Pseudomonas aeruginosa Planktonic- and Biofilm-Conditioned Media Elicit Discrete Metabolic Responses in Human Macrophages

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Primary Human Monocytes
2.2. In vitro Differentiation of Primary Human Monocyte-Derived MΦs
2.3. Fluorescent-Conjugated Antibody Staining and Flow Cytometry Analysis
2.4. Biofilm-Conditioned Medium (BCM)
2.5. Planktonic-Conditioned Medium (PCM)
2.6. Exposure of Primary Human Monocyte-Derived MΦs to PCM, BCM, and Control Media
2.7. Intra- and Extracellular Metabolite Extraction
2.8. Protein Assay
2.9. NMR Sample Preparation
2.10. NMR Data Acquisition and Preprocessing
2.11. Analysis of NMR Data
2.12. Statistical Analysis
3. Results
3.1. NMR Analysis of Metabolite Extracts Derived from MΦs Exposed to PCM, BCM, or Control Media Reveals Spectral Pattern Differences
3.2. Multivariate Statistical Analysis of Quantitative Metabolic Data Identifies Metabolic Differences between MΦ Exposure Groups
3.3. BCM-Exposed MΦs Exhibit Distinct Metabolic Characteristics Relative to PCM-Exposed MΦs
3.4. Disparate Metabolic Patterns are Presented by PCM- and BCM-Exposed MΦs Compared to Control MΦs
3.5. BCM- and PCM-Exposed MΦs Display Similar Metabolic Responses Compared to Control MΦs
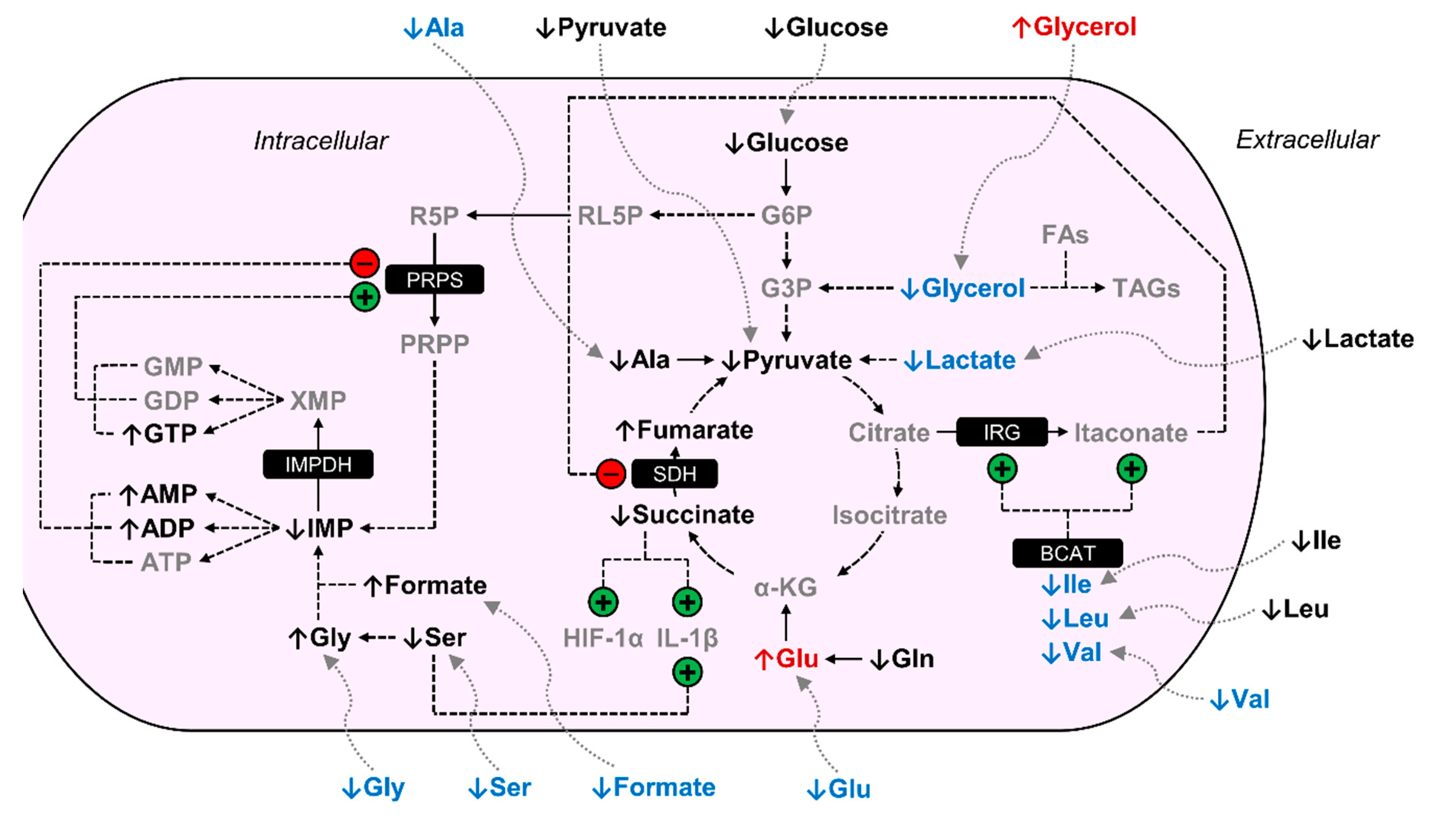
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clarke, T.B. Microbial programming of systemic innate immunity and resistance to infection. PLoS Pathog. 2014, 10, e1004506. [Google Scholar] [CrossRef] [Green Version]
- Aderem, A. Phagocytosis and the inflammatory response. J. Infect. Dis. 2003, 187, S340–S345. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Leibovich, S.J. Regulation of macrophage polarization and wound healing. Adv. Wound Care (New Rochelle) 2012, 1, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, C.C. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 138–152. [Google Scholar] [CrossRef]
- Frykberg, R.G.; Banks, J. Challenges in the treatment of chronic wounds. Adv. Wound Care (New Rochelle) 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baum, C.L.; Arpey, C.J. Normal cutaneous wound healing: Clinical correlation with cellular and molecular events. Dermatol. Surg. 2005, 31, 674–686, discussion 686. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in chronic wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [Green Version]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Ghigo, J.M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen 2009, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Goren, I.; Müller, E.; Schiefelbein, D.; Christen, U.; Pfeilschifter, J.; Mühl, H.; Frank, S. Systemic anti-TNFalpha treatment restores diabetes-impaired skin repair in ob/ob mice by inactivation of macrophages. J. Investig. Dermatol. 2007, 127, 2259–2267. [Google Scholar] [CrossRef] [Green Version]
- Loots, M.A.; Lamme, E.N.; Zeegelaar, J.; Mekkes, J.R.; Bos, J.D.; Middelkoop, E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J. Investig. Dermatol. 1998, 111, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- James, G.A.; Swogger, E.; Wolcott, R.; Pulcini, E.d.; Secor, P.; Sestrich, J.; Costerton, J.W.; Stewart, P.S. Biofilms in chronic wounds. Wound Repair Regen. 2008, 16, 37–44. [Google Scholar] [CrossRef]
- Davis, S.C.; Ricotti, C.; Cazzaniga, A.; Welsh, E.; Eaglstein, W.H.; Mertz, P.M. Microscopic and physiologic evidence for biofilm-associated wound colonization in vivo. Wound Repair Regen. 2008, 16, 23–29. [Google Scholar] [CrossRef]
- Zhao, G.; Hochwalt, P.C.; Usui, M.L.; Underwood, R.A.; Singh, P.K.; James, G.A.; Stewart, P.S.; Fleckman, P.; Olerud, J.E. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: A model for the study of chronic wounds. Wound Repair Regen. 2010, 18, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Dowd, S.E.; Sun, Y.; Secor, P.R.; Rhoads, D.D.; Wolcott, B.M.; James, G.A.; Wolcott, R.D. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurice, N.M.; Bedi, B.; Sadikot, R.T. Pseudomonas aeruginosa biofilms: Host response and clinical implications in lung infections. Am. J. Respir. Cell Mol. Biol. 2018, 58, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.M.; Armstrong, D.S.; Carzino, R.; Carlin, J.B.; Olinsky, A.; Robertson, C.F.; Grimwood, K. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J. Pediatr. 2001, 138, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Kirker, K.R.; Secor, P.R.; James, G.A.; Fleckman, P.; Olerud, J.E.; Stewart, P.S. Loss of viability and induction of apoptosis in human keratinocytes exposed to Staphylococcus aureus biofilms in vitro. Wound Repair Regen. 2009, 17, 690–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirker, K.R.; James, G.A.; Fleckman, P.; Olerud, J.E.; Stewart, P.S. Differential effects of planktonic and biofilm MRSA on human fibroblasts. Wound Repair Regen. 2012, 20, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Riboldi, E.; Ippolito, A.; Sica, A. Molecular and epigenetic basis of macrophage polarized activation. Semin. Immunol. 2015, 27, 237–248. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Buck, M.D.; Sowell, R.T.; Kaech, S.M.; Pearce, E.L. Metabolic instruction of immunity. Cell 2017, 169, 570–586. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Emwas, A.M.; Salek, R.M.; Griffin, J.L.; Merzaban, J. NMR-based metabolomics in human disease diagnosis: Applications, limitations, and recommendations. Metabolomics 2013, 9, 1048–1072. [Google Scholar] [CrossRef]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, J.K.; Lindon, J.C. Systems biology: Metabonomics. Nature 2008, 455, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O. Metabolomics--the link between genotypes and phenotypes. Plant. Mol. Biol. 2002, 48, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprogramming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.L.; Schiller, S.M.; Keegan, W.J.; Ammons, M.C.B.; Eilers, B.; Tripet, B.; Copié, V. Quantitative 1H NMR metabolomics reveal distinct metabolic adaptations in human macrophages following differential activation. Metabolites 2019, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Ammons, M.C.; Tripet, B.P.; Carlson, R.P.; Kirker, K.R.; Gross, M.A.; Stanisich, J.J.; Copié, V. Quantitative NMR metabolite profiling of methicillin-resistant and methicillin-susceptible Staphylococcus aureus discriminates between biofilm and planktonic phenotypes. J. Proteome Res. 2014, 13, 2973–2985. [Google Scholar] [CrossRef] [Green Version]
- García-Cañaveras, J.C.; López, S.; Castell, J.V.; Donato, M.T.; Lahoz, A. Extending metabolome coverage for untargeted metabolite profiling of adherent cultured hepatic cells. Anal. Bioanal. Chem. 2016, 408, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Fathi, F.; Brun, A.; Rott, K.H.; Falco Cobra, P.; Tonelli, M.; Eghbalnia, H.R.; Caviedes-Vidal, E.; Karasov, W.H.; Markley, J.L. NMR-based identification of metabolites in polar and non-polar extracts of avian liver. Metabolites 2017, 7, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramm Sander, P.; Peer, M.; Grandl, M.; Bogdahn, U.; Schmitz, G.; Kalbitzer, H.R. NMR spectroscopy of macrophages loaded with native, oxidized or enzymatically degraded lipoproteins. PLoS ONE 2013, 8, e56360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.J.; Peters, T.R.; Tripet, B.; Van Vuren, A.; Rakesh; Lee, R.E.; Copié, V.; Teintze, M. Exposure of methicillin-resistant Staphylococcus aureus to low levels of the antibacterial THAM-3ΦG generates a small colony drug-resistant phenotype. Sci. Rep. 2018, 8, 9850. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.L.; Weaver Jr, A.J.; Tripet, B.P.; Ammons, M.C.B.; Teintze, M.; Copié, V. Characterization of the antibacterial activity of Bald’s eyesalve against drug resistant Staphylococcus aureus and Pseudomonas aeruginosa. PLoS ONE 2018, 13, e0208108. [Google Scholar] [CrossRef]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton NMR spectra. J. Biomol. NMR 2011, 49, 307–323. [Google Scholar] [CrossRef]
- Emwas, A.H.; Saccenti, E.; Gao, X.; McKay, R.T.; Dos Santos, V.A.P.M.; Roy, R.; Wishart, D.S. Recommended strategies for spectral processing and post-processing of 1D 1H-NMR data of biofluids with a particular focus on urine. Metabolomics 2018, 14, 31. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics 2011. [Google Scholar] [CrossRef]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Hames, C.; Halbedel, S.; Hoppert, M.; Frey, J.; Stülke, J. Glycerol metabolism is important for cytotoxicity of Mycoplasma pneumoniae. J. Bacteriol. 2009, 191, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Kang, Y.; Norris, M.H.; Troyer, R.M.; Son, M.S.; Schweizer, H.P.; Dow, S.W.; Hoang, T.T. Blocking phosphatidylcholine utilization in Pseudomonas aeruginosa, via mutagenesis of fatty acid, glycerol and choline degradation pathways, confirms the importance of this nutrient source in vivo. PLoS ONE 2014, 9, e103778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemesh, M.; Chai, Y. A combination of glycerol and manganese promotes biofilm formation in Bacillus subtilis via histidine kinase KinD signaling. J. Bacteriol. 2013, 195, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Scoffield, J.; Silo-Suh, L. Glycerol metabolism promotes biofilm formation by Pseudomonas aeruginosa. Can. J. Microbiol. 2016, 62, 704–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalai Chelvam, K.; Yap, K.P.; Chai, L.C.; Thong, K.L. Variable responses to carbon utilization between planktonic and biofilm cells of a human carrier strain of Salmonella enterica serovar typhi. PLoS ONE 2015, 10, e0126207. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages II. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and ATP generation. J. Exp. Med. 1976, 144, 1484–1493. [Google Scholar] [CrossRef]
- Pavlou, S.; Wang, L.; Xu, H.; Chen, M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J. Inflamm. (Lond) 2017, 14, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diskin, C.; Pålsson-McDermott, E.M. Metabolic modulation in macrophage effector function. Front. Immunol 2018, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Ždralević, M.; Brand, A.; Di lanni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, T.C.; Coelho, R.G.; Da Silva, D.; Coelho, W.S.; Marinho-Carvalho, M.M.; Sola-Penna, M. Lactate downregulates the glycolytic enzymes hexokinase and phosphofructokinase in diverse tissues from mice. FEBS Lett. 2011, 585, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: Balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef]
- Deretic, V.; Kimura, T.; Timmins, G.; Moseley, P.; Chauhan, S.; Mandell, M. Immunologic manifestations of autophagy. J. Clin. Investig. 2015, 125, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8, 16040. [Google Scholar] [CrossRef]
- Luan, H.H.; Medzhitov, R. Food fight: Role of itaconate and other metabolites in antimicrobial defense. Cell Metab. 2016, 24, 379–387. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [Green Version]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine metabolism supports macrophage IL-1β production. Cell Metab. 2019, 29, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidal, K.G.; Munson, K.L.; Denning, G.M. Small molecular weight secretory factors from Pseudomonas aeruginosa have opposite effects on IL-8 and RANTES expression by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001, 25, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Ciszek-Lenda, M.; Strus, M.; Walczewska, M.; Majka, G.; Machul-Żwirbla, A.; Mikołajczyk, D.; Górska, S.; Gamian, A.; Chain, B.; Marcinkiewicz, J. Pseudomonas aeruginosa biofilm is a potent inducer of phagocyte hyperinflammation. Inflamm. Res. 2019, 68, 397–413. [Google Scholar] [CrossRef] [Green Version]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.C.; Scicluna, B.P.; Arts, R.J.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J. Vis. Exp. 2015, 105, 53424. [Google Scholar] [CrossRef] [Green Version]
- Hovi, T.; Allison, A.C.; Raivio, K.; Vaheri, A. Purine metabolism and control of cell proliferation. Ciba Found. Symp. 1977, 48, 225–248. [Google Scholar] [CrossRef]
- Fairbanks, L.D.; Bofill, M.; Ruckemann, K.; Simmonds, H.A. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. Disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J. Biol. Chem. 1995, 270, 29682–29689. [Google Scholar]
- Allison, A.C.; Eugui, E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 2000, 47, 85–118. [Google Scholar] [CrossRef]
- Keppeke, G.D.; Chang, C.C.; Peng, M.; Chen, L.Y.; Lin, W.C.; Pai, L.M.; Andrade, L.E.C.; Sung, L.Y.; Liu, J.L. IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div. 2018, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, R.C.; Leoni, P.; Allison, A.C. Control of phosphoribosylpyrophosphate synthesis in human lymphocytes. Biochem. Biophys. Res. Commun. 1977, 77, 1067–1073. [Google Scholar] [CrossRef]
- Jia, Y.; Schurmans, S.; Luo, H.R. Regulation of innate immunity by inositol 1,3,4,5-tetrakisphosphate. Cell Cycle 2008, 7, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Ellson, C.; Hawkins, P. Roles of PI3Ks in leukocyte chemotaxis and phagocytosis. Curr. Opin. Cell Biol. 2002, 14, 203–213. [Google Scholar] [CrossRef]
- Xu, J.; Wang, F.; Van Keymeulen, A.; Herzmark, P.; Straight, A.; Kelly, K.; Takuwa, Y.; Sugimoto, N.; Mitchison, T.; Bourne, H.R. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 2003, 114, 201–214. [Google Scholar] [CrossRef]
- Marshall, J.G.; Booth, J.W.; Stambolic, V.; Mak, T.; Balla, T.; Schreiber, A.D.; Meyer, T.; Grinstein, S. Restricted accumulation of phosphatidylinositol 3-kinase products in a plasmalemmal subdomain during Fc gamma receptor-mediated phagocytosis. J. Cell Biol. 2001, 153, 1369–1380. [Google Scholar] [CrossRef] [Green Version]
- Kanai, F.; Liu, H.; Field, S.J.; Akbary, H.; Matsuo, T.; Brown, G.E.; Cantley, L.C.; Yaffe, M.B. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol. 2001, 3, 675–678. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Metabolite | PCM MΦs 2 | BCM MΦs 2 | BCM vs. PCM MΦs | |||
|---|---|---|---|---|---|---|
| FC | p-Value | FC | p-Value | FC | p-Value | |
| Acetate | −1.46 | * | −1.53 | * | −1.05 | NS |
| ADP | −1.98 | ** | −1.89 | ** | 1.05 | NS |
| AMP | −1.38 | * | −1.27 | * | 1.09 | NS |
| Arginine | −1.82 | ** | −2.62 | ** | −1.44 | NS |
| Asparagine | −1.91 | ** | −2.69 | ** | −1.41 | ** |
| Betaine | −2.45 | * | −2.06 | * | 1.19 | NS |
| Choline | −1.83 | * | 1.09 | NS | 2.00 | ** |
| Formate | −2.08 | * | −1.73 | * | 1.21 | NS |
| Fructose | −3.18 | * | −2.89 | NS | 1.10 | NS |
| Fumarate | −1.45 | * | −1.87 | ** | −1.28 | NS |
| Glucose | −3.34 | ** | −4.17 | ** | −1.25 | NS |
| Glutamate | −1.30 | * | −1.04 | NS | 1.25 | * |
| Glutamine | −2.88 | * | −3.57 | * | −1.24 | NS |
| Glycerol | 5.62 | * | −1.40 | NS | −7.86 | * |
| Glycine | −1.27 | * | −1.18 | ** | 1.08 | NS |
| IMP | 9.25 3 | ** | 8.67 3 | *** | −1.07 | NS |
| Isoleucine | −1.55 | * | −2.14 | * | −1.38 | * |
| Lactate | −2.30 | * | −3.27 | * | −1.42 | * |
| Leucine | −1.04 | NS | −1.45 | NS | −1.39 | * |
| Methionine | −1.26 | NS | −1.94 | * | −1.54 | NS |
| myo-Inositol | −1.11 | NS | −1.28 | * | −1.15 | NS |
| O-Phosphocholine | 1.38 | * | 1.29 | NS | −1.07 | NS |
| Proline | −1.42 | NS | −1.59 | * | −1.12 | NS |
| Propionate | −1.19 | NS | −1.26 | * | −1.06 | NS |
| Pyroglutamate | −2.39 | * | −3.15 | * | −1.32 | ** |
| Pyruvate | −2.04 | * | −2.24 | * | −1.10 | NS |
| Serine | −1.74 | * | −1.93 | ** | −1.11 | NS |
| Succinate | −2.00 | * | −1.92 | * | 1.05 | NS |
| Taurine | −1.11 | NS | 1.20 | * | 1.34 | NS |
| Tyrosine | −1.07 | NS | −1.39 | NS | −1.30 | * |
| UMP | −1.45 | NS | −1.88 | ** | −1.30 | NS |
| Valine | 1.43 | * | −1.13 | NS | −1.62 | * |
| Metabolite | Concentration (mean ± SD) | p-value | ||||
|---|---|---|---|---|---|---|
| Control MΦs | PCM MΦs | BCM MΦs | PCM 2 | BCM 2 | BCM vs. PCM | |
| 3-Hydroxybutyrate | 7.94 ± 8.71 | −65.98 ± 2.54 | −21.07 ± 2.44 | ** | * | **** |
| 3-Hydroxyisobutyrate | 6.90 ± 2.29 | 0.80 ± 2.41 | −20.64 ± 6.81 | * | * | * |
| 4-Hydroxyproline | −46.62 ± 26.71 | −215.88 ± 104.10 | −534.62 ± 143.50 | NS | * | * |
| Acetate | 25.43 ± 18.11 | −34.91 ± 48.50 | −302.70 ± 100.15 | NS | * | * |
| Alanine | −20.93 ± 36.91 | −158.21 ± 81.82 | −829.65 ± 237.35 | NS | * | * |
| Arginine | −568.00 ± 138.16 | −1980.38 ± 613.21 | −3106.32 ± 846.80 | NS | * | NS |
| Asparagine | −226.11 ± 114.24 | −193.57 ± 243.09 | −637.79 ± 183.49 | NS | * | NS |
| Aspartate | −249.34 ± 46.71 | −635.40 ± 142.92 | −1589.74 ± 483.01 | * | * | NS |
| Choline | −0.81 ± 4.71 | −64.97 ± 5.81 | −152.90 ± 63.85 | *** | NS | NS |
| Creatine | −7.32 ± 12.01 | −81.20 ± 29.44 | −71.00 ± 19.09 | * | * | NS |
| Formate | 43.92 ± 43.80 | −39.01 ± 31.66 | −236.62 ± 66.66 | NS | ** | * |
| Fructose | −890.66 ± 324.03 | −2232.88 ± 532.77 | −2737.16 ± 825.08 | * | * | NS |
| Glucose | −7480.88 ± 1804.55 | −23845.20 ± 4976.92 | −32361.26 ± 8719.05 | * | * | NS |
| Glutamate | −50.04 ± 75.38 | −344.05 ± 268.89 | −954.86 ± 223.92 | NS | * | * |
| Glycerol | −676.16 ± 416.36 | −1265.91 ± 398.51 | 125.28 ± 124.64 | NS | NS | * |
| Glycine | −82.41 ± 74.53 | −75.85 ± 133.27 | −844.93 ± 248.20 | NS | * | * |
| Histidine | 13.29 ± 15.51 | −170.28 ± 73.99 | −290.03 ± 90.29 | * | * | NS |
| Isoleucine | −128.19 ± 54.32 | −950.15 ± 222.44 | −1331.88 ± 370.25 | * | * | NS |
| Lactate | 1487.13 ± 589.49 | −886.91 ± 725.71 | −1869.35 ± 839.64 | * | ** | NS |
| Leucine | −84.52 ± 21.47 | −1059.98 ± 148.64 | −2042.80 ± 569.71 | ** | * | NS |
| Lysine | 2.98 ± 27.87 | −256.78 ± 22.48 | −858.23 ± 247.75 | *** | * | NS |
| Mannose | −99.02 ± 17.95 | −40.48 ± 10.90 | −95.68 ± 20.40 | * | NS | * |
| Methionine | −45.48 ± 23.01 | −141.62 ± 62.69 | −367.03 ± 87.79 | NS | * | * |
| myo-Inositol | −187.26 ± 52.51 | −773.68 ± 187.85 | −916.76 ± 246.42 | * | * | NS |
| O-Phosphocholine | −0.23 ± 8.71 | −92.94 ± 6.27 | −19.47 ± 10.81 | *** | NS | ** |
| Phenylalanine | −1.22 ± 20.74 | −139.58 ± 69.60 | −371.54 ± 103.40 | NS | * | * |
| Proline | −155.96 ± 73.49 | −705.16 ± 251.50 | −1373.44 ± 429.60 | NS | * | NS |
| Pyroglutamate | −563.35 ± 340.29 | −2993.47 ± 597.73 | −3236.09 ± 925.54 | ** | * | NS |
| Pyruvate | −41.17 ± 19.08 | −747.06 ± 25.79 | −773.20 ± 421.83 | **** | NS | NS |
| Serine | −417.67 ± 177.14 | −380.27 ± 113.26 | −1468.66 ± 441.28 | NS | * | * |
| Threonine | −66.08 ± 61.60 | 373.43 ± 26.50 | −484.36 ± 84.08 | ** | ** | ** |
| Tyrosine | −55.89 ± 53.16 | −265.40 ± 141.53 | −579.01 ± 150.66 | NS | * | NS |
| Urea | 43.01 ± 151.53 | −3410.97 ± 1002.71 | −2291.28 ± 454.86 | * | ** | NS |
| Valine | −69.95 ± 23.42 | −604.31 ± 175.38 | −1446.36 ± 397.74 | * | * | * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuchs, A.L.; Miller, I.R.; Schiller, S.M.; Ammons, M.C.B.; Eilers, B.; Tripet, B.; Copié, V. Pseudomonas aeruginosa Planktonic- and Biofilm-Conditioned Media Elicit Discrete Metabolic Responses in Human Macrophages. Cells 2020, 9, 2260. https://doi.org/10.3390/cells9102260
Fuchs AL, Miller IR, Schiller SM, Ammons MCB, Eilers B, Tripet B, Copié V. Pseudomonas aeruginosa Planktonic- and Biofilm-Conditioned Media Elicit Discrete Metabolic Responses in Human Macrophages. Cells. 2020; 9(10):2260. https://doi.org/10.3390/cells9102260
Chicago/Turabian StyleFuchs, Amanda L., Isaac R. Miller, Sage M. Schiller, Mary Cloud B. Ammons, Brian Eilers, Brian Tripet, and Valérie Copié. 2020. "Pseudomonas aeruginosa Planktonic- and Biofilm-Conditioned Media Elicit Discrete Metabolic Responses in Human Macrophages" Cells 9, no. 10: 2260. https://doi.org/10.3390/cells9102260