Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins


Abstract
:1. Introduction
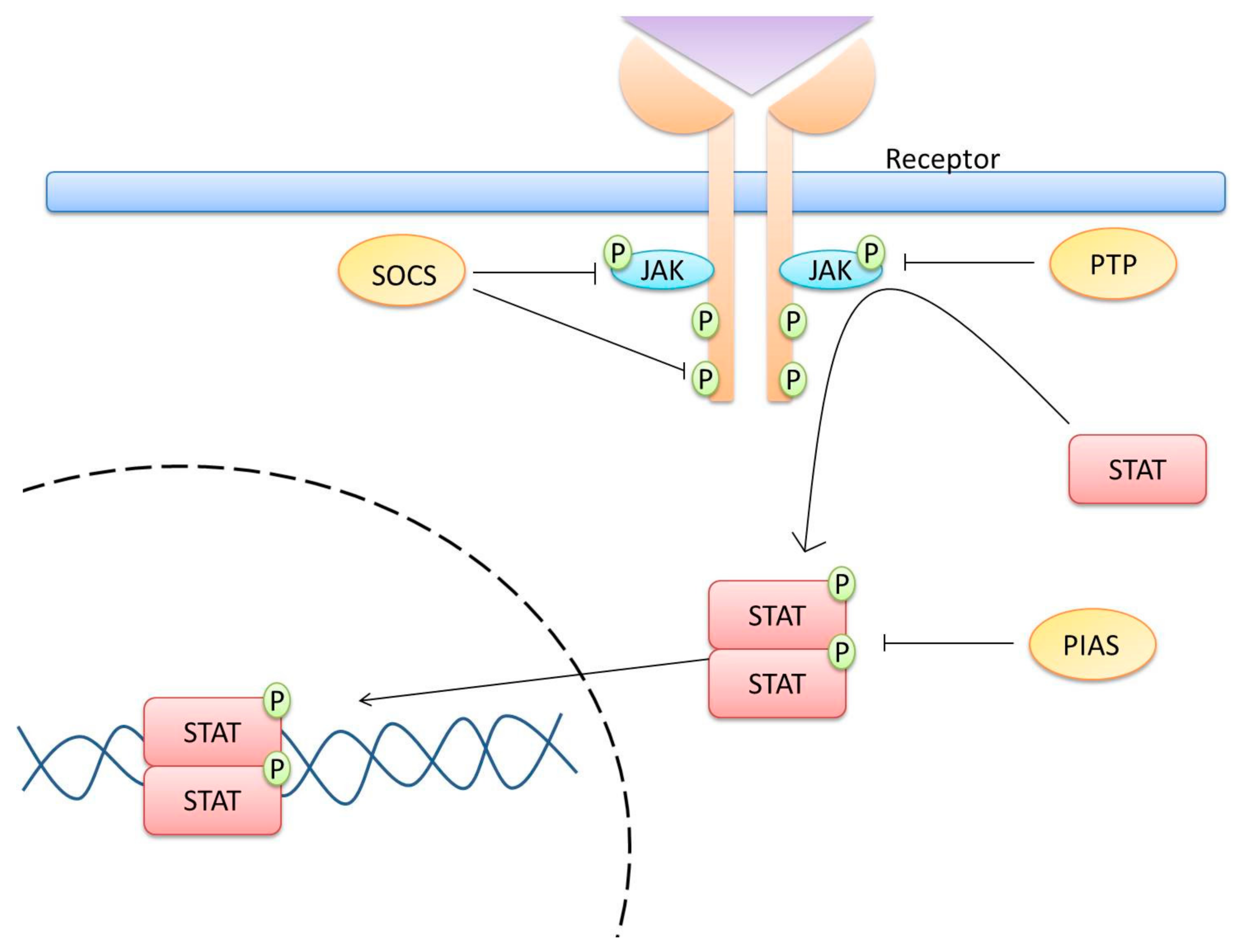
2. JAK/STAT Pathway
3. Central Players for the Activation and Regulation of the JAK/STAT Pathway
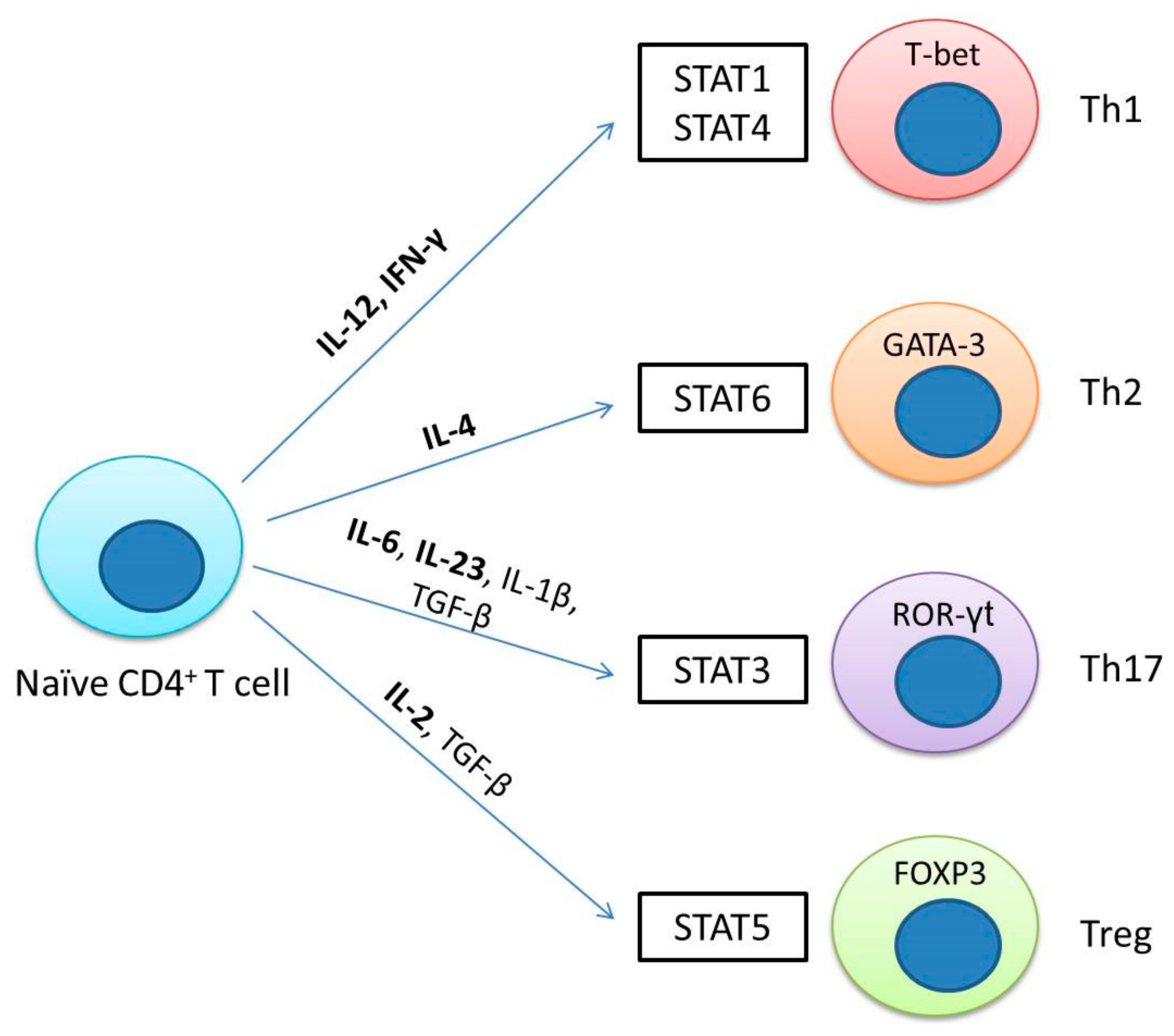
4. The JAK/STAT Pathway Is Involved in T Helper Cell Differentiation
5. JAK/STAT Pathway and Cervical Cancer
5.1. Role of STAT Proteins in Cervical Cancer
5.1.1. STAT1
5.1.2. STAT2
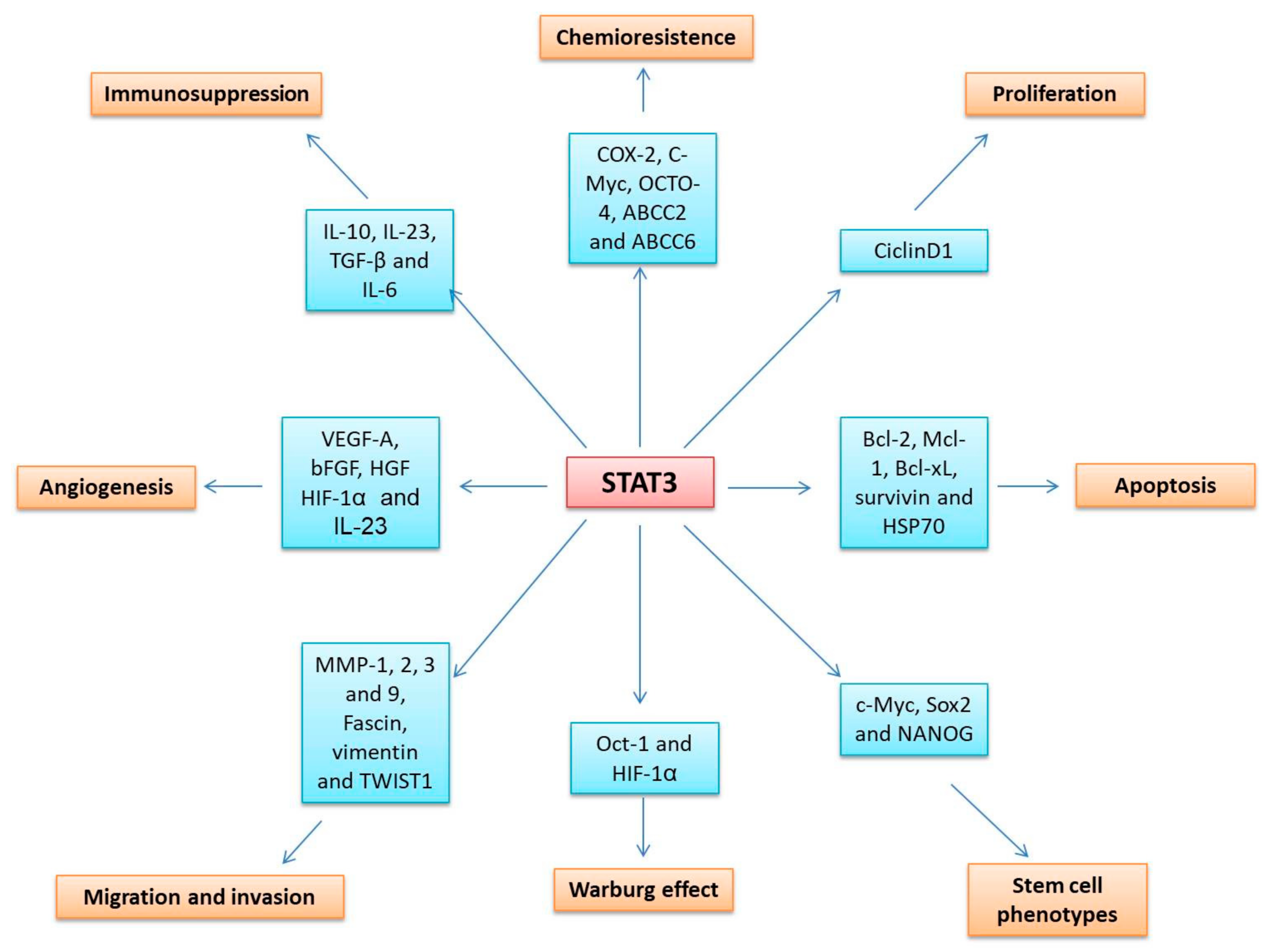
5.1.3. STAT3
5.1.4. STAT4
5.1.5. STAT5
5.1.6. STAT6
6. SOCS and Cervical Cancer
7. Inhibition of the JAK/STAT Pathway as a Therapeutic Target in Cervical Cancer
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sanjose, S.; Quint, W.G.V.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Wang, R.; Pan, W.; Jin, L.; Huang, W.; Li, Y.; Wu, D.; Gao, C.; Ma, D.; Liao, S. Human papillomavirus vaccine against cervical cancer: Opportunity and challenge. Cancer Lett. 2020, 471, 88–102. [Google Scholar] [CrossRef]
- Drolet, M.; Bénard, É.; Pérez, N.; Brisson, M.; Ali, H.; Boily, M.C.; Baldo, V.; Brassard, P.; Brotherton, J.M.L.; Callander, D.; et al. Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: Updated systematic review and meta-analysis. Lancet 2019, 394, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Songock, W.K.; Kim, S.M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [Green Version]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-risk human papillomaviral oncogenes E6 and E7 target key cellular pathways to achieve oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2019, 94. [Google Scholar] [CrossRef]
- Zheng, Z.M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2011, 1809, 668–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, R.R.; Matunis, E.L. JAK-STAT Signaling in Stem Cells. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2013; Volume 786, pp. 247–267. [Google Scholar]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAK-STAT 2012, 1, 184–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Livnah, O.; Stura, E.A.; Middleton, S.A.; Johnson, D.L.; Jolliffe, L.K.; Wilson, I.A. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 1999, 283, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Adams, J.J.; Pelekanos, R.A.; Wan, Y.; McKinstry, W.J.; Palethorpe, K.; Seeber, R.M.; Monks, T.A.; Eidne, K.A.; Parke, M.W.; et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 2005, 12, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.J. Minireview: Receptor dimerization in GH and erythropoietin action-It takes two to Tango, but how? Endocrinology 2002, 143, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Dai, W.; O’Mara, M.L.; Abankwa, D.; Chhabra, Y.; Pelekanos, R.A.; Gardon, O.; Tunny, K.A.; Blucher, K.M.; Morton, C.J.; et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 2014, 344. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.X.; Zheng, Z.; Chen, X.; Perrimon, N. The JAK/STAT pathway in model organisms: Emerging roles in cell movement. Dev. Cell 2002, 3, 765–778. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Premkumar Reddy, E. JAK/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Danial, N.N.; Losman, J.A.; Lu, T.; Yip, N.; Krishnan, K.; Krolewski, J.; Goff, S.P.; Wang, J.Y.J.; Rothman, P.B. Direct Interaction of Jak1 and v-Abl Is Required for v-Abl-Induced Activation of STATs and Proliferation. Mol. Cell. Biol. 1998, 18, 6795–6804. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Turkson, J.; Carter-Su, C.; Smithgall, T.; Levitzki, A.; Kraker, A.; Krolewski, J.J.; Medveczky, P.; Jove, R. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J. Biol. Chem. 2000, 275, 24935–24944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Uddin, S.; Majchrzak, B.; Huynh, T.; Proudfoot, A.E.I.; Platanias, L.C.; Fish, E.N. RANTES Activates Jak2 and Jak3 to Regulate Engagement of Multiple Signaling Pathways in T Cells. J. Biol. Chem. 2001, 276, 11427–11431. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Kantarjian, H.; Cortes, J.; Verstovsek, S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat. Rev. Drug Discov. 2011, 10, 127–140. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Lahesmaa, R.; Vahedi, G.; Laurence, A.; Kanno, Y. Genomic views of STAT function in CD4 + T helper cell differentiation. Nat. Rev. Immunol. 2011, 11, 239–250. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.G.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [Green Version]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation and cancer. JAK-STAT 2013, 2, e24053. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.J.; Xu, J.D.; Yuan, W.J.; Sun, J.J.; Yang, M.C.; He, Z.H.; Zhao, X.F.; Wang, J.X. Protein inhibitor of activated STAT (PIAS) negatively regulates the JAK/STAT pathway by inhibiting STAT phosphorylation and translocation. Front. Immunol. 2018, 9, 2392. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell. Res. 2006, 16, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehender, A.; Huang, J.; Györfi, A.H.; Matei, A.E.; Trinh-Minh, T.; Xu, X.; Li, Y.N.; Chen, C.W.; Lin, J.; Dees, C.; et al. The tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to regulate fibroblast activation and fibrosis. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Niu, R. Functions of Shp2 in cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef]
- Liu, Q.; Qu, J.; Zhao, M.; Xu, Q.; Sun, Y. Targeting SHP2 as a promising strategy for cancer immunotherapy. Pharmacol. Res. 2020, 152, 104595. [Google Scholar] [CrossRef]
- Liu, W.; Yu, W.M.; Zhang, J.; Chan, R.J.; Loh, M.L.; Zhang, Z.; Bunting, K.D.; Qu, C.K. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 2017, 31, 1415–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Xiang, D.M.; Sun, W.; Liu, N.; Sun, H.L.; Wen, W.; Shen, W.F.; Wang, R.Y.; Chen, C.; Wang, X.; et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 2015, 63, 651–660. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y. Understanding the development and function of T follicular helper cells. Cell. Mol. Immunol. 2010, 7, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Magee, C.N.; Boenisch, O.; Najafian, N. The role of costimulatory molecules in directing the functional differentiation of alloreactive T helper cells. Am. J. Transplant. 2012, 12, 2588–2600. [Google Scholar] [CrossRef] [PubMed]
- Wertek, F.; Xu, C. Digital response in T cells: To be or not to be. Cell Res. 2014, 24, 265–266. [Google Scholar] [CrossRef] [Green Version]
- Gee, K.; Guzzo, C.; Mat, N.F.C.; Ma, W.; Kumar, A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm. Allergy Drug Targets 2009, 8, 40–52. [Google Scholar] [CrossRef]
- Delespine-Carmagnat, M.; Bouvier, G.; Bertoglio, J. Association of STAT1, STAT3 and STAT5 proteins with the IL-2 receptor involves different subdomains of the IL-2 receptor β chain. Eur. J. Immunol. 2000, 30, 59–68. [Google Scholar] [CrossRef]
- Tormo, A.J.; Letellier, M.C.; Sharma, M.; Elson, G.; Crabé, S.; Gauchat, J.F. IL-6 activates STAT5 in T cells. Cytokine 2012, 60, 575–582. [Google Scholar] [CrossRef]
- Leung, S.; Liu, X.; Fang, L.; Chen, X.; Guo, T.; Zhang, J. The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell. Mol. Immunol. 2010, 7, 182–189. [Google Scholar] [CrossRef]
- Freudenberg, K.; Lindner, N.; Dohnke, S.; Garbe, A.I.; Schallenberg, S.; Kretschmer, K. Critical role of TGF-β and IL-2 receptor signaling in Foxp3 induction by an inhibitor of DNA methylation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Sanchez, M.E.; Huerta, L.; Alvarez-Buylla, E.R.; Luján, C.V. Role of cytokine combinations on CD4+ T cell differentiation, partial polarization, and plasticity: Continuous network modeling approach. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 2010, 238, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Thieu, V.T.; Yu, Q.; Chang, H.C.; Yeh, N.; Nguyen, E.T.; Sehra, S.; Kaplan, M.H. Signal Transducer and Activator of Transcription 4 Is Required for the Transcription Factor T-bet to Promote T Helper 1 Cell-Fate Determination. Immunity 2008, 29, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Huang, H.; Huang, Z. STAT1 signaling is required for optimal Th1 cell differentiation in mice. Chin. Sci. Bull. 2010, 55, 1032–1040. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Suto, A.; Iwamoto, T.; Kashiwakuma, D.; Kagami, S.I.; Suzuki, K.; Takatori, H.; Tamachi, T.; Hirose, K.; Onodera, A.; et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORγt induction as downstream targets of Stat3. J. Exp. Med. 2014, 211, 1857–1874. [Google Scholar] [CrossRef] [PubMed]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 Receptor β-Dependent STAT5 Activation is Required for the Development of Foxp3 + Regulatory T Cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.H.; Lai, A.G. An immunoevasive strategy through clinically-relevant pan-cancer genomic and transcriptomic alterations of JAK-STAT signaling components. Mol. Med. 2019, 25. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [Green Version]
- Walboomers, J.M.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.F.; Peto, J.; Meijer, C.J.L.M.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Chen, J. Signaling pathways in HPV-associated cancers and therapeutic implications. Rev. Med. Virol. 2015, 25, 24–53. [Google Scholar] [CrossRef] [PubMed]
- Campos-Parra, A.D.; Padua-Bracho, A.; Pedroza-Torres, A.; Figueroa-González, G.; Fernández-Retana, J.; Millan-Catalan, O.; Peralta-Zaragoza, O.; Cantú de León, D.; Herrera, L.A.; Pérez-Plasencia, C. Comprehensive transcriptome analysis identifies pathways with therapeutic potential in locally advanced cervical cancer. Gynecol. Oncol. 2016, 143, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordziel, C.; Bratsch, J.; Moriggl, R.; Knösel, T.; Friedrich, K. Both STAT1 and STAT3 are favourable prognostic determinants in colorectal carcinoma. Br. J. Cancer 2013, 109, 138–146. [Google Scholar] [CrossRef]
- Zhang, Y.; Molavi, O.; Su, M.; Lai, R. The clinical and biological significance of STAT1 in esophageal squamous cell carcinoma. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [Green Version]
- Dimco, G.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010, 9, 4638–4649. [Google Scholar] [CrossRef] [Green Version]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lee, M.S. STAT1 as a key modulator of cell death. Cell. Signal. 2007, 19, 454–465. [Google Scholar] [CrossRef]
- Sironi, J.J.; Ouchi, T. STAT1-induced Apoptosis Is Mediated by Caspases 2, 3, and 7. J. Biol. Chem. 2004, 279, 4066–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.K.; Gimeno, R.; Levy, D.E. Differential regulation of constitutive major histocompatibility complex class I expression in T and B lymphocytes. J. Exp. Med. 1999, 190, 1451–1463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jin, G.; Zhang, J.; Mi, R.; Zhou, Y.; Fan, W.; Cheng, S.; Song, W.; Zhang, B.; Ma, M.; et al. Overexpression of STAT1 suppresses angiogenesis under hypoxia by regulating VEGF-A in human glioma cells. Biomed. Pharmacother. 2018, 104, 566–575. [Google Scholar] [CrossRef]
- Battle, T.E.; Lynch, R.A.; Frank, D.A. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006, 66, 3649–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, C.W.; Willey, C.D.; Zhi, D.; Cui, X.; Harris, J.J.; Vaughan, L.K.; Mehta, T.; McCubrey, R.O.; Khodarev, N.N.; Weichselbaum, R.R.; et al. Expression Signature of IFN/STAT1 Signaling Genes Predicts Poor Survival Outcome in Glioblastoma Multiforme in a Subtype-Specific Manner. PLoS ONE 2012, 7, e29653. [Google Scholar] [CrossRef] [PubMed]
- Khodarev, N.; Ahmad, R.; Rajabi, H.; Pitroda, S.; Kufe, T.; McClary, C.; Joshi, M.D.; MacDermed, D.; Weichselbaum, R.; Kufe, D. Cooperativity of the MUC1 oncoprotein and STAT1 pathway in poor prognosis human breast cancer. Oncogene 2010, 29, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hix, L.M.; Karavitis, J.; Khan, M.W.; Shi, Y.H.; Khazaie, K.; Zhang, M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J. Biol. Chem. 2013, 288, 11676–11688. [Google Scholar] [CrossRef] [Green Version]
- Khodarev, N.N.; Roach, P.; Pitroda, S.P.; Golden, D.W.; Bhayani, M.; Shao, M.Y.; Darga, T.E.; Beveridge, M.G.; Sood, R.F.; Sutton, H.G.; et al. STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernandez, N.; Metodiev, M.V. Stat1 and CD74 overexpression is co-dependent and linked to increased invasion and lymph node metastasis in triple-negative breast cancer. J. Proteom. 2012, 75, 3031–3040. [Google Scholar] [CrossRef]
- Zimmerman, M.A.; Rahman, N.T.; Yang, D.; Lahat, G.; Lazar, A.J.; Pollock, R.E.; Lev, D.; Liu, K. Unphosphorylated STAT1 promotes sarcoma development through repressing expression of fas and bad and conferring apoptotic resistance. Cancer Res. 2012, 72, 4724–4732. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yamazaki, T.; Pietrocola, F.; Zhou, H.; Zitvogel, L.; Ma, Y.; Kroemer, G. STAT3 inhibition enhances the therapeutic efficacy of immunogenic chemotherapy by stimulating type 1 interferon production by cancer cells. Cancer Res. 2015, 75, 3812–3822. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, T.; Sabitha, K.; Vijayalakshmi, N.; Shirley, S.; Bose, M.V.; Gopal, G.; Selvaluxmy, G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Yang, M.; She, S.; Min, H.; Xv, X.; Ran, X.; Wu, Y.; Wang, W.; Wang, L.; Yi, L.; et al. ITRAQ-based quantitative proteomic analysis of cervical cancer. Int. J. Oncol. 2015, 46, 1748–1758. [Google Scholar] [CrossRef]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol. Lett. 2020, 19, 3316–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.E.; Laimins, L.A. Microarray Analysis Identifies Interferon-Inducible Genes and Stat-1 as Major Transcriptional Targets of Human Papillomavirus Type 31. J. Virol. 2000, 74, 4174–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 Expression by Human Papillomaviruses Is Necessary for Differentiation-Dependent Genome Amplification and Plasmid Maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus Type 16 Oncogenes Downregulate Expression of Interferon-Responsive Genes and Upregulate Proliferation-Associated and NF-κB-Responsive Genes in Cervical Keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, R.L.; Xiao, S.S.; Xue, M. Cisplatin inhibits proliferation of cervical carcinoma cell line by up-regulating Stat1 expression. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 88–92. [Google Scholar] [CrossRef]
- Buttarelli, M.; Babini, G.; Raspaglio, G.; Filippetti, F.; Battaglia, A.; Ciucci, A.; Ferrandina, G.; Petrillo, M.; Marino, C.; Mancuso, M.; et al. A combined ANXA2-NDRG1-STAT1 gene signature predicts response to chemoradiotherapy in cervical cancer. J. Exp. Clin. Cancer Res. 2019, 38, 279. [Google Scholar] [CrossRef] [Green Version]
- Gamero, A.M.; Young, M.R.; Mentor-Marcel, R.; Bobe, G.; Scarzello, A.J.; Wise, J.; Colburn, N.H. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev. Res. 2010, 3, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Yue, C.; Xu, J.; Tan Estioko, M.D.; Kotredes, K.P.; Lopez-Otalora, Y.; Hilliard, B.A.; Baker, D.P.; Gallucci, S.; Gamero, A.M. Host STAT2/type i interferon axis controls tumor growth. Int. J. Cancer 2015, 136, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Pelzel, C.; Begitt, A.; Mee, M.; Elsheikha, H.M.; Scott, D.J.; Vinkemeier, U. STAT2 Is a Pervasive Cytokine Regulator due to Its Inhibition of STAT1 in Multiple Signaling Pathways. PLoS Biol. 2016, 14, e2000117. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Gao, L.H.; Cao, L.J.; Feng, D.Y.; Cao, Y.; Luo, Q.Z.; Yu, P.; Li, M. Detection of STAT2 in early stage of cervical premalignancy and in cervical cancer. Asian Pac. J. Trop. Med. 2012, 5, 738–742. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Labrecque, S.; Gauzzi, M.C.; Cuddihy, A.R.; Wong, A.H.T.; Pellegrini, S.; Matlashewski, G.J.; Koromilas, A.E. The human papilloma virus (HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs Jak-STAT activation by interferon-α. Oncogene 1999, 18, 5727–5737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonsson, A.; Payne, E.; Hengst, K.; McMillan, D.N.A.J. The Human Papillomavirus Type 16 E7 Protein Binds Human Interferon Regulatory Factor-9 via a Novel PEST Domain Required for Transformation. J. Interf. Cytokine Res. 2006, 26, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Végran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation. JAK-STAT 2013, 2, e23010. [Google Scholar] [CrossRef] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports RasDependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Lo, H.W. STAT3 target genes relevant to human cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, C.; Huang, M.; Jin, X.; Cho, K.; Lilja, J.; Reynolds, R.K.; Lin, J. Elevated phosphorylation of AKT and Stat3 in prostate, breast, and cervical cancer cells. Int. J. Oncol. 2000, 17, 23–28. [Google Scholar] [CrossRef]
- Shukla, S.; Mahata, S.; Shishodia, G.; Pandey, A.; Tyagi, A.; Vishnoi, K.; Basir, S.F.; Das, B.C.; Bharti, A.C. Functional Regulatory Role of STAT3 in HPV16-Mediated Cervical Carcinogenesis. PLoS ONE 2013, 8, 67849. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in hpv positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca Suarez, A.A.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Jadli, M.; Thakur, K.; Shishodia, G.; Mahata, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Level of phospho-STAT3 (Tyr705) correlates with copy number and physical state of human papillomavirus 16 genome in cervical precancer and cancer lesions. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, I.; Grattendick, K.G.; Tyring, S.K. Interleukin-10 induces transcription of the early promoter of human papillomavirus type 16 (HPV16) through the 5′-segment of the upstream regulatory region (URR). Antiviral Res. 2002, 55, 331–339. [Google Scholar] [CrossRef]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 activation by E6 is essential for the differentiation-dependent HPV18 life cycle. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, Z. The clinical value of HPV E6/E7 and STAT3 mRNA detection in cervical cancer screening. Pathol. Res. Pract. 2018, 214, 767–775. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3 and Stat4: Members of the family of signal transducers and activators of transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 4806–4810. [Google Scholar] [CrossRef] [Green Version]
- Wurster, A.L.; Tanaka, T.; Grusby, M.J. The biology of Stat4 and Stat6. Oncogene 2000, 19, 2577–2584. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ji, G.; Le, X.; Luo, Z.; Wang, C.; Feng, M.; Xu, L.; Zhang, Y.; Lau, W.B.; Lau, B.; et al. An integrated analysis identifies STAT4 as a key regulator of ovarian cancer metastasis. Oncogene 2017, 36, 3384–3396. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Ryan, N.; Volpedo, G.; Varikuti, S.; Satoskar, A.R.; Oghumu, S. Immune Suppression Mediated by STAT4 Deficiency Promotes Lymphatic Metastasis in HNSCC. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, M.; Batsaikhan, B.E.; Yoshikawa, K.; Higashijima, J.; Tokunaga, T.; Takasu, C.; Kashihara, H.; Ishikawa, D.; Shimada, M. High STAT4 Expression Indicates Better Disease-free Survival in Patients with Gastric Cancer. Anticancer Res. 2017, 37, 6723–6729. [Google Scholar] [CrossRef]
- Núñez-Marrero, A. Assessing the Role of the Interleukin-12/STAT4 Axis in Breast Cancer by a Bioinformatics Approach. Int. J. Sci. Basic Appl. Res. 2019, 48, 38–52. [Google Scholar] [PubMed]
- Luo, J.; Huang, Q.; Lin, X.; Wei, K.; Ling, Y.; Su, S.; Cao, Y.; Luo, J.; Pan, D.; Dang, Y.; et al. STAT4 expression is correlated with clinicopathological characteristics of cervical lesions. Int. J. Clin. Exp. Pathol. 2016, 9, 3751–3758. [Google Scholar]
- Ibarra Sierra, E.; Díaz Chávez, J.; Cortés-Malagón, E.M.; Uribe-Figueroa, L.; Hidalgo-Miranda, A.; Lambert, P.F.; Gariglio, P. Differential gene expression between skin and cervix induced by the E7 oncoprotein in a transgenic mouse model. Virology 2012, 433, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadreev, I.I.; Chen, M.Z.Q.; Umezawa, Y.; Biktashev, V.N.; Kemper, C.; Salakhieva, D.V.; Welsh, G.I.; Kotov, N.V. The competitive nature of signal transducer and activator of transcription complex formation drives phenotype switching of T cells. Immunology 2018, 153, 488–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yang, X.; Wang, Y.; Zhang, H.; Guo, Z. Interleukin-35 is associated with the tumorigenesis and progression of prostate cancer. Oncol. Lett. 2019, 17, 5094–5102. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Chen, X.; Wang, F.; Shao, Q.; Liu, J.; Zhao, H.; Yuan, C.; Ren, H.; Mao, H. Breast cancer cell-derived IL-35 promotes tumor progression via induction of IL-35-producing induced regulatory T cells. Carcinogenesis 2018, 39, 1488–1496. [Google Scholar] [CrossRef] [Green Version]
- Bitar, M.; Boldt, A.; Freitag, M.-T.; Gruhn, B.; Köhl, U.; Sack, U. Evaluating STAT5 Phosphorylation as a Mean to Assess T Cell Proliferation. Front. Immunol. 2019, 10, 722. [Google Scholar] [CrossRef] [Green Version]
- Hassel, J.C.; Winnemöller, D.; Schartl, M.; Wellbrock, C. STAT5 contributes to antiapoptosis in melanoma. Melanoma Res. 2008, 18, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Lui, V.W.Y.; Man, D.; Xi, S.; Chai, R.L.; Nelson, E.; Tobey, A.B.J.; Grandis, J.R. Constitutive activation of signal transducer and activator of transcription 5 contributes to tumor growth, epithelial-mesenchymal transition, and resistance to epidermal growth factor receptor targeting. Clin. Cancer Res. 2008, 14, 7682–7690. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Addya, S.; Fortina, P.; Dasgupta, A.; Hyslop, T.; et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am. J. Pathol. 2010, 176, 1959–1972. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Glass, A.; Zellweger, T.; Gehan, E.; Bubendorf, L.; Gelmann, E.P.; Nevalainen, M.T. Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin. Cancer Res. 2005, 11, 5863–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaller, J.; Parganas, E.; Wang, D.; Cain, D.; Aster, J.C.; Williams, I.R.; Lee, C.K.; Gerthner, R.; Kitamura, T.; Frantsve, J.; et al. Stat5 is essential for the myelo-and lymphoproliferative disease induced by TEL/JAK2. Mol. Cell 2000, 6, 693–704. [Google Scholar] [CrossRef]
- Li, H.; Ahonen, T.J.; Alanen, K.; Xie, J.; LeBaron, M.J.; Pretlow, T.G.; Ealley, E.L.; Zhang, Y.; Nurmi, M.; Singh, B.; et al. Activation of signal transducer and activator of transcription 5 in human prostate cancer is associated with high histological grade. Cancer Res. 2004, 64, 4774–4782. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Cao, S. Activation of STAT5 contributes to proliferation in U87 human glioblastoma multiforme cells. Mol. Med. Rep. 2014, 10, 203–210. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of Bcr/Abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Xiong, H.; Su, W.Y.; Liang, Q.C.; Zhang, Z.G.; Chen, H.M.; Du, W.; Chen, Y.X.; Fang, J.Y. Inhibition of STAT5 induces G1 cell cycle arrest and reduces tumor cell invasion in human colorectal cancer cells. Lab. Investig. 2009, 89, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.W.; Chou, C.Y.; Wu, Y.H.; Hsueh, W.T.; Hsu, C.H.; Guo, H.R.; Lee, W.Y.; Su, W.C. Constitutive STAT5 activation correlates with better survival in cervical cancer patients treated with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 658–666. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Z.; Hofstetter, W.L.; Zhou, Y.; Hu, W.; Guo, C.; Wang, L.; Guo, W.; Pataer, A.; Correa, A.M.; et al. Aberrant expression of proteins involved in signal transduction and DNA repair pathways in lung cancer and their association with clinical parameters. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Wang, S.; Yu, L.; Shi, W.; Li, X.; Yu, L. Prognostic roles of signal transducers and activators of transcription family in human breast cancer. Biosci. Rep. 2018, 38, 20171175. [Google Scholar] [CrossRef]
- Sultan, A.S.; Xie, J.; LeBaron, M.J.; Ealley, E.L.; Nevalainen, M.T.; Rui, H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene 2005, 24, 746–760. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, H.; Nishio, M.; Ando, Y.; Zhang, Z.; Hamaguchi, M.; Mita, K.; Kobayashi, S.; Fujii, Y.; Iwase, H. Stat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocr. Relat. Cancer 2006, 13, 885–893. [Google Scholar] [CrossRef]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Bharadwaj, M.; Das, B.C. Deregulation of STAT-5 isoforms in the development of HPV-mediated cervical carcinogenesis. J. Recept. Signal Transduct. 2010, 30, 178–188. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. JAK2 Inhibition Impairs Proliferation and Sensitises Cervical Cancer Cells to Cisplatin-Induced Cell Death. Cancers 2019, 11, 1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Laimins, L.A. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Corona, R.; Corona-Ortega, T.; Soto-Cruz, I.; López-Labra, A.; Pablo-Arcos, T.; Torres-Guarneros, C.F.; Weiss-Steider, B. Evidence that cervical cancer cells secrete IL-2, which becomes an autocrine growth factor. Cytokine 2010, 50, 273–277. [Google Scholar] [CrossRef]
- Valle-Mendiola, A.; Weiss-Steider, B.; Rocha-Zavaleta, L.; Soto-Cruz, I. IL-2 enhances cervical cancer cells proliferation and JAK3/STAT5 phosphorylation at low doses, while at high doses IL-2 has opposite effects. Cancer Investig. 2014, 32, 115–125. [Google Scholar] [CrossRef]
- Lagunas-Cruz, M.D.C.; Valle-Mendiola, A.; Trejo-Huerta, J.; Rocha-Zavaleta, L.; Mora-García, M.D.L.; Gutiérrez-Hoya, A.; Weiss-Steider, B.; Soto-Cruz, I. IL-2 induces transient arrest in the G1 phase to protect cervical cancer cells from entering apoptosis. J. Oncol. 2019, 2019, 7475295. [Google Scholar] [CrossRef]
- Lopez, T.V.; Lappin, T.R.J.; Maxwell, P.; Shi, Z.; Lopez-Marure, R.; Aguilar, C.; Rocha-Zavaleta, L. Autocrine/paracrine erythropoietin signalling promotes JAK/STAT-dependent proliferation of human cervical cancer cells. Int. J. Cancer 2011, 129, 2566–2576. [Google Scholar] [CrossRef]
- Novak, U.; Mui, A.; Miyajima, A.; Paradiso, L. Formation of STAT5-containing DNA Binding Complexes in Response to Colony-stimulating Factor-1 and Platelet-derived Growth Factor. J. Biol. Chem. 1996, 271, 18350–18354. [Google Scholar] [CrossRef] [Green Version]
- Goenka, S.; Kaplan, M.H. Transcriptional regulation by STAT6. Immunol. Res. 2011, 50, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Vignali, D.A.A. STAT heterodimers in immunity. JAK-STAT 2013, 2, e23060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Lin, C.W.; Lin, Y.J.; Sheu, J.J.C.; Chen, B.H.; Liao, C.C.; Tsai, Y.; Lin, W.Y.; Lai, C.H.; Tsai, F.J. Type i IFN induced IL1-Ra expression in hepatocytes is mediated by activating STAT6 through the formation of STAT2: STAT6 heterodimer. J. Cell. Mol. Med. 2008, 12, 876–888. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Jiang, M.; Pernis, A.B. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. J. Immunol. 1999, 163, 3834–3841. [Google Scholar] [PubMed]
- Xu, B.I.N.; Lu, X.; Zhao, Y.; Liu, C.; Huang, X.; Chen, S.; Zhu, W.; Zhang, L.; Chen, M. MicroRNA-135a induces prostate cancer cell apoptosis via inhibition of STAT6. Oncol. Lett. 2019, 17, 1889–1895. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Roth, C.P.; Wasson, L.M.; Vishwanatha, J.K. Signal transducer and activator of transcription-6 (STAT6) is a constitutively expressed survival factor in human prostate cancer. Prostate 2007, 67, 1550–1564. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 axis is required for IL-4-induced epithelial-mesenchymal transition of colorectal cancer cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, J.; Liu, H.; Wan, L.; Zhang, H.; Huang, Q.; Xu, E.; Lai, M. IL-13/STAT6 signaling plays a critical role in the epithelialmesenchymal transition of colorectal cancer cells. Oncotarget 2016, 7, 61183–61198. [Google Scholar] [CrossRef] [Green Version]
- Merk, B.C.; Owens, J.L.; Lopes, M.B.S.; Silva, C.M.; Hussaini, I.M. STAT6 expression in glioblastoma promotes invasive growth. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [Green Version]
- Salguero-Aranda, C.; Sancho-Mensat, D.; Canals-Lorente, B.; Sultan, S.; Reginald, A.; Chapman, L. STAT6 knockdown using multiple siRNA sequences inhibits proliferation and induces apoptosis of human colorectal and breast cancer cell lines. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- Lee, H.L.; Park, M.H.; Song, J.K.; Jung, Y.Y.; Kim, Y.; Kim, K.B.; Hwang, D.Y.; Yoon, D.Y.; Song, M.J.; Han, S.B.; et al. Tumor growth suppressive effect of IL-4 through p21-mediated activation of STAT6 in IL-4Rα overexpressed melanoma models. Oncotarget 2016, 7, 23425–23438. [Google Scholar] [CrossRef] [Green Version]
- Gooch, J.L.; Christy, B.; Yee, D. STAT6 mediates interleukin-4 growth inhibition in human breast cancer cells. Neoplasia 2002, 4, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Guan, Y.Q.; Liu, J.M. The role of STAT-6 as a key transcription regulator in HeLa cell death induced by IFN-γ/TNF-α co-immobilized on nanoparticles. Biomaterials 2014, 35, 5016–5027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, X.; Hu, L.; Ma, Y.; Xiu, Z.; Huang, B.; Feng, Y.; Tang, X. Overexpression of human papillomavirus type 16 oncoproteins enhances epithelial-mesenchymal transition via STAT3 signaling pathway in non-small cell lung cancer cells. Oncol. Res. 2017, 25, 843–852. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.-S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef] [Green Version]
- Neuwirt, H.; Puhr, M.; Santer, F.R.; Susani, M.; Doppler, W.; Marcias, G.; Rauch, V.; Brugger, M.; Hobisch, A.; Kenner, L.; et al. Suppressor of cytokine signaling (SOCS)-1 is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am. J. Pathol. 2009, 174, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chen, H.; Zhou, L.; Huang, X.; Su, F.; Wang, P. Identification of SOCS family members with prognostic values in human ovarian cancer. Am. J. Transl. Res. 2020, 12, 1824–1838. [Google Scholar] [PubMed]
- Lv, Y.; Song, G.E.; Li, P. Correlation of SOCS-1 gene with onset and prognosis of breast cancer. Oncol. Lett. 2018, 16, 383–387. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, M.S.; Kim, W.; Kang, M.A.; Cacalano, N.A.; Kang, S.B.; Shin, Y.J.; Jeong, J.H. Suppressor of cytokine signaling (SOCS) genes are silenced by DNA hypermethylation and histone deacetylation and regulate response to radiotherapy in cervical cancer cells. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Nijhawan, R.; Bharti, A.C.; Bharadwaj, M.; Das, B.C. Aberrant promoter methylation and loss of Suppressor of Cytokine Signalling-1 gene expression in the development of uterine cervical carcinogenesis. Cell. Oncol. 2011, 34, 533–543. [Google Scholar] [CrossRef]
- Kamio, M.; Yoshida, T.; Ogata, H.; Douchi, T.; Nagata, Y.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Yoshimura, A. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 2004, 23, 3107–3115. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef]
- Das, R.; Bhattacharya, K.; Samanta, S.K.; Pal, B.C.; Mandal, C. Improved chemosensitivity in cervical cancer to cisplatin: Synergistic activity of mahanine through STAT3 inhibition. Cancer Lett. 2014, 351, 81–90. [Google Scholar] [CrossRef]
- Sun, C.Y.; Nie, J.; Huang, J.P.; Zheng, G.J.; Feng, B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed. Pharmacother. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.L.; Rao, W. SiRNA interfering STAT3 enhances DDP sensitivity in cervical cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4098–4106. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lu, Y.; Pang, Y.; Li, M.; Cheng, X.; Chen, J. Propofol enhances the cisplatin-induced apoptosis on cervical cancer cells via EGFR/JAK2/STAT3 pathway. Biomed. Pharmacother. 2017, 86, 324–333. [Google Scholar] [CrossRef]
- Yao, X.; Zhu, F.; Zhao, Z.; Liu, C.; Luo, L.; Yin, Z. Arctigenin enhances chemosensitivity of cancer cells to cisplatin through inhibition of the STAT3 signaling pathway. J. Cell. Biochem. 2011, 112, 2837–2849. [Google Scholar] [CrossRef]
- Gotthardt, D.; Sexl, V. STATs in NK-Cells: The good, the bad, and the ugly. Front. Immunol. 2017, 7, 694. [Google Scholar] [CrossRef] [Green Version]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- McLornan, D.P.; Khan, A.A.; Harrison, C.N. Immunological Consequences of JAK Inhibition: Friend or Foe? Curr. Hematol. Malig. Rep. 2015, 10, 370–379. [Google Scholar] [CrossRef]
- Damele, L.; Ottonello, S.; Mingari, M.C.; Pietra, G.; Vitale, C. Targeted therapies: Friends or foes for patient’s NK cell-mediated tumor immune-surveillance? Cancers 2020, 12, 774. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.S.; Ferreira, D.; Paige, E.; Gedye, C.; Boyle, M. Infectious complications of biological and small molecule targeted immunomodulatory therapies. Clin. Microbiol. Rev. 2020, 33, 1–117. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Type I Receptors | Shared γc subunit | IL-2R, IL-4R, IL-7R, IL-9R, IL-15R, and IL-21R | JAK1, JAK2, and JAK3 |
| Shared gp130 subunit | IL6R, IL-11R, OSMR, LIFR, CNTFR, and IL-27R | JAK1, JAK2, and TYK2 | |
| Shared βc subunit | IL-3R, IL-5R, and GM-CSFR | JAK2 | |
| Shared IL-12Rβ1 subunit | IL-12R and IL-23R | TYK2 and JAK2 | |
| Homodimeric cytokine receptors | EPOR, G-CSFR, GHR, PRLR, TPOR, and LEPR | JAK1 and JAK2 | |
| Type II Receptors | Interferon receptors | IFNα, IFNβ, IFNγ, IL-10R, IL-19R, IL-20R, IL-22R, IL-24R, IL-28R, and IL-29R | JAK1, JAK2, and TYK2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutiérrez-Hoya, A.; Soto-Cruz, I. Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins. Cells 2020, 9, 2297. https://doi.org/10.3390/cells9102297
Gutiérrez-Hoya A, Soto-Cruz I. Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins. Cells. 2020; 9(10):2297. https://doi.org/10.3390/cells9102297
Chicago/Turabian StyleGutiérrez-Hoya, Adriana, and Isabel Soto-Cruz. 2020. "Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins" Cells 9, no. 10: 2297. https://doi.org/10.3390/cells9102297
APA StyleGutiérrez-Hoya, A., & Soto-Cruz, I. (2020). Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins. Cells, 9(10), 2297. https://doi.org/10.3390/cells9102297