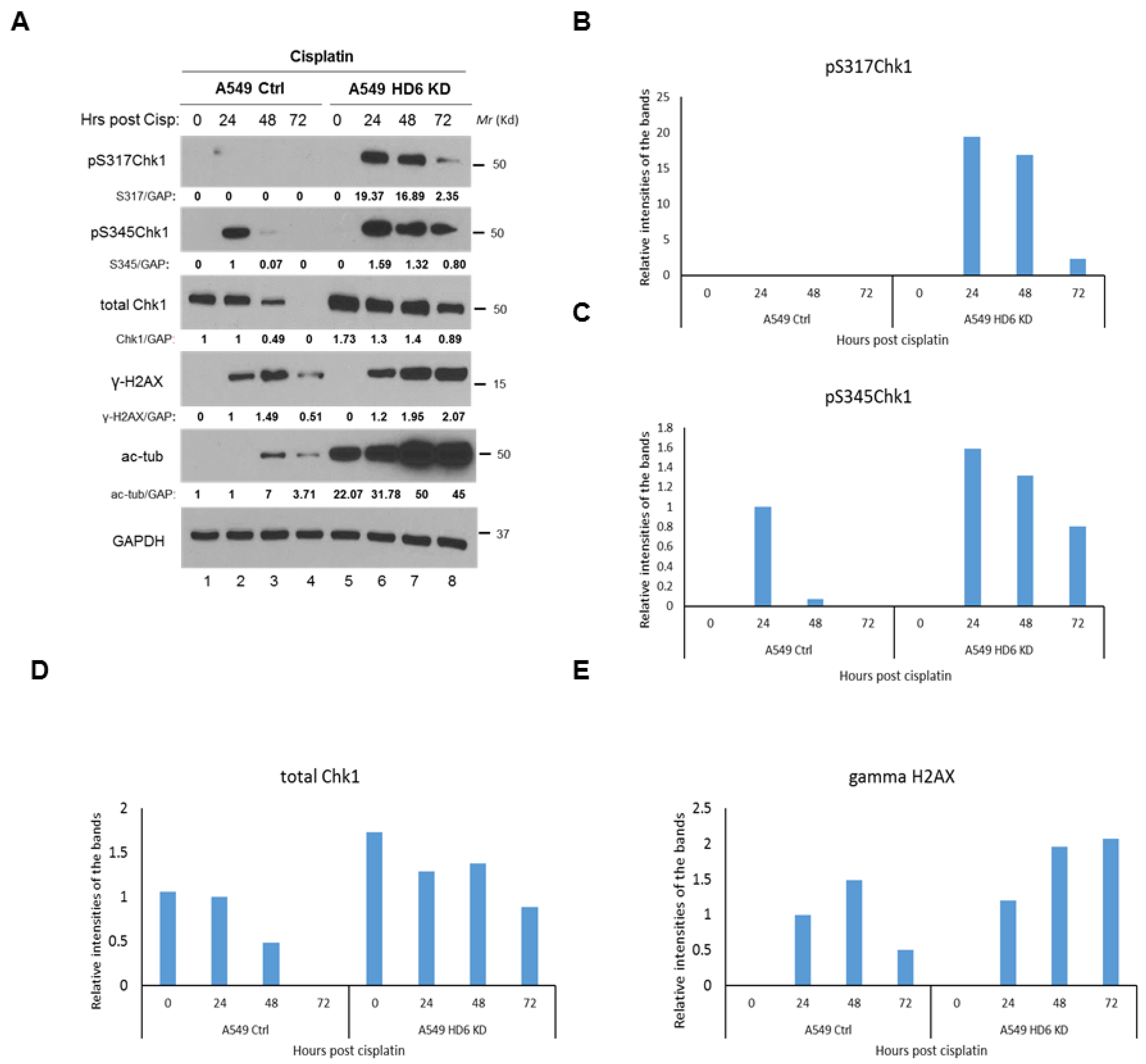
3.3. Chk1 Is Constitutively Active in HDAC6 Knockdown Cells Post-DNA Damage
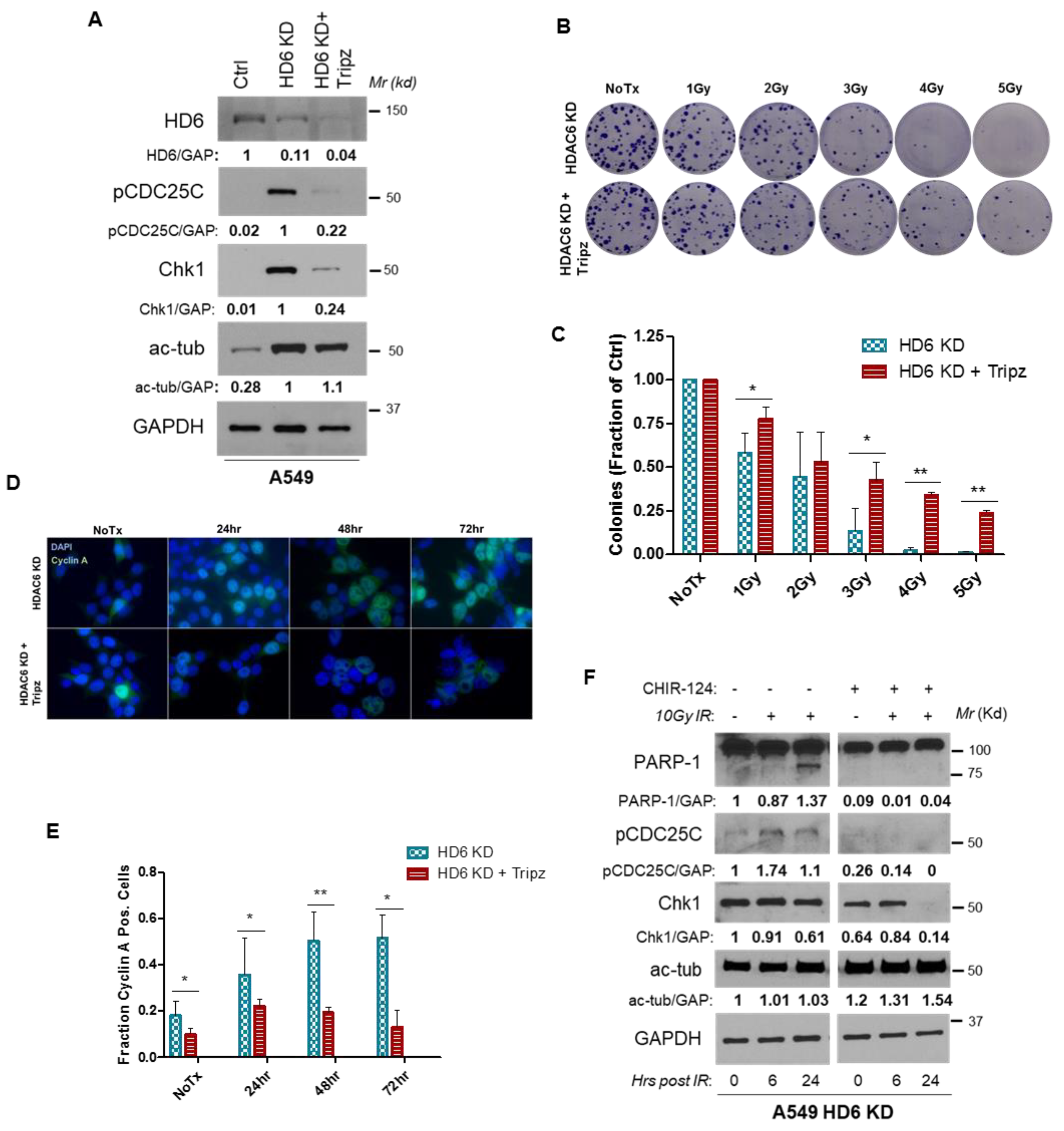
3.4. Radiosensitivity of HDAC6 Knockdown Cells Is Dependent on Increased Chk1 Activity
3.5. HDAC6 Influences Chk1 Protein Stability and Ubiquitinates Chk1
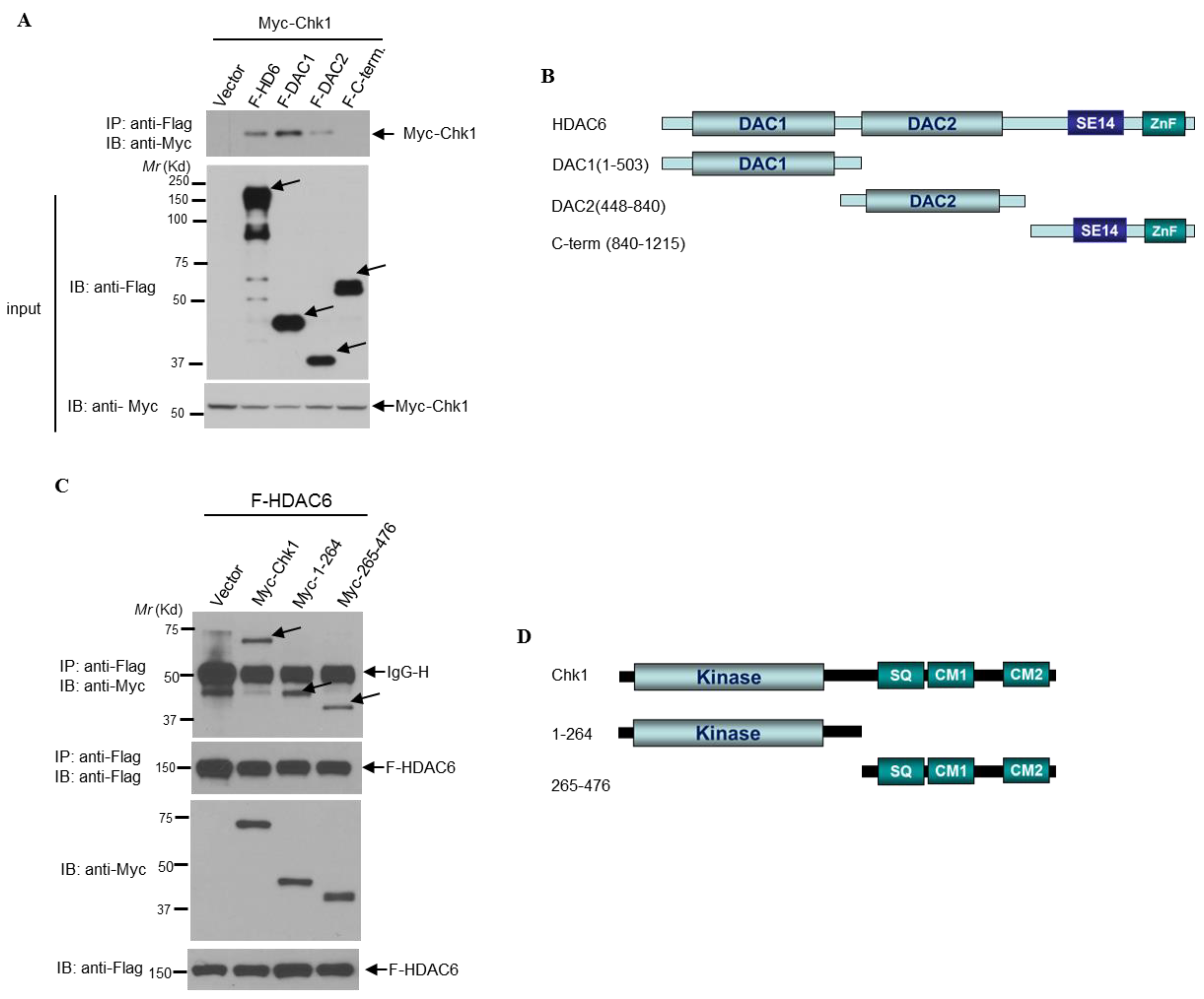
3.6. HDAC6 and Chk1 Physically Interact
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verdel, A.; Khochbin, S. Identification of a New Family of Higher Eukaryotic Histone Deacetylases. J. Biol. Chem. 1999, 274, 2440–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.-J.; Dent, S.R.; et al. HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nat. Cell Biol. 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activation of Glucocorticoid Receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.-I.; Iwase, H. HDAC6 Expression Is Correlated with Better Survival in Breast Cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Iii, F.J.R.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Hackanson, B.; Rimmele, L.; Benkisser, M.; Abdelkarim, M.; Fliegauf, M.; Jung, M.; Lubbert, M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk. Res. 2012, 36, 1055–1062. [Google Scholar] [CrossRef]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 sustain DNA double strand break repair and stem-like phenotype by promoting radioresistance in glioblastoma cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, M.; Moses, N.; Hu, C.-L.; Polin, L.; Chen, W.; Jang, H.; Heyza, J.; Malysa, A.; Caruso, J.A.; et al. The USP10-HDAC6 axis confers cisplatin resistance in non-small cell lung cancer lacking wild-type p53. Cell Death Dis. 2020, 11, 328. [Google Scholar] [CrossRef]
- Lemoine, M.; Younes, A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov. Med. 2010, 10, 462–470. [Google Scholar] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of Histone Deacetylase Inhibitors in Cancer Therapy: Isoform Selectivity or Multitargeted Strategy. Med. Res. Rev. 2014, 35, 63–84. [Google Scholar] [CrossRef]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.-L.; Chua, K.; et al. Mice Lacking Histone Deacetylase 6 Have Hyperacetylated Tubulin but Are Viable and Develop Normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [Green Version]
- Perego, P.; Zuco, V.; Gatti, L.; Zunino, F. Sensitization of tumor cells by targeting histone deacetylases. Biochem. Pharmacol. 2012, 83, 987–994. [Google Scholar] [CrossRef]
- Ryu, H.-W.; Shin, N.-H.; Lee, N.H.; Won, H.-R.; Kwon, S. A potent hydroxamic acid-based, small-molecule inhibitor A452 preferentially inhibits HDAC6 activity and induces cytotoxicity toward cancer cells irrespective of p53 status. Carcinogenesis 2018, 39, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Won, H.-R.; Ryu, H.-W.; Shin, D.-H.; Yeon, S.-K.; Lee, D.H.; Kwon, S.H. A452, an HDAC6-selective inhibitor, synergistically enhances the anticancer activity of chemotherapeutic agents in colorectal cancer cells. Mol. Carcinog. 2018, 57, 1383–1395. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef]
- Wang, L.; Xiang, S.; Williams, K.A.; Dong, H.; Bai, W.; Nicosia, S.V.; Khochbin, S.; Bepler, G.; Zhang, X. Depletion of HDAC6 Enhances Cisplatin-Induced DNA Damage and Apoptosis in Non-Small Cell Lung Cancer Cells. PLoS ONE 2012, 7, e44265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xiang, S.; Joo, H.-Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2013, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-W.; Otterness, D.M.; Chiang, G.G.; Xie, W.; Liu, Y.-C.; Mercurio, F.; Abraham, R.T. Genotoxic Stress Targets Human Chk1 for Degradation by the Ubiquitin-Proteasome Pathway. Mol. Cell 2005, 19, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Collis, S.J.; Barber, L.J.; Clark, A.J.; Martin, J.S.; Ward, J.D.; Boulton, S.J. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat. Cell Biol. 2007, 9, 391–401. [Google Scholar] [CrossRef]
- Feng, J.-M.; Zhu, H.; Zhang, X.-W.; Ding, J.; Miao, Z.-H. Proteasome-dependent degradation of Chk1 kinase induced by the topoisomerase II inhibitor R16 contributes to its anticancer activity. Cancer Biol. Ther. 2008, 7, 1726–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 Mediates S and G2 Arrests through Cdc25A Degradation in Response to DNA-damaging Agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-S.; Ryan, C.E.; Piwnica-Worms, H. Chk1 Kinase Negatively Regulates Mitotic Function of Cdc25A Phosphatase through 14-3-3 Binding. Mol. Cell. Biol. 2003, 23, 7488–7497. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation Through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Kaneko, Y.; Watanabe, N.; Morisaki, H.; Akita, H.; Fujimoto, A.; Tominaga, K.; Terasawa, M.; Tachibana, A.; Ikeda, K.; Nakanishi, M. Cell cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 1999, 18, 3673–3681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-W.; Brognard, J.; Coughlin, C.; You, Z.; Dolled-Filhart, M.; Aslanian, A.; Manning, G.; Abraham, R.T.; Hunter, T. The F Box Protein Fbx6 Regulates Chk1 Stability and Cellular Sensitivity to Replication Stress. Mol. Cell 2009, 35, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Leung-Pineda, V.; Huh, J.; Piwnica-Worms, H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009, 69, 2630–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.; Piwnica-Worms, H. CRL4CDT2 Targets CHK1 for PCNA-Independent Destruction. Mol. Cell. Biol. 2012, 33, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, K.B.; Bang, S.; Kurokawa, M.; Gerber, S.A. Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1. FEBS J. 2019, 287, 1985–1999. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; D’Andrea, A. Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef]
- Hock, A.K.; Vousden, K.H. Regulation of the p53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 2010, 42, 1618–1621. [Google Scholar] [CrossRef]
- Tho, L.M.; Libertini, S.; Rampling, R.; Sansom, O.J.; Gillespie, D.A. Chk1 is essential for chemical carcinogen-induced mouse skin tumorigenesis. Oncogene 2011, 31, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, L.; Vanden Bempt, I.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; De Wolf-Peeters, C.; Christiaens, M.-R.; Michiels, L.; Bouillon, R.; et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor /progesterone receptor /HER-2 breast carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Han, X.; Zhang, Y. Autoregulatory mechanisms of phosphorylation of checkpoint kinase 1. Cancer Res. 2012, 72, 3786–3794. [Google Scholar] [CrossRef] [Green Version]
- Kajstura, M.; Halicka, H.D.; Pryjma, J.; Darzynkiewicz, Z. Discontinuous fragmentation of nuclear DNA during apoptosis revealed by discrete “sub-G1” peaks on DNA content histograms. Cytom. Part A 2007, 71, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Donehower, L.A. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Develop. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Cho, C.-Y.; Kuo, P.-L.; Huang, Y.-T.; Lin, C.-C. Plumbagin (5-Hydroxy-2-methyl-1,4-naphthoquinone) Induces Apoptosis and Cell Cycle Arrest in A549 Cells through p53 Accumulation via c-Jun NH2-Terminal Kinase-Mediated Phosphorylation at Serine 15 in Vitro and in Vivo. J. Pharmacol. Exp. Ther. 2006, 318, 484–494. [Google Scholar] [CrossRef]
- Archie, N.T.; Rendahl, K.G.; Sheikh, T.; Cheema, H.; Aardalen, K.; Embry, M.; Hibner, B. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 2007, 13, 591–602. [Google Scholar]
- Graves, P.R.; Yu, L.; Schwarz, J.K.; Gales, J.; Sausville, E.A.; O’Connor, P.M.; Piwnica-Worms, H. The Chk1 Protein Kinase and the Cdc25C Regulatory Pathways Are Targets of the Anticancer Agent UCN-01. J. Biol. Chem. 2000, 275, 5600–5605. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2017, 293, 1976–1993. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.; Ito, A.; Yao, T.-P. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Han, X.; Feng, X.; Wang, Z.; Zhang, Y. Coupling Cellular Localization and Function of Checkpoint Kinase 1 (Chk1) in Checkpoints and Cell Viability. J. Biol. Chem. 2012, 287, 25501–25509. [Google Scholar] [CrossRef] [Green Version]
- Gatei, M.; Sloper, K.; Sørensen, C.S.; Syljuasen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.-B.; Bartek, J.J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent Phosphorylation of Chk1 on Ser-317 in Response to Ionizing Radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [Green Version]
- Dent, P.; Tang, Y.; Yacoub, A.; Dai, Y.; Fisher, P.B.; Grant, S. CHK1 Inhibitors in Combination Chemotherapy. Mol. Interv. 2011, 11, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smits, V.A.J.; Gillespie, D.A. DNA damage control: Regulation and functions of checkpoint kinase 1. FEBS J. 2015, 282, 3681–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Pereira, E.; Maxfield, M.; Russell, B.; Goudelock, D.M.; Sanchez, Y. Regulation of Chk1 Includes Chromatin Association and 14-3-3 Binding following Phosphorylation on Ser-345. J. Biol. Chem. 2003, 278, 25207–25217. [Google Scholar] [CrossRef] [Green Version]
- Leung-Pineda, V.; Ryan, C.E.; Piwnica-Worms, H. Phosphorylation of Chk1 by ATR Is Antagonized by a Chk1-Regulated Protein Phosphatase 2A Circuit. Mol. Cell. Biol. 2006, 26, 7529–7538. [Google Scholar] [CrossRef] [Green Version]
- Saji, S.; Kawakami, M.; Hayashi, S.-I.; Yoshida, N.; Hirose, M.; Horiguchi, S.-I.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar] [CrossRef] [Green Version]
- Okuda, K.; Ito, A.; Uehara, T. Regulation of Histone Deacetylase 6 Activity via S-Nitrosylation. Biol. Pharm. Bull. 2015, 38, 1434–1437. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Ci, Y.; Dai, X.; Wu, F.; Guo, J.; Liu, D.; North, B.J.; Huo, J.; Zhang, J. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget 2017, 8, 47890–47901. [Google Scholar] [CrossRef] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moses, N.; Zhang, M.; Wu, J.-Y.; Hu, C.; Xiang, S.; Geng, X.; Chen, Y.; Bai, W.; Zhang, Y.-W.; Bepler, G.; et al. HDAC6 Regulates Radiosensitivity of Non-Small Cell Lung Cancer by Promoting Degradation of Chk1. Cells 2020, 9, 2237. https://doi.org/10.3390/cells9102237
Moses N, Zhang M, Wu J-Y, Hu C, Xiang S, Geng X, Chen Y, Bai W, Zhang Y-W, Bepler G, et al. HDAC6 Regulates Radiosensitivity of Non-Small Cell Lung Cancer by Promoting Degradation of Chk1. Cells. 2020; 9(10):2237. https://doi.org/10.3390/cells9102237
Chicago/Turabian StyleMoses, Niko, Mu Zhang, Jheng-Yu Wu, Chen Hu, Shengyan Xiang, Xinran Geng, Yue Chen, Wenlong Bai, You-Wei Zhang, Gerold Bepler, and et al. 2020. "HDAC6 Regulates Radiosensitivity of Non-Small Cell Lung Cancer by Promoting Degradation of Chk1" Cells 9, no. 10: 2237. https://doi.org/10.3390/cells9102237
APA StyleMoses, N., Zhang, M., Wu, J.-Y., Hu, C., Xiang, S., Geng, X., Chen, Y., Bai, W., Zhang, Y.-W., Bepler, G., & Zhang, X. M. (2020). HDAC6 Regulates Radiosensitivity of Non-Small Cell Lung Cancer by Promoting Degradation of Chk1. Cells, 9(10), 2237. https://doi.org/10.3390/cells9102237