Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments


Abstract
1. Introduction
2. Role of Cholesterol in Normal Cells and Cancer
2.1. Role of Cholesterol in Normal Cells
2.2. Role of Cholesterol in Cancer
3. Physiological Role of FDFT1
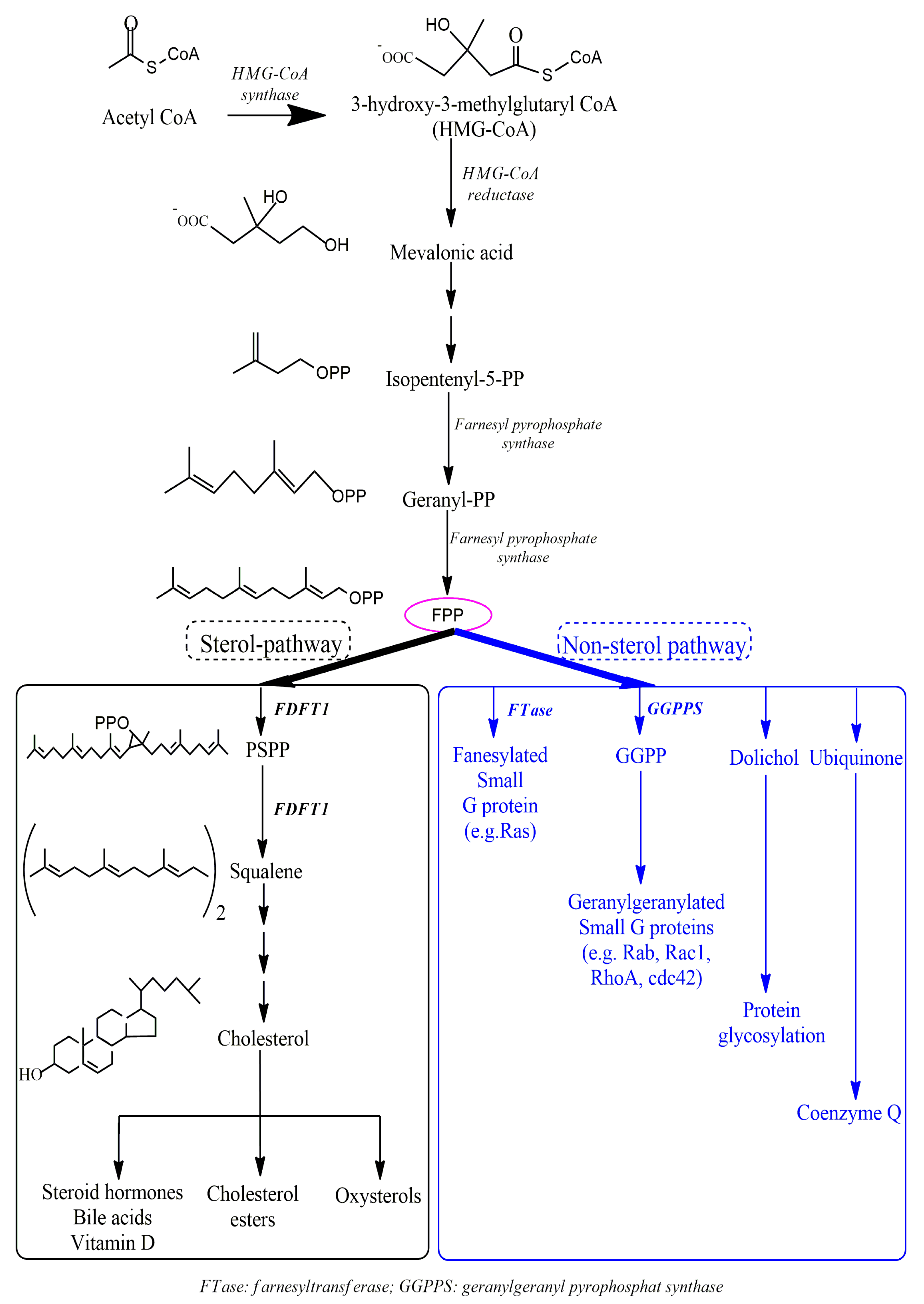
3.1. The Roles of FDFT1 in the Cholesterol Biosynthesis Pathway
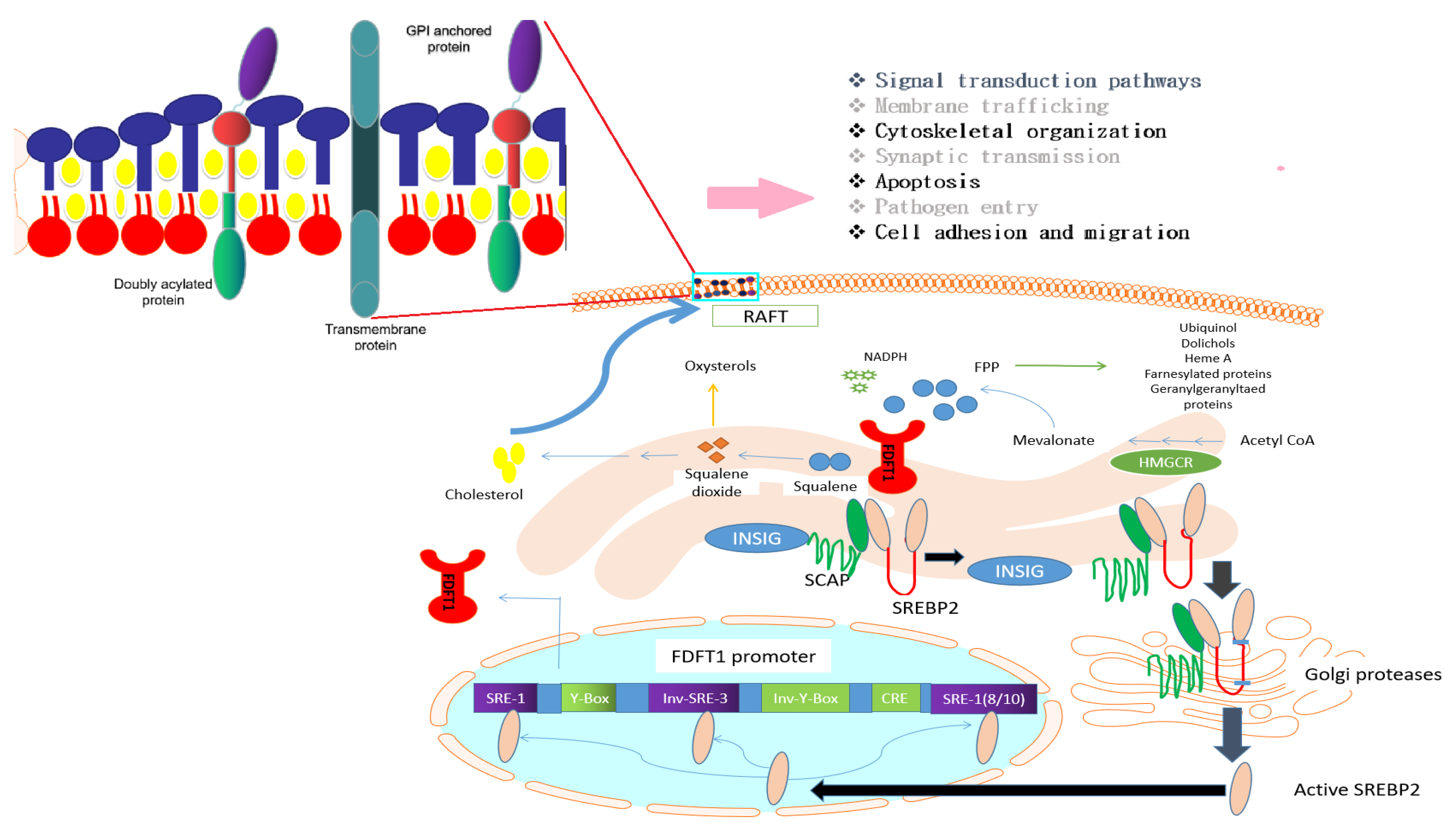
3.2. Transcriptional Regulation of FDFT1
3.3. Binding Partners of FDFT1
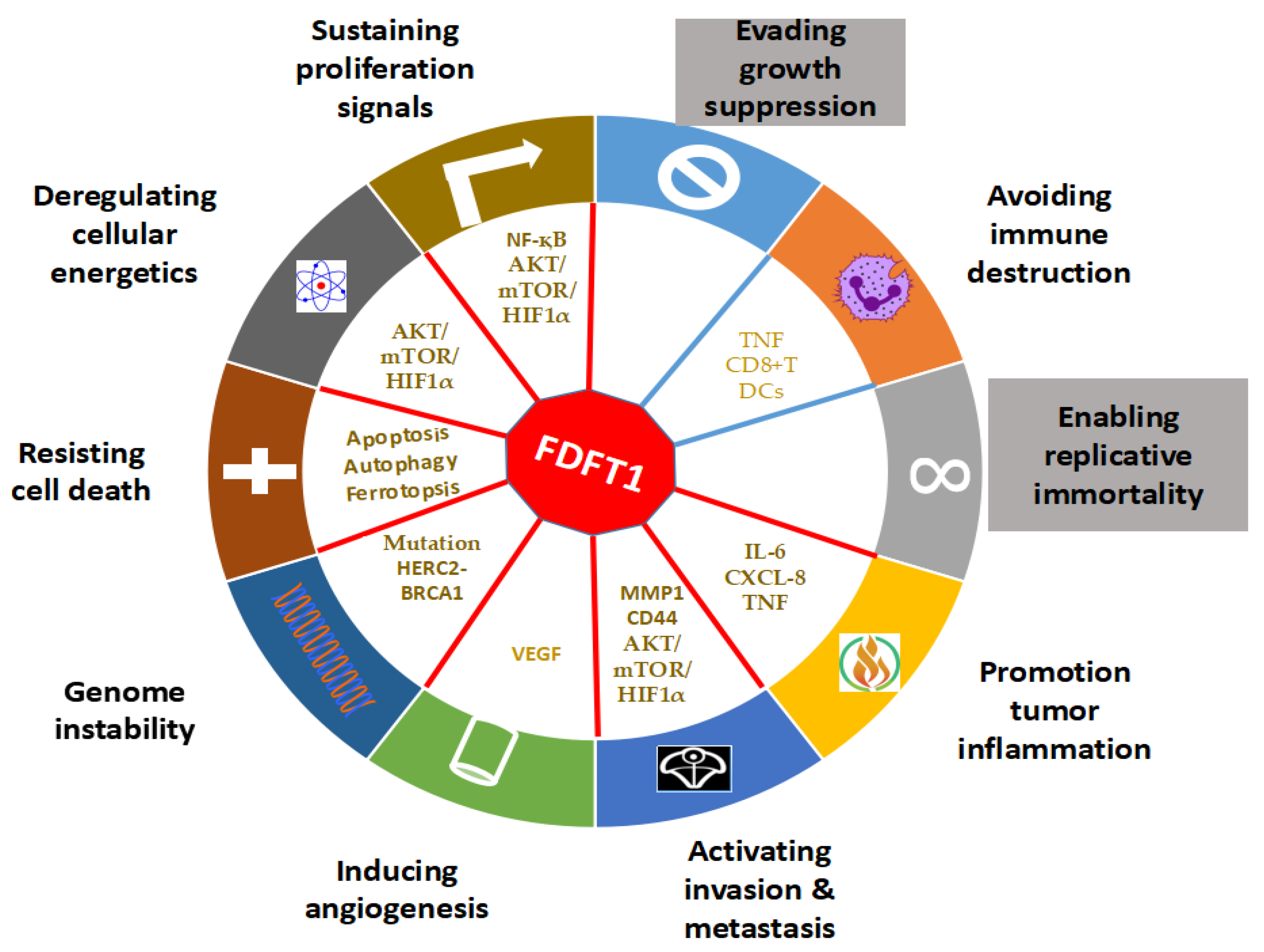
4. The Role of FDFT1 in Cancer
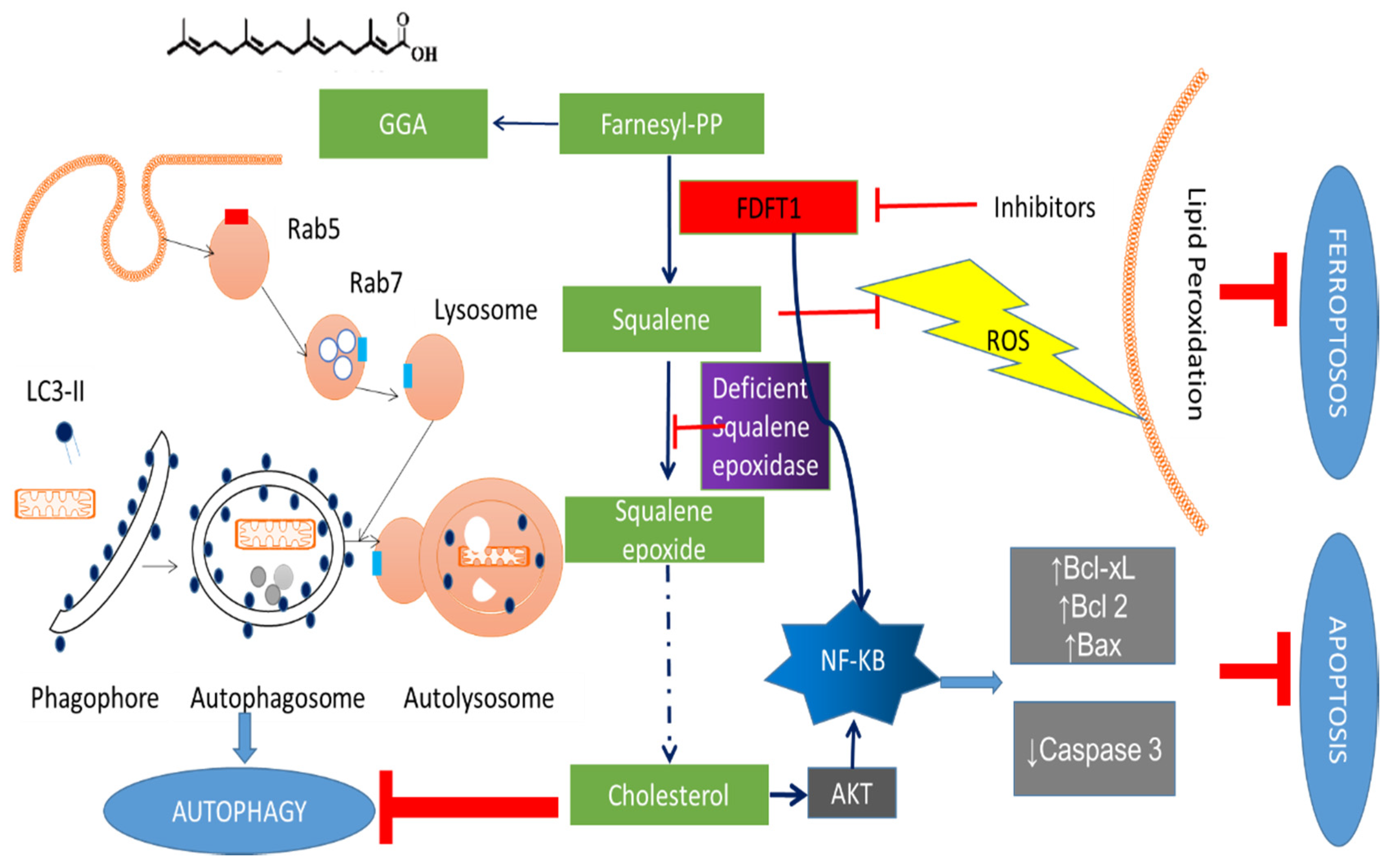
4.1. Effects of FDFT1 on Proliferation and Cell Death
4.2. Effects of FDFT1 on Genomic Instability
4.3. Effects of FDFT1 on Invasion and Metastasis
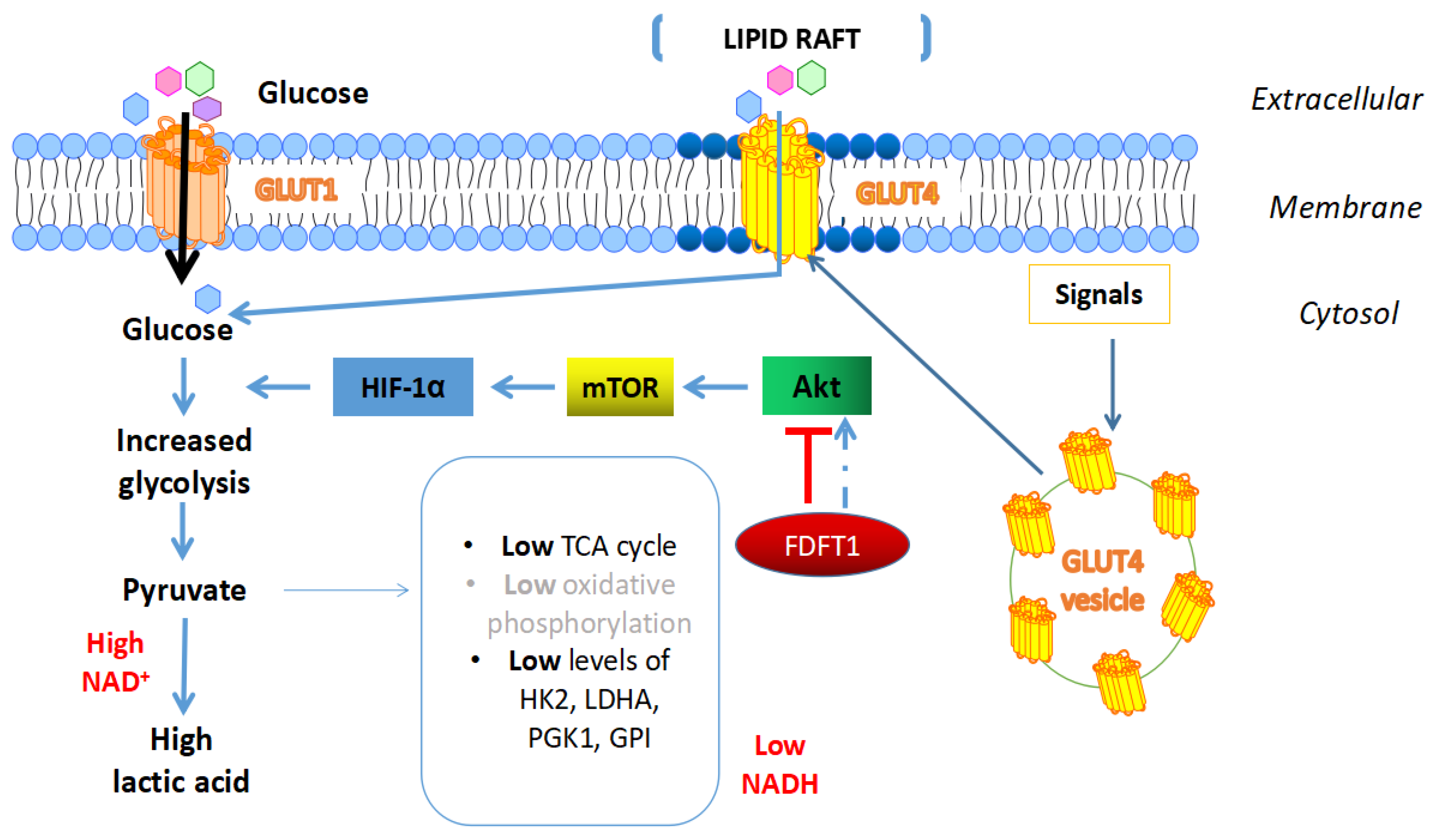
4.4. Effects of FDFT1 on Cancer Metabolism
4.5. Effects of FDFT1 on Angiogenesis and Inflammation
4.6. Effects of FDFT1 on Immune Evasion and Neuronal Contribution
5. FDFT1 Inhibitors as Anticancer Agents
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelissen:, I.C.; Brown, A.J. An overview of cholesterol homeostasis. Methods Mol. Biol. 2017, 1583, 1–6. [Google Scholar] [CrossRef]
- Lin, C.J.; Lai, C.K.; Kao, M.C.; Wu, L.T.; Lo, U.G.; Lin, L.C.; Chen, Y.A.; Lin, H.; Hsieh, J.T.; Lai, C.H.; et al. Impact of cholesterol on disease progression. Biomedicine 2015, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.M.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Yang, Y.F.; Chang, Y.C.; Jan, Y.H.; Yang, C.J.; Huang, M.S.; Hsiao, M. Squalene synthase promotes the invasion of lung cancer cells via the osteopontin/ERK pathway. Oncogenesis 2020, 9, 78. [Google Scholar] [CrossRef]
- Weng, M.L.; Chen, W.K.; Chen, X.Y.; Lu, H.; Sun, Z.R.; Yu, Q.; Sun, P.F.; Xu, Y.J.; Zhu, M.M.; Jiang, N.; et al. Fasting inhibits aerobic glycolysis and proliferation in colorectal cancer via the Fdft1-mediated AKT/mTOR/HIF1alpha pathway suppression. Nat. Commun. 2020, 11, 1869. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Wang, Y.; Rogers, P.M.; Su, C.; Varga, G.; Stayrook, K.R.; Burris, T.P. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J. Biol. Chem. 2008, 283, 26332–26339. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, C.M.; Brumell, J.H.; Finlay, B.B. Microbial pathogenesis: Lipid rafts as pathogen portals. Curr. Biol. 2000, 10, R823–R825. [Google Scholar] [CrossRef]
- Sebastiao, A.M.; Colino-Oliveira, M.; Assaife-Lopes, N.; Dias, R.B.; Ribeiro, J.A. Lipid rafts, synaptic transmission and plasticity: Impact in age-related neurodegenerative diseases. Neuropharmacology 2013, 64, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118, quiz 1404-1105. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hager, M.H.; Solomon, K.R.; Freeman, M.R. The role of cholesterol in prostate cancer. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 379–385. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Moreno-Bueno, G. Updating the role of obesity and cholesterol in breast cancer. Breast Cancer Res. 2019, 21, 35. [Google Scholar] [CrossRef]
- Abdeen, S.K.; Ben-David, U.; Shweiki, A.; Maly, B.; Aqeilan, R.I. Somatic loss of WWOX is associated with TP53 perturbation in basal-like breast cancer. Cell Death Dis. 2018, 9, 832. [Google Scholar] [CrossRef]
- Hu, J.; La Vecchia, C.; de Groh, M.; Negri, E.; Morrison, H.; Mery, L.; Canadian Cancer Registries Epidemiology Research, G. Dietary cholesterol intake and cancer. Ann. Oncol. 2012, 23, 491–500. [Google Scholar] [CrossRef]
- Jarvinen, R.; Knekt, P.; Hakulinen, T.; Rissanen, H.; Heliovaara, M. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br. J. Cancer 2001, 85, 357–361. [Google Scholar] [CrossRef]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep 2017, 18, 2228–2242. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular cholesterol directly activates smoothened in hedgehog signaling. Cell 2016, 166, 1176–1187.e1114. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, G.; Sircar, R.; Kong, J.H.; Nachtergaele, S.; Sagner, A.; Byrne, E.F.; Covey, D.F.; Siebold, C.; Rohatgi, R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Guixa-Gonzalez, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Marti-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martin, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 14505. [Google Scholar] [CrossRef]
- Sheng, R.; Chen, Y.; Yung Gee, H.; Stec, E.; Melowic, H.R.; Blatner, N.R.; Tun, M.P.; Kim, Y.; Kallberg, M.; Fujiwara, T.K.; et al. Cholesterol modulates cell signaling and protein networking by specifically interacting with PDZ domain-containing scaffold proteins. Nat. Commun. 2012, 3, 1249. [Google Scholar] [CrossRef]
- Vaquero, J.; Nguyen Ho-Bouldoires, T.H.; Claperon, A.; Fouassier, L. Role of the PDZ-scaffold protein NHERF1/EBP50 in cancer biology: From signaling regulation to clinical relevance. Oncogene 2017, 36, 3067–3079. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, J. Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol. Biol. Cell 2008, 19, 4366–4373. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef]
- Attard, G.; Cooper, C.S.; de Bono, J.S. Steroid hormone receptors in prostate cancer: A hard habit to break? Cancer Cell 2009, 16, 458–462. [Google Scholar] [CrossRef]
- Finlay-Schultz, J.; Sartorius, C.A. Steroid hormones, steroid receptors, and breast cancer stem cells. J. Mammary Gland Biol. Neoplasia 2015, 20, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Thysell, E.; Surowiec, I.; Hornberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic characterization of human prostate cancer bone metastases reveals increased levels of cholesterol. PLoS ONE 2010, 5, e14175. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Smith, B.; Land, H. Anticancer activity of the cholesterol exporter ABCA1 gene. Cell Rep. 2012, 2, 580–590. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Raccosta, L.; Zhou, D.; Fontana, R.; Maggioni, D.; Negro, A.; Sanvito, F.; Ponzoni, M.; Valentinis, B.; Bregni, M.; et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 2010, 16, 98–105. [Google Scholar] [CrossRef]
- Baek, H.S.; Park, N.; Kwon, Y.J.; Ye, D.J.; Shin, S.; Chun, Y.J. Annexin A5 suppresses cyclooxygenase-2 expression by downregulating the protein kinase C-zeta-nuclear factor-kappaB signaling pathway in prostate cancer cells. Oncotarget 2017, 8, 74263–74275. [Google Scholar] [CrossRef] [PubMed]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef] [PubMed]
- Tansey, T.R.; Shechter, I. Squalene synthase: Structure and regulation. Prog. Nucleic. Acid. Res. Mol. Biol. 2001, 65, 157–195. [Google Scholar] [CrossRef] [PubMed]
- Shechter, I.; Conrad, D.G.; Hart, I.; Berger, R.C.; McKenzie, T.L.; Bleskan, J.; Patterson, D. Localization of the squalene synthase gene (FDFT1) to human chromosome 8p22-p23.1. Genomics 1994, 20, 116–118. [Google Scholar] [CrossRef]
- Do, R.; Kiss, R.S.; Gaudet, D.; Engert, J.C. Squalene synthase: A critical enzyme in the cholesterol biosynthesis pathway. Clin. Genet. 2009, 75, 19–29. [Google Scholar] [CrossRef]
- Grunler, J.; Ericsson, J.; Dallner, G. Branch-point reactions in the biosynthesis of cholesterol, dolichol, ubiquinone and prenylated proteins. Biochim. Biophys. Acta 1994, 1212, 259–277. [Google Scholar] [CrossRef]
- Okazaki, H.; Tazoe, F.; Okazaki, S.; Isoo, N.; Tsukamoto, K.; Sekiya, M.; Yahagi, N.; Iizuka, Y.; Ohashi, K.; Kitamine, T.; et al. Increased cholesterol biosynthesis and hypercholesterolemia in mice overexpressing squalene synthase in the liver. J. Lipid. Res. 2006, 47, 1950–1958. [Google Scholar] [CrossRef]
- Charlton-Menys, V.; Durrington, P.N. Squalene synthase inhibitors: Clinical pharmacology and cholesterol-lowering potential. Drugs 2007, 67, 11–16. [Google Scholar] [CrossRef]
- Faust, J.R.; Goldstein, J.L.; Brown, M.S. Synthesis of ubiquinone and cholesterol in human fibroblasts: Regulation of a branched pathway. Arch. Biochem. Biophys. 1979, 192, 86–99. [Google Scholar] [CrossRef]
- Friesen, J.A.; Rodwell, V.W. The 3-hydroxy-3-methylglutaryl coenzyme-A (HMG-CoA) reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef]
- Seiki, S.; Frishman, W.H. Pharmacologic inhibition of squalene synthase and other downstream enzymes of the cholesterol synthesis pathway: A new therapeutic approach to treatment of hypercholesterolemia. Cardiol. Rev. 2009, 17, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.B.; Casey, P.J. The role of prenylation in G-protein assembly and function. Cell Signal. 1996, 8, 433–437. [Google Scholar] [CrossRef]
- Waller, D.D.; Park, J.; Tsantrizos, Y.S. Inhibition of farnesyl pyrophosphate (FPP) and/or geranylgeranyl pyrophosphate (GGPP) biosynthesis and its implication in the treatment of cancers. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Baranyi, M.; Buday, L.; Hegedus, B. K-Ras prenylation as a potential anticancer target. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Edwards, P.A.; Kast, H.R.; Anisfeld, A.M. BAREing it all: The adoption of LXR and FXR and their roles in lipid homeostasis. J. Lipid. Res. 2002, 43, 2–12. [Google Scholar]
- Bonnans, C.; Levy, B.D. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am. J. Respir. Cell Mol. Biol. 2007, 36, 201–205. [Google Scholar] [CrossRef]
- Corey, E.; Grogan, M.J. Stereocontrolled syntheses of 24 (S), 25-epoxycholesterol and related oxysterols for studies on the activation of LXR receptors. Tetrahedron Lett. 1998, 39, 9351–9354. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518. [Google Scholar] [CrossRef]
- Horton, J.D.; Shimomura, I.; Brown, M.S.; Hammer, R.E.; Goldstein, J.L.; Shimano, H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Investig. 1998, 101, 2331–2339. [Google Scholar] [CrossRef]
- Liao, J.K. Squalene synthase inhibitor lapaquistat acetate: Could anything be better than statins? Circulation 2011, 123, 1925–1928. [Google Scholar] [CrossRef]
- Park, J.; Matralis, A.N.; Berghuis, A.M.; Tsantrizos, Y.S. Human isoprenoid synthase enzymes as therapeutic targets. Front. Chem. 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell. Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.E.; Ioannou, Y.A. Apoptosis-induced release of mature sterol regulatory element-binding proteins activates sterol-responsive genes. J. Lipid. Res. 2001, 42, 1939–1946. [Google Scholar]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Faust, T.B.; Li, Y.; Bacon, C.W.; Jang, G.M.; Weiss, A.; Jayaraman, B.; Newton, B.W.; Krogan, N.J.; D’Orso, I.; Frankel, A.D. The HIV-1 Tat protein recruits a ubiquitin ligase to reorganize the 7SK snRNP for transcriptional activation. eLife 2018, 7. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B.; et al. A reference map of the human binary protein interactome. Nature 2020, 580, 402–408. [Google Scholar] [CrossRef]
- Liu, Z.; Chu, S.; Yao, S.; Li, Y.; Fan, S.; Sun, X.; Su, L.; Liu, X. CD74 interacts with CD44 and enhances tumorigenesis and metastasis via RHOA-mediated cofilin phosphorylation in human breast cancer cells. Oncotarget 2016, 7, 68303–68313. [Google Scholar] [CrossRef]
- Foerster, S.; Kacprowski, T.; Dhople, V.M.; Hammer, E.; Herzog, S.; Saafan, H.; Bien-Moller, S.; Albrecht, M.; Volker, U.; Ritter, C.A. Characterization of the EGFR interactome reveals associated protein complex networks and intracellular receptor dynamics. Proteomics 2013, 13, 3131–3144. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Rubin Grandis, J.; Melhem, M.F.; Gooding, W.E.; Day, R.; Holst, V.A.; Wagener, M.M.; Drenning, S.D.; Tweardy, D.J. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J. Natl. Cancer Inst. 1998, 90, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed. Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef]
- Giurato, G.; Nassa, G.; Salvati, A.; Alexandrova, E.; Rizzo, F.; Nyman, T.A.; Weisz, A.; Tarallo, R. Quantitative mapping of RNA-mediated nuclear estrogen receptor beta interactome in human breast cancer cells. Sci. Data 2018, 5, 180031. [Google Scholar] [CrossRef] [PubMed]
- Hiramitsu, S.; Ishikawa, T.; Lee, W.R.; Khan, T.; Crumbley, C.; Khwaja, N.; Zamanian, F.; Asghari, A.; Sen, M.; Zhang, Y.; et al. Estrogen Receptor Beta-Mediated Modulation of Lung Cancer Cell Proliferation by 27-Hydroxycholesterol. Front. Endocrinol. 2018, 9, 470. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Bass, M.D.; Craig, S.E.; Pinney, J.W.; Knight, D.; Humphries, M.J. Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci. Signal. 2009, 2, ra51. [Google Scholar] [CrossRef]
- Xiao, J.; Yang, W.; Xu, B.; Zhu, H.; Zou, J.; Su, C.; Rong, J.; Wang, T.; Chen, Z. Expression of fibronectin in esophageal squamous cell carcinoma and its role in migration. BMC Cancer 2018, 18, 976. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Liu, C.; Zhang, T.N.; Zhu, Y.W.; Dong, X.; Xue, P. Down-regulation of FN1 inhibits colorectal carcinogenesis by suppressing proliferation, migration, and invasion. J. Cell Biochem. 2018, 119, 4717–4728. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nakayama, H.; Nagata, M.; Yoshida, R.; Kawahara, K.; Hirosue, A.; Tanaka, T.; Yuno, A.; Matsuoka, Y.; Kojima, T.; et al. Overexpression of fibronectin confers cell adhesion-mediated drug resistance (CAM-DR) against 5-FU in oral squamous cell carcinoma cells. Int. J. Oncol. 2014, 44, 1376–1384. [Google Scholar] [CrossRef]
- Wang, J.; Deng, L.; Huang, J.; Cai, R.; Zhu, X.; Liu, F.; Wang, Q.; Zhang, J.; Zheng, Y. High expression of Fibronectin 1 suppresses apoptosis through the NF-kappaB pathway and is associated with migration in nasopharyngeal carcinoma. Am. J. Transl. Res. 2017, 9, 4502–4511. [Google Scholar]
- Sun, Y.; Zhao, C.; Ye, Y.; Wang, Z.; He, Y.; Li, Y.; Mao, H. High expression of fibronectin 1 indicates poor prognosis in gastric cancer. Oncol. Lett. 2020, 19, 93–102. [Google Scholar] [CrossRef]
- Galligan, J.T.; Martinez-Noel, G.; Arndt, V.; Hayes, S.; Chittenden, T.W.; Harper, J.W.; Howley, P.M. Proteomic analysis and identification of cellular interactors of the giant ubiquitin ligase HERC2. J. Proteome. Res. 2015, 14, 953–966. [Google Scholar] [CrossRef]
- Bonanno, L.; Costa, C.; Majem, M.; Sanchez, J.J.; Rodriguez, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Vergnenegre, A.; Massuti, B.; Favaretto, A.; et al. Combinatory effect of BRCA1 and HERC2 expression on outcome in advanced non-small-cell lung cancer. BMC Cancer 2016, 16, 312. [Google Scholar] [CrossRef]
- Montano, M.M.; Yeh, I.J.; Chen, Y.; Hernandez, C.; Kiselar, J.G.; de la Fuente, M.; Lawes, A.M.; Nieman, M.T.; Kiser, P.D.; Jacobberger, J.; et al. Inhibition of the histone demethylase, KDM5B, directly induces re-expression of tumor suppressor protein HEXIM1 in cancer cells. Breast Cancer Res. 2019, 21, 138. [Google Scholar] [CrossRef]
- Ogba, N.; Doughman, Y.Q.; Chaplin, L.J.; Hu, Y.; Gargesha, M.; Watanabe, M.; Montano, M.M. HEXIM1 modulates vascular endothelial growth factor expression and function in breast epithelial cells and mammary gland. Oncogene 2010, 29, 3639–3649. [Google Scholar] [CrossRef]
- Yu, L.; Jearawiriyapaisarn, N.; Lee, M.P.; Hosoya, T.; Wu, Q.; Myers, G.; Lim, K.C.; Kurita, R.; Nakamura, Y.; Vojtek, A.B.; et al. BAP1 regulation of the key adaptor protein NCoR1 is critical for gamma-globin gene repression. Genes Dev. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Bai, J.; Yeh, S.; Qiu, X.; Hu, L.; Zeng, J.; Cai, Y.; Zuo, L.; Li, G.; Yang, G.; Chang, C. TR4 nuclear receptor promotes clear cell renal cell carcinoma (ccRCC) vasculogenic mimicry (VM) formation and metastasis via altering the miR490-3p/vimentin signals. Oncogene 2018, 37, 5901–5912. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Ma, Y.; Chen, J.; Dong, B.; Zhao, W.; Wang, X.; Zheng, Q.; Fang, F.; Yang, Y. Testicular orphan receptor 4 (TR4) is a marker for metastasis and poor prognosis in non-small cell lung cancer that drives the EMT phenotype. Lung Cancer 2015, 89, 320–328. [Google Scholar] [CrossRef]
- Qiu, X.; Zhu, J.; Sun, Y.; Fan, K.; Yang, D.R.; Li, G.; Yang, G.; Chang, C. TR4 nuclear receptor increases prostate cancer invasion via decreasing the miR-373-3p expression to alter TGFbetaR2/p-Smad3 signals. Oncotarget 2015, 6, 15397–15409. [Google Scholar] [CrossRef]
- Ding, X.; Yang, D.R.; Lee, S.O.; Chen, Y.L.; Xia, L.; Lin, S.J.; Yu, S.; Niu, Y.J.; Li, G.; Chang, C. TR4 nuclear receptor promotes prostate cancer metastasis via upregulation of CCL2/CCR2 signaling. Int. J. Cancer 2015, 136, 955–964. [Google Scholar] [CrossRef]
- Shi, G.; Liu, C.; Yang, Y.; Song, L.; Liu, X.; Wang, C.; Peng, Z.; Li, H.; Zhong, L. Panx1 promotes invasion-metastasis cascade in hepatocellular carcinoma. J. Cancer 2019, 10, 5681–5688. [Google Scholar] [CrossRef]
- Lai, C.P.; Bechberger, J.F.; Thompson, R.J.; MacVicar, B.A.; Bruzzone, R.; Naus, C.C. Tumor-suppressive effects of pannexin 1 in C6 glioma cells. Cancer Res. 2007, 67, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Jalaleddine, N.; El-Hajjar, L.; Dakik, H.; Shaito, A.; Saliba, J.; Safi, R.; Zibara, K.; El-Sabban, M. Pannexin1 Is Associated with Enhanced Epithelial-To-Mesenchymal Transition in Human Patient Breast Cancer Tissues and in Breast Cancer Cell Lines. Cancers 2019, 11, 1967. [Google Scholar] [CrossRef] [PubMed]
- Asperger, H.; Stamm, N.; Gierke, B.; Pawlak, M.; Hofmann, U.; Zanger, U.M.; Marton, A.; Katona, R.L.; Buhala, A.; Vizler, C.; et al. Progesterone receptor membrane component 1 regulates lipid homeostasis and drives oncogenic signaling resulting in breast cancer progression. Breast Cancer Res. 2020, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Ewing, R.M.; Chu, P.; Elisma, F.; Li, H.; Taylor, P.; Climie, S.; McBroom-Cerajewski, L.; Robinson, M.D.; O’Connor, L.; Li, M.; et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Lauscher, J.C.; Loddenkemper, C.; Kosel, L.; Grone, J.; Buhr, H.J.; Huber, O. Increased pontin expression in human colorectal cancer tissue. Hum. Pathol. 2007, 38, 978–985. [Google Scholar] [CrossRef]
- Huber, O.; Menard, L.; Haurie, V.; Nicou, A.; Taras, D.; Rosenbaum, J. Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res. 2008, 68, 6873–6876. [Google Scholar] [CrossRef]
- Grigoletto, A.; Lestienne, P.; Rosenbaum, J. The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim. Biophys. Acta 2011, 1815, 147–157. [Google Scholar] [CrossRef]
- Lu, C.; Fang, S.; Weng, Q.; Lv, X.; Meng, M.; Zhu, J.; Zheng, L.; Hu, Y.; Gao, Y.; Wu, X.; et al. Integrated analysis reveals critical glycolytic regulators in hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 97. [Google Scholar] [CrossRef]
- Christianson, J.C.; Olzmann, J.A.; Shaler, T.A.; Sowa, M.E.; Bennett, E.J.; Richter, C.M.; Tyler, R.E.; Greenblatt, E.J.; Harper, J.W.; Kopito, R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2011, 14, 93–105. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic. Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. Embo J. 2007, 26, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Hubel, P.; Urban, C.; Bergant, V.; Schneider, W.M.; Knauer, B.; Stukalov, A.; Scaturro, P.; Mann, A.; Brunotte, L.; Hoffmann, H.H.; et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol. 2019, 20, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Wagai, S.; Kasamatsu, A.; Iyoda, M.; Hayashi, F.; Hiroshima, K.; Yoshimura, S.; Miyamoto, I.; Nakashima, D.; Endo-Sakamoto, Y.; Shiiba, M.; et al. UNC93B1 promotes tumoral growth by controlling the secretion level of granulocyte macrophage colony-stimulating factor in human oral cancer. Biochem. Biophys. Res. Commun. 2019, 513, 81–87. [Google Scholar] [CrossRef]
- Gourley, C.; Paige, A.J.; Taylor, K.J.; Ward, C.; Kuske, B.; Zhang, J.; Sun, M.; Janczar, S.; Harrison, D.J.; Muir, M. WWOX gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin α3. Cancer Res. 2009, 69, 4835–4842. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.J.; Glover, T.W.; Zhu, X.X.; Kuick, R.; Thoraval, D.; Orringer, M.B.; Beer, D.G.; Hanash, S. A novel amplicon at 8p22-23 results in overexpression of cathepsin B in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 12410–12415. [Google Scholar] [CrossRef]
- Nupponen, N.N.; Kakkola, L.; Koivisto, P.; Visakorpi, T. Genetic alterations in hormone-refractory recurrent prostate carcinomas. Am. J. Pathol. 1998, 153, 141–148. [Google Scholar] [CrossRef]
- Zhao, H.; Kim, Y.; Wang, P.; Lapointe, J.; Tibshirani, R.; Pollack, J.R.; Brooks, J.D. Genome-wide characterization of gene expression variations and DNA copy number changes in prostate cancer cell lines. Prostate 2005, 63, 187–197. [Google Scholar] [CrossRef]
- Wolf, M.; Mousses, S.; Hautaniemi, S.; Karhu, R.; Huusko, P.; Allinen, M.; Elkahloun, A.; Monni, O.; Chen, Y.; Kallioniemi, A.; et al. High-resolution analysis of gene copy number alterations in human prostate cancer using CGH on cDNA microarrays: Impact of copy number on gene expression. Neoplasia 2004, 6, 240–247. [Google Scholar] [CrossRef]
- Levy, A.; Dang, U.C.; Bookstein, R. High-density screen of human tumor cell lines for homozygous deletions of loci on chromosome arm 8p. Genes Chromosomes Cancer 1999, 24, 42–47. [Google Scholar] [CrossRef]
- Dehghani, M.; Samani, Z.; Abidi, H.; Manzouri, L.; Mahmoudi, R.; Hosseini Teshnizi, S.; Nikseresht, M. Relationship of SNP rs2645429 in Farnesyl-Diphosphate Farnesyltransferase 1 Gene Promoter with Susceptibility to Lung Cancer. Int. J. Genom. 2018, 2018, 4863757. [Google Scholar] [CrossRef] [PubMed]
- Stattermayer, A.F.; Rutter, K.; Beinhardt, S.; Wrba, F.; Scherzer, T.M.; Strasser, M.; Hofer, H.; Steindl-Munda, P.; Trauner, M.; Ferenci, P. Role of FDFT1 polymorphism for fibrosis progression in patients with chronic hepatitis C. Liver Int. 2014, 34, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, Y.; Matsui, H.; Koike, H.; Sekine, Y.; Shechter, I.; Ohtake, N.; Nakata, S.; Ito, K.; Suzuki, K. Role of squalene synthase in prostate cancer risk and the biological aggressiveness of human prostate cancer. Prostate. Cancer Prostatic. Dis. 2012, 15, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, Y.; Uchida, T.; Karnan, S.; Noguchi, T.; Nguyen, L.T.; Tanigawa, M.; Takeuchi, I.; Matsuura, K.; Hijiya, N.; Nakada, C.; et al. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. J. Pathol. 2008, 216, 471–482. [Google Scholar] [CrossRef]
- Ma, Y.S.; Wu, Z.J.; Zhang, H.W.; Cai, B.; Huang, T.; Long, H.D.; Xu, H.; Zhao, Y.Z.; Yin, Y.Z.; Xue, S.B.; et al. Dual Regulatory Mechanisms of Expression and Mutation Involving Metabolism-Related Genes FDFT1 and UQCR5 during CLM. Mol. Oncolytics. 2019, 14, 172–178. [Google Scholar] [CrossRef]
- Yang, Y.F.; Jan, Y.H.; Liu, Y.P.; Yang, C.J.; Su, C.Y.; Chang, Y.C.; Lai, T.C.; Chiou, J.; Tsai, H.Y.; Lu, J.; et al. Squalene synthase induces tumor necrosis factor receptor 1 enrichment in lipid rafts to promote lung cancer metastasis. Am. J. Respir. Crit. Care Med. 2014, 190, 675–687. [Google Scholar] [CrossRef]
- Brusselmans, K.; Timmermans, L.; Van de Sande, T.; Van Veldhoven, P.P.; Guan, G.; Shechter, I.; Claessens, F.; Verhoeven, G.; Swinnen, J.V. Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J. Biol. Chem. 2007, 282, 18777–18785. [Google Scholar] [CrossRef]
- Tuzmen, S.; Hostetter, G.; Watanabe, A.; Ekmekci, C.; Carrigan, P.E.; Shechter, I.; Kallioniemi, O.; Miller, L.J.; Mousses, S. Characterization of farnesyl diphosphate farnesyl transferase 1 (FDFT1) expression in cancer. Per. Med. 2019, 16, 51–65. [Google Scholar] [CrossRef]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e3926. [Google Scholar] [CrossRef]
- Liu, M.; Xia, Y.; Ding, J.; Ye, B.; Zhao, E.; Choi, J.H.; Alptekin, A.; Yan, C.; Dong, Z.; Huang, S.; et al. Transcriptional Profiling Reveals a Common Metabolic Program in High-Risk Human Neuroblastoma and Mouse Neuroblastoma Sphere-Forming Cells. Cell Rep. 2016, 17, 609–623. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.L.; Francescangeli, F.; Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers 2019, 11, 1569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Dai, X.; Wang, Y. 5-Aza-2’-deoxycytidine induced growth inhibition of leukemia cells through modulating endogenous cholesterol biosynthesis. Mol. Cell Proteom. 2012, 11, M111-016915. [Google Scholar] [CrossRef]
- Yamasaki, M.; Nishimura, M.; Sakakibara, Y.; Suiko, M.; Morishita, K.; Nishiyama, K. Delta-tocotrienol induces apoptotic cell death via depletion of intracellular squalene in ED40515 cells. Food Funct. 2014, 5, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.J.; Alves, G.; Viennois, E.; Bernard, S.; Communal, Y.; Sion, B.; Marceau, G.; Damon, C.; Mouzat, K.; Caira, F.; et al. Liver X Receptor activation downregulates AKT survival signaling in lipid rafts and induces apoptosis of prostate cancer cells. Oncogene 2010, 29, 2712–2723. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Baudrier, L.; Bayraktar, E.C.; Shen, Y.; La, K.; Guarecuco, R.; Yucel, B.; Fiore, D.; Tavora, B.; Freinkman, E.; et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 2019, 567, 118–122. [Google Scholar] [CrossRef]
- Okamoto, K.; Sakimoto, Y.; Imai, K.; Senoo, H.; Shidoji, Y. Induction of an incomplete autophagic response by cancer-preventive geranylgeranoic acid (GGA) in a human hepatoma-derived cell line. Biochem. J. 2011, 440, 63–71. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef]
- Hussain, T.; Lee, J.; Abba, M.C.; Chen, J.; Aldaz, C.M. Delineating WWOX Protein Interactome by Tandem Affinity Purification-Mass Spectrometry: Identification of Top Interactors and Key Metabolic Pathways Involved. Front. Oncol. 2018, 8, 591. [Google Scholar] [CrossRef]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.C.; Green, R.; Khalil, R.; Foran, E.; Quarni, W.; Nair, R.; Stevens, S.; Grinchuk, A.; Hanna, A.; Mohapatra, S.; et al. Lung cancer cells survive epidermal growth factor receptor tyrosine kinase inhibitor exposure through upregulation of cholesterol synthesis. Faseb. Bioadv. 2020, 2, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.; Giurato, G.; Saggese, P.; Pecoraro, G.; Lamberti, J.; Ravo, M.; Rizzo, F.; Rocco, D.; Tarallo, R.; Nyman, T.A.; et al. Interaction proteomics identifies ERbeta association with chromatin repressive complexes to inhibit cholesterol biosynthesis and exert an oncosuppressive role in triple-negative breast cancer. Mol. Cell Proteom. 2020, 19, 245–260. [Google Scholar] [CrossRef]
- Alexandrova, E.; Lamberti, J.; Saggese, P.; Pecoraro, G.; Memoli, D.; Cappa, V.M.; Ravo, M.; Iorio, R.; Tarallo, R.; Rizzo, F.; et al. Small non-coding RNA profiling identifies miR-181a-5p as a mediator of estrogen receptor beta-induced inhibition of cholesterol biosynthesis in triple-negative breast cancer. Cells 2020, 9, 874. [Google Scholar] [CrossRef]
- Molica, F.; Meens, M.J.; Dubrot, J.; Ehrlich, A.; Roth, C.L.; Morel, S.; Pelli, G.; Vinet, L.; Braunersreuther, V.; Ratib, O.; et al. Pannexin1 links lymphatic function to lipid metabolism and atherosclerosis. Sci. Rep. 2017, 7, 13706. [Google Scholar] [CrossRef]
- Crespo Yanguas, S.; Willebrords, J.; Johnstone, S.R.; Maes, M.; Decrock, E.; De Bock, M.; Leybaert, L.; Cogliati, B.; Vinken, M. Pannexin1 as mediator of inflammation and cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 51–61. [Google Scholar] [CrossRef]
- Puri, T.; Wendler, P.; Sigala, B.; Saibil, H.; Tsaneva, I.R. Dodecameric structure and ATPase activity of the human TIP48/TIP49 complex. J. Mol. Biol. 2007, 366, 179–192. [Google Scholar] [CrossRef]
- Vassileva, I.; Yanakieva, I.; Peycheva, M.; Gospodinov, A.; Anachkova, B. The mammalian INO80 chromatin remodeling complex is required for replication stress recovery. Nucleic. Acids Res. 2014, 42, 9074–9086. [Google Scholar] [CrossRef]
- Wang, J.; Gao, S.; Peng, X.; Wu, K.; Yang, S. Roles of the INO80 and SWR1 chromatin remodeling complexes in plants. Int. J. Mol. Sci. 2019, 20, 4591. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Taras, D.; Grigoletto, A.; Haurie, V.; Nicou, A.; Dugot-Senant, N.; Costet, P.; Rousseau, B.; Rosenbaum, J. In vivo silencing of Reptin blocks the progression of human hepatocellular carcinoma in xenografts and is associated with replicative senescence. J. Hepatol. 2010, 52, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Amano, T.; Yamasaki, S.; Yagishita, N.; Tsuchimochi, K.; Shin, H.; Kawahara, K.; Aratani, S.; Fujita, H.; Zhang, L.; Ikeda, R.; et al. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003, 17, 2436–2449. [Google Scholar] [CrossRef]
- Yewdell, J.W. Not such a dismal science: The economics of protein synthesis, folding, degradation and antigen processing. Trends Cell Biol. 2001, 11, 294–297. [Google Scholar] [CrossRef]
- Fukui, R.; Saitoh, S.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef]
- Koupenova, M.; Mick, E.; Mikhalev, E.; Benjamin, E.J.; Tanriverdi, K.; Freedman, J.E. Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors. Arter. Thromb. Vasc. Biol. 2015, 35, 1030–1037. [Google Scholar] [CrossRef]
- Iatan, I.; Choi, H.Y.; Ruel, I.; Reddy, M.V.; Kil, H.; Lee, J.; Odeh, M.A.; Salah, Z.; Abu-Remaileh, M.; Weissglas-Volkov, D.; et al. The WWOX gene modulates high-density lipoprotein and lipid metabolism. Circ. Cardiovasc. Genet. 2014, 7, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Salah, Z.; Aqeilan, R.; Huebner, K. WWOX gene and gene product: Tumor suppression through specific protein interactions. Future Oncol. 2010, 6, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Hazan, I.; Hofmann, T.G.; Aqeilan, R.I. Tumor Suppressor Genes within Common Fragile Sites Are Active Players in the DNA Damage Response. PLoS Genet. 2016, 12, e1006436. [Google Scholar] [CrossRef][Green Version]
- Aqeilan, R.I.; Croce, C.M. WWOX in biological control and tumorigenesis. J. Cell Physiol. 2007, 212, 307–310. [Google Scholar] [CrossRef]
- Chang, N.S.; Hsu, L.J.; Lin, Y.S.; Lai, F.J.; Sheu, H.M. WW domain-containing oxidoreductase: A candidate tumor suppressor. Trends Mol. Med. 2007, 13, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Aqeilan, R.I.; Trapasso, F.; Hussain, S.; Costinean, S.; Marshall, D.; Pekarsky, Y.; Hagan, J.P.; Zanesi, N.; Kaou, M.; Stein, G.S.; et al. Targeted deletion of Wwox reveals a tumor suppressor function. Proc. Natl. Acad. Sci. USA 2007, 104, 3949–3954. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dai, W. Genomic instability and cancer. J. Carcinog. Mutagenesis 2014, 5, 1000165. [Google Scholar]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef]
- Kavanaugh, G.; Ye, F.; Mohni, K.N.; Luzwick, J.W.; Glick, G.; Cortez, D. A whole genome RNAi screen identifies replication stress response genes. DNA Repair. 2015, 35, 55–62. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Kang, T.H.; Lindsey-Boltz, L.A.; Reardon, J.T.; Sancar, A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2010, 107, 4890–4895. [Google Scholar] [CrossRef]
- Kang, T.H.; Reardon, J.T.; Sancar, A. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic. Acids. Res. 2011, 39, 3176–3187. [Google Scholar] [CrossRef]
- Wu, W.; Sato, K.; Koike, A.; Nishikawa, H.; Koizumi, H.; Venkitaraman, A.R.; Ohta, T. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 2010, 70, 6384–6392. [Google Scholar] [CrossRef]
- Cubillos-Rojas, M.; Amair-Pinedo, F.; Peiro-Jordan, R.; Bartrons, R.; Ventura, F.; Rosa, J.L. The E3 ubiquitin protein ligase HERC2 modulates the activity of tumor protein p53 by regulating its oligomerization. J. Biol. Chem. 2014, 289, 14782–14795. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Yang, D.R.; Yang, G.; Lin, C.Y.; Chang, H.C.; Li, G.; Chang, C. TR2 and TR4 Orphan Nuclear Receptors: An Overview. Curr. Top. Dev. Biol. 2017, 125, 357–373. [Google Scholar] [CrossRef]
- Kim, E.; Xie, S.; Yeh, S.D.; Lee, Y.F.; Collins, L.L.; Hu, Y.C.; Shyr, C.R.; Mu, X.M.; Liu, N.C.; Chen, Y.T.; et al. Disruption of TR4 orphan nuclear receptor reduces the expression of liver apolipoprotein E/C-I/C-II gene cluster. J. Biol. Chem. 2003, 278, 46919–46926. [Google Scholar] [CrossRef]
- Alpy, F.; Rousseau, A.; Schwab, Y.; Legueux, F.; Stoll, I.; Wendling, C.; Spiegelhalter, C.; Kessler, P.; Mathelin, C.; Rio, M.C.; et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 2013, 126, 5500–5512. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Liu, S.; Liu, N.C.; Wang, R.S.; Chen, L.M.; Lin, W.J.; Ting, H.J.; Ho, H.C.; Li, G.; Puzas, E.J.; et al. Premature aging with impaired oxidative stress defense in mice lacking TR4. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E91–E98. [Google Scholar] [CrossRef][Green Version]
- Rajendra, E.; Garaycoechea, J.I.; Patel, K.J.; Passmore, L.A. Abundance of the Fanconi anaemia core complex is regulated by the RuvBL1 and RuvBL2 AAA+ ATPases. Nucleic. Acids Res. 2014, 42, 13736–13748. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.C.; Dadhania, V.; Zhang, L.; Majewski, T.; Bondaruk, J.; Sykulski, M.; Wronowska, W.; Gambin, A.; Wang, Y.; Zhang, S.; et al. Gene Expression Profile of the Clinically Aggressive Micropapillary Variant of Bladder Cancer. Eur. Urol. 2016, 70, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e907. [Google Scholar] [CrossRef]
- Bellosta, P.; Hulf, T.; Balla Diop, S.; Usseglio, F.; Pradel, J.; Aragnol, D.; Gallant, P. Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc. Natl. Acad. Sci. USA 2005, 102, 11799–11804. [Google Scholar] [CrossRef]
- McBride, K.M.; Kil, H.; Mu, Y.; Plummer, J.B.; Lee, J.; Zelazowski, M.J.; Sebastian, M.; Abba, M.C.; Aldaz, C.M. Wwox Deletion in Mouse B Cells Leads to Genomic Instability, Neoplastic Transformation, and Monoclonal Gammopathies. Front Oncol. 2019, 9, 517. [Google Scholar] [CrossRef]
- Alpy, F.; Tomasetto, C.L. STARD3: A lipid transfer protein in breast cancer and cholesterol trafficking. In Cholesterol Transporters of the START Domain Protein Family in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2014; pp. 119–138. [Google Scholar]
- Murai, T.; Maruyama, Y.; Mio, K.; Nishiyama, H.; Suga, M.; Sato, C. Low cholesterol triggers membrane microdomain-dependent CD44 shedding and suppresses tumor cell migration. J. Biol. Chem. 2011, 286, 1999–2007. [Google Scholar] [CrossRef]
- Ramprasad, O.G.; Srinivas, G.; Rao, K.S.; Joshi, P.; Thiery, J.P.; Dufour, S.; Pande, G. Changes in cholesterol levels in the plasma membrane modulate cell signaling and regulate cell adhesion and migration on fibronectin. Cell Motil. Cytoskelet. 2007, 64, 199–216. [Google Scholar] [CrossRef]
- Hirata, H.; Tatsumi, H.; Sokabe, M. Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J. Cell Sci. 2008, 121, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Beswick, E.J.; Reyes, V.E. CD74 in antigen presentation, inflammation, and cancers of the gastrointestinal tract. World J. Gastroenterol. 2009, 15, 2855–2861. [Google Scholar] [CrossRef]
- Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.B.; Zhang, H.; Xu, T.; Lei, S.F.; Zhang, Y.H. Identification of important genes associated with total cholesterol using bioinformatics analysis. Pharmacogenomics 2016, 17, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; De Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol. Cell Endocrinol. 2016, 431, 123–132. [Google Scholar] [CrossRef]
- Waalkes, S.; Atschekzei, F.; Kramer, M.W.; Hennenlotter, J.; Vetter, G.; Becker, J.U.; Stenzl, A.; Merseburger, A.S.; Schrader, A.J.; Kuczyk, M.A.; et al. Fibronectin 1 mRNA expression correlates with advanced disease in renal cancer. BMC Cancer 2010, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Dhar-Mascareno, M.; Rozenberg, I.; Iqbal, J.; Hussain, M.M.; Beckles, D.; Mascareno, E. Hexim1 heterozygosity stabilizes atherosclerotic plaque and decreased steatosis in ApoE null mice fed atherogenic diet. Int. J. Biochem. Cell Biol. 2017, 83, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dhar-Mascareno, M.; Ramirez, S.N.; Rozenberg, I.; Rouille, Y.; Kral, J.G.; Mascareno, E.J. Hexim1, a Novel Regulator of Leptin Function, Modulates Obesity and Glucose Disposal. Mol. Endocrinol. 2016, 30, 314–324. [Google Scholar] [CrossRef]
- Jin, R.; Lin, H.; Li, G.; Xu, J.; Shi, L.; Chang, C.; Cai, X. TR4 nuclear receptor suppresses HCC cell invasion via downregulating the EphA2 expression. Cell Death Dis. 2018, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yuan, M.; Yao, Y.; Wu, D.; Dong, S.; Tong, X. In vitro effect of Pannexin 1 channel on the invasion and migration of I-10 testicular cancer cells via ERK1/2 signaling pathway. Biomed. Pharm. 2019, 117, 109090. [Google Scholar] [CrossRef]
- Tan, X.; He, X.; Fan, Z. Upregulation of HRD1 promotes cell migration and invasion in colon cancer. Mol. Cell Biochem. 2019, 454, 1–9. [Google Scholar] [CrossRef]
- Liu, L.; Long, H.; Wu, Y.; Li, H.; Dong, L.; Zhong, J.L.; Liu, Z.; Yang, X.; Dai, X.; Shi, L.; et al. HRD1-mediated PTEN degradation promotes cell proliferation and hepatocellular carcinoma progression. Cell Signal. 2018, 50, 90–99. [Google Scholar] [CrossRef]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 22. [Google Scholar] [CrossRef]
- Ugawa, T.; Kakuta, H.; Moritani, H.; Matsuda, K.; Ishihara, T.; Yamaguchi, M.; Naganuma, S.; Iizumi, Y.; Shikama, H. YM-53601, a novel squalene synthase inhibitor, reduces plasma cholesterol and triglyceride levels in several animal species. Br. J. Pharm. 2000, 131, 63–70. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, Q.; Ishida, Y.; Hajjar, S.; Tang, X.; Shi, H.; Dang, C.V.; Le, A.D. EGF induces epithelial-mesenchymal transition and cancer stem-like cell properties in human oral cancer cells via promoting Warburg effect. Oncotarget 2017, 8, 9557–9571. [Google Scholar] [CrossRef]
- Yang, R.; Li, X.; Wu, Y.; Zhang, G.; Liu, X.; Li, Y.; Bao, Y.; Yang, W.; Cui, H. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 2020, 39, 2975–2986. [Google Scholar] [CrossRef]
- De Rosa, V.; Iommelli, F.; Monti, M.; Fonti, R.; Votta, G.; Stoppelli, M.P.; Del Vecchio, S. Reversal of Warburg Effect and Reactivation of Oxidative Phosphorylation by Differential Inhibition of EGFR Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 5110–5120. [Google Scholar] [CrossRef]
- Carito, V.; Bonuccelli, G.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Caroleo, M.C.; Cione, E.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Metabolic remodeling of the tumor microenvironment: Migration stimulating factor (MSF) reprograms myofibroblasts toward lactate production, fueling anabolic tumor growth. Cell Cycle 2012, 11, 3403–3414. [Google Scholar] [CrossRef] [PubMed]
- Molina, H.; Azocar, L.; Ananthanarayanan, M.; Arrese, M.; Miquel, J.F. Localization of the Sodium-Taurocholate cotransporting polypeptide in membrane rafts and modulation of its activity by cholesterol in vitro. Biochim. Biophys. Acta 2008, 1778, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Roscam Abbing, R.L.P.; Slijepcevic, D.; Donkers, J.M.; Havinga, R.; Duijst, S.; Paulusma, C.C.; Kuiper, J.; Kuipers, F.; Groen, A.K.; Oude Elferink, R.P.J.; et al. Blocking Sodium-Taurocholate Cotransporting Polypeptide Stimulates Biliary Cholesterol and Phospholipid Secretion in Mice. Hepatology 2020, 71, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Ribeiro, V. The expression of the solute carriers NTCP and OCT-1 is regulated by cholesterol in HepG2 cells. Fundam. Clin. Pharm. 2007, 21, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Binh, M.T.; Hoan, N.X.; Van Tong, H.; Sy, B.T.; Trung, N.T.; Bock, C.T.; Toan, N.L.; Song, L.H.; Bang, M.H.; Meyer, C.G.; et al. NTCP S267F variant associates with decreased susceptibility to HBV and HDV infection and decelerated progression of related liver diseases. Int. J. Infect. Dis. 2019, 80, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.I.; Choo, A.; Lee, C.S.; Dayan, S.; O’Keefe, L. WWOX, the chromosomal fragile site FRA16D spanning gene: Its role in metabolism and contribution to cancer. Exp. Biol. Med. 2015, 240, 338–344. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Khalaileh, A.; Pikarsky, E.; Aqeilan, R.I. WWOX controls hepatic HIF1alpha to suppress hepatocyte proliferation and neoplasia. Cell Death Dis. 2018, 9, 511. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Shim, J.S. Cholesterol Trafficking: An Emerging Therapeutic Target for Angiogenesis and Cancer. Cells 2019, 8, 389. [Google Scholar] [CrossRef]
- Vincent, L.; Chen, W.; Hong, L.; Mirshahi, F.; Mishal, Z.; Mirshahi-Khorassani, T.; Vannier, J.P.; Soria, J.; Soria, C. Inhibition of endothelial cell migration by cerivastatin, an HMG-CoA reductase inhibitor: Contribution to its anti-angiogenic effect. FEBS Lett. 2001, 495, 159–166. [Google Scholar] [CrossRef]
- Healey, G.D.; Collier, C.; Griffin, S.; Schuberth, H.J.; Sandra, O.; Smith, D.G.; Mahan, S.; Dieuzy-Labaye, I.; Sheldon, I.M. Mevalonate Biosynthesis Intermediates Are Key Regulators of Innate Immunity in Bovine Endometritis. J. Immunol. 2016, 196, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Varshney, P.; Yadav, V.; Saini, N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Lizarbe, M.A.; Barrasa, J.I.; Olmo, N.; Gavilanes, F.; Turnay, J. Annexin-phospholipid interactions. Functional implications. Int. J. Mol. Sci. 2013, 14, 2652–2683. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Draeger, A. Annexins in cell membrane dynamics. Ca(2+)-regulated association of lipid microdomains. J. Cell Biol. 2000, 150, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Lin, Y.L.; Kuo, C.F. Effect of high-fat diet on hepatic proteomics of hamsters. J. Agric. Food Chem. 2015, 63, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Grewal, T.; Koese, M.; Rentero, C.; Enrich, C. Annexin A6-regulator of the EGFR/Ras signalling pathway and cholesterol homeostasis. Int. J. Biochem. Cell Biol. 2010, 42, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.W.; Wahafu, W.; Song, L.; Ping, H.; Wang, M.; Yang, F.; Niu, Y.; Qing, W.; Xing, N. Expression of CD74 in bladder cancer and its suppression in association with cancer proliferation, invasion and angiogenesis in HT-1376 cells. Oncol. Lett. 2018, 15, 7631–7638. [Google Scholar] [CrossRef]
- Sharma, A.K.; Charles, E.J.; Zhao, Y.; Narahari, A.K.; Baderdinni, P.K.; Good, M.E.; Lorenz, U.M.; Kron, I.L.; Bayliss, D.A.; Ravichandran, K.S.; et al. Pannexin-1 channels on endothelial cells mediate vascular inflammation during lung ischemia-reperfusion injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 315, L301–L312. [Google Scholar] [CrossRef]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Lu, Y.; Qiu, Y.; Chen, P.; Chang, H.; Guo, L.; Zhang, F.; Ma, L.; Zhang, C.; Zheng, X.; Xiao, J.; et al. ER-localized Hrd1 ubiquitinates and inactivates Usp15 to promote TLR4-induced inflammation during bacterial infection. Nat. Microbiol. 2019, 4, 2331–2346. [Google Scholar] [CrossRef]
- Seabra, M.A.L.; Candido, E.B.; Vidigal, P.V.T.; Lamaita, R.M.; Rodrigues, A.N.; Silva Filho, A.L.D. Immunohistochemical WWOX Expression and Association with Angiogenesis, p53 Expression, Cell Proliferation and Clinicopathological Parameters in Cervical Cancer. Rev. Bras. Ginecol. Obs. 2018, 40, 79–85. [Google Scholar] [CrossRef]
- Wen, J.; Xu, Z.; Li, J.; Zhang, Y.; Fan, W.; Wang, Y.; Lu, M.; Li, J. Decreased WWOX expression promotes angiogenesis in osteosarcoma. Oncotarget 2017, 8, 60917–60932. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lanterna, C.; Musumeci, A.; Raccosta, L.; Corna, G.; Moresco, M.; Maggioni, D.; Fontana, R.; Doglioni, C.; Bordignon, C.; Traversari, C.; et al. The administration of drugs inhibiting cholesterol/oxysterol synthesis is safe and increases the efficacy of immunotherapeutic regimens in tumor-bearing mice. Cancer Immunol. Immunother. 2016, 65, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Zhang, Y.; Li, N.; Liu, X.; Dong, J. CD74: A potential novel target for triple-negative breast cancer. Tumour Biol. 2012, 33, 2273–2277. [Google Scholar] [CrossRef]
- Ji, S.Q.; Su, X.L.; Cheng, W.L.; Zhang, H.J.; Zhao, Y.Q.; Han, Z.X. Down-regulation of CD74 inhibits growth and invasion in clear cell renal cell carcinoma through HIF-1alpha pathway. Urol. Oncol. 2014, 32, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, M.; Tanese, K.; Masugi, Y.; Hayashi, M.; Sakamoto, M. Macrophage migration inhibitory factor-CD74 interaction regulates the expression of programmed cell death ligand 1 in melanoma cells. Cancer Sci. 2019, 110, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascal, D. Tumor progression: The neuronal input. Ann. Transl. Med. 2018, 6, 89. [Google Scholar] [CrossRef]
- Lee, C.H.; Cho, J.; Lee, K. Tumour Regression via Integrative Regulation of Neurological, Inflammatory, and Hypoxic Tumour Microenvironment. Biomolther 2020, 28, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Tozawa, R.; Ishibashi, S.; Osuga, J.; Yagyu, H.; Oka, T.; Chen, Z.; Ohashi, K.; Perrey, S.; Shionoiri, F.; Yahagi, N.; et al. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J. Biol. Chem. 1999, 274, 30843–30848. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Salmona, M.; Diomede, L.; Williams, A. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J. Biol. Chem. 2004, 279, 14983–14990. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Squalestatin protects neurons and reduces the activation of cytoplasmic phospholipase A2 by Abeta(1-42). Neuropharmacology 2007, 53, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Coman, D.; Vissers, L.; Riley, L.G.; Kwint, M.P.; Hauck, R.; Koster, J.; Geuer, S.; Hopkins, S.; Hallinan, B.; Sweetman, L.; et al. Squalene Synthase Deficiency: Clinical, Biochemical, and Molecular Characterization of a Defect in Cholesterol Biosynthesis. Am. J. Hum. Genet. 2018, 103, 125–130. [Google Scholar] [CrossRef]
- Yoshida, E.; Atkinson, T.G.; Chakravarthy, B. Neuroprotective gene expression profiles in ischemic cortical cultures preconditioned with IGF-1 or bFGF. Brain Res. Mol. Brain Res. 2004, 131, 33–50. [Google Scholar] [CrossRef]
- Koide, N.; Yamada, T.; Shibata, R.; Mori, T.; Fukuma, M.; Yamazaki, K.; Aiura, K.; Shimazu, M.; Hirohashi, S.; Nimura, Y.; et al. Establishment of perineural invasion models and analysis of gene expression revealed an invariant chain (CD74) as a possible molecule involved in perineural invasion in pancreatic cancer. Clin. Cancer Res. 2006, 12, 2419–2426. [Google Scholar] [CrossRef]
- Bergstrom, J.D.; Kurtz, M.M.; Rew, D.J.; Amend, A.M.; Karkas, J.D.; Bostedor, R.G.; Bansal, V.S.; Dufresne, C.; VanMiddlesworth, F.L.; Hensens, O.D.; et al. Zaragozic acids: A family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 80–84. [Google Scholar] [CrossRef]
- Hiyoshi, H.; Yanagimachi, M.; Ito, M.; Ohtsuka, I.; Yoshida, I.; Saeki, T.; Tanaka, H. Effect of ER-27856, a novel squalene synthase inhibitor, on plasma cholesterol in rhesus monkeys: Comparison with 3-hydroxy-3-methylglutaryl-coa reductase inhibitors. J. Lipid. Res. 2000, 41, 1136–1144. [Google Scholar]
- Amin, D.; Rutledge, R.Z.; Needle, S.N.; Galczenski, H.F.; Neuenschwander, K.; Scotese, A.C.; Maguire, M.P.; Bush, R.C.; Hele, D.J.; Bilder, G.E.; et al. RPR 107393, a potent squalene synthase inhibitor and orally effective cholesterol-lowering agent: Comparison with inhibitors of HMG-CoA reductase. J. Pharm. Exp. 1997, 281, 746–752. [Google Scholar]
- Ugawa, T.; Kakuta, H.; Moritani, H.; Inagaki, O.; Shikama, H. YM-53601, a novel squalene synthase inhibitor, suppresses lipogenic biosynthesis and lipid secretion in rodents. Br. J. Pharm. 2003, 139, 140–146. [Google Scholar] [CrossRef]
- Tanimoto, T.; Onodera, K.; Hosoya, T.; Takamatsu, Y.; Kinoshita, T.; Tago, K.; Kogen, H.; Fujioka, T.; Hamano, K.; Tsujita, Y. Schizostatin, a novel squalene synthase inhibitor produced by the mushroom, Schizophyllum commune. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1996, 49, 617–623. [Google Scholar] [CrossRef]
- Memon, R.A.; Shechter, I.; Moser, A.; Shigenaga, J.K.; Grunfeld, C.; Feingold, K. Endotoxin, tumor necrosis factor, and interleukin-1 decrease hepatic squalene synthase activity, protein, and mRNA levels in Syrian hamsters. J. Lipid. Res. 1997, 38, 1620–1629. [Google Scholar]
- Vaidya, S.; Bostedor, R.; Kurtz, M.M.; Bergstrom, J.D.; Bansal, V.S. Massive production of farnesol-derived dicarboxylic acids in mice treated with the squalene synthase inhibitor zaragozic acid A. Arch. Biochem. Biophys. 1998, 355, 84–92. [Google Scholar] [CrossRef]
- Bostedor, R.G.; Karkas, J.D.; Arison, B.H.; Bansal, V.S.; Vaidya, S.; Germershausen, J.I.; Kurtz, M.M.; Bergstrom, J.D. Farnesol-derived dicarboxylic acids in the urine of animals treated with zaragozic acid A or with farnesol. J. Biol. Chem. 1997, 272, 9197–9203. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Ishikawa, E.; Anayama, H.; Hamajyo, H.; Nagai, H.; Hirakata, M.; Tozawa, R. Protective effects of a squalene synthase inhibitor, lapaquistat acetate (TAK-475), on statin-induced myotoxicity in guinea pigs. Toxicol. Appl. Pharm. 2007, 223, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Teshima, K.; Kondo, T.; Tagawa, Y.; Moriwaki, T.; Asahi, S. Pharmacokinetics of TAK-475, a Squalene Synthase Inhibitor, in Rats and Dogs. Drug Res. 2016, 66, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Takeuchi, T.; Moriya, Y.; Tagawa, Y.; Kondo, T.; Moriwaki, T.; Asahi, S. Characterization of Transporters in the Hepatic Uptake of TAK-475 M-I, a Squalene Synthase Inhibitor, in Rats and Humans. Drug Res. 2016, 66, 316–323. [Google Scholar] [CrossRef]
- Biller, S.A.; Forster, C.; Gordon, E.M.; Harrity, T.; Rich, L.C.; Marretta, J.; Ciosek, C.P., Jr. Isoprenyl phosphinylformates: New inhibitors of squalene synthetase. J. Med. Chem. 1991, 34, 1912–1914. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Kori, M.; Mabuchi, H.; Banno, H.; Tozawa, R.; Nakamura, M.; Itokawa, S.; Sugiyama, Y.; Yukimasa, H. Novel 4,1-benzoxazepine derivatives with potent squalene synthase inhibitory activities. Bioorg. Med. Chem. 2002, 10, 401–414. [Google Scholar] [CrossRef]
- Poulter, C.D.; Capson, T.L.; Thompson, M.D.; Bard, R.S. Squalene synthetase, inhibition by ammonium analogs of carbocationic intermediates in the conversion of presqualene diphosphate to squalene. J. Am. Chem. Soc. 1989, 111, 3734–3739. [Google Scholar] [CrossRef]
- Prashad, M.; Kathawala, F.; Scallen, T. N-(arylalkyl) farnesylamines: New potent squalene synthetase inhibitors. J. Med. Chem. 1993, 36, 1501–1504. [Google Scholar] [CrossRef]
- Koohang, A.; Bailey, J.L.; Coates, R.M.; Erickson, H.K.; Owen, D.; Poulter, C.D. Enantioselective inhibition of squalene synthase by aziridine analogues of presqualene diphosphate. J. Org. Chem. 2010, 75, 4769–4777. [Google Scholar] [CrossRef][Green Version]
- Iwasawa, Y.; Hayashi, M.; Nomoto, T.; Shibata, J.; Mitsuya, M.; Hirota, K.; Yonemoto, M.; Kamei, T.; Miura, K.; Tomimoto, K. Synthesis and biological activity of J-104,118, a novel, potent inhibitor of squalene synthase. Biorgan. Med. Chem. Lett. 1995, 5, 1989–1994. [Google Scholar] [CrossRef]
- Iwasawa, Y.; Shibata, J.; Mitsuya, M.; Masaki, H.; Hayashi, M.; Kanno, T.; Sawasaki, Y.; Hisaka, A.; Kamei, T.; Tomimoto, K. J-104,123, a novel and orally-active inhibitor of squalene synthase: Stereoselective synthesis and cholesterol lowering effects in dogs. Biorgan. Med. Chem. Lett. 1996, 6, 463–466. [Google Scholar] [CrossRef]
- Fung, A.K.; Baker, W.R.; Fakhoury, S.; Stein, H.H.; Cohen, J.; Donner, B.G.; Garvey, D.S.; Spina, K.P.; Rosenberg, S.H. (1α, 2β, 3β, 4α)-1, 2-Bis [[N-propyl-N-(4-phen-oxybenzyl) amino] carbonyl] cyclobutane-3, 4-dicarboxylic Acid (A-87049): A Novel Potent Squalene Synthase Inhibitor. J. Med. Chem. 1997, 40, 2123–2125. [Google Scholar] [CrossRef]
- Ishihara, T.; Kakuta, H.; Moritani, H.; Ugawa, T.; Yanagisawa, I. Synthesis and biological evaluation of novel propylamine derivatives as orally active squalene synthase inhibitors. Biorgan. Med. Chem. 2004, 12, 5899–5908. [Google Scholar] [CrossRef]
- McTaggart, F.; Brown, G.R.; Davidson, R.G.; Freeman, S.; Holdgate, G.A.; Mallion, K.B.; Mirrlees, D.J.; Smith, G.J.; Ward, W.H. Inhibition of squalene synthase of rat liver by novel 3′ substituted quinuclidines. Biochem. Pharmacol. 1996, 51, 1477–1487. [Google Scholar] [CrossRef]
- Tavridou, A.; Kaklamanis, L.; Megaritis, G.; Kourounakis, A.P.; Papalois, A.; Roukounas, D.; Rekka, E.A.; Kourounakis, P.N.; Charalambous, A.; Manolopoulos, V.G. Pharmacological characterization in vitro of EP2306 and EP2302, potent inhibitors of squalene synthase and lipid biosynthesis. Eur. J. Pharmacol. 2006, 535, 34–42. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Partners of FDFT1 | Effects on Cancer Hallmarks | Cancer Types |
|---|---|---|
| ANXA5 [66] | Angiogenesis and inflammation | Prostate cancer [41] |
| CD74 [67] | Metastasis, immune evasion | Breast cancer [68] |
| EGFR [69] | Proliferation and cell death, cancer metabolism | Breast cancer, head-and-neck cancer, non-small-cell lung cancer (NSCLC), renal cancer, ovarian cancer, colon cancer [70,71,72] |
| ESR2 (ERβ) [73] | Metastasis | Triple-negative breast cancer (TNBC), lung cancer [74] |
| FN1 [75] | Metastasis, cancer metabolism | Oesophageal squamous cell carcinoma, oral cell carcinoma, colorectal, ovarian, renal, gastric cancer [76,77,78,79,80] |
| HERC2 [81] | Genomic instability | NSCLC [82] |
| HEXIM1 [66] | Metastasis, angiogenesis, and inflammation | Breast cancer, prostate cancer, melanomas, and acute myeloid leukaemia (AML) [83,84] |
| NR2C2 (TR4) [85] | Metastasis, angiogenesis and inflammation | Clear cell renal cell carcinoma, prostate cancer, hepatocellular carcinoma (HCC) [86,87,88,89] |
| PANX1 [67] | Proliferation and cell death, metastasis, angiogenesis, and inflammation | HCC, glioma, breast cancer [90,91,92] |
| PGRMC1 [93] | Proliferation and cell death | Breast cancer [93] |
| RUVBL1/2 [94] | Proliferation and cell death | Liver, breast, colorectal cancer, NSCLC [95,96,97] |
| SLC10A1 [67] | Cancer metabolism | HCC [98] |
| SYVN1 [99,100] | Proliferation and cell death, metastasis | Colon cancer, HCC [101] |
| UNC93B1 [102,103] | Proliferation and cell death | Oral cancer [104] |
| WWOX [102] | Proliferation and cell death, cancer metabolism | Ovarian cancer [105] |
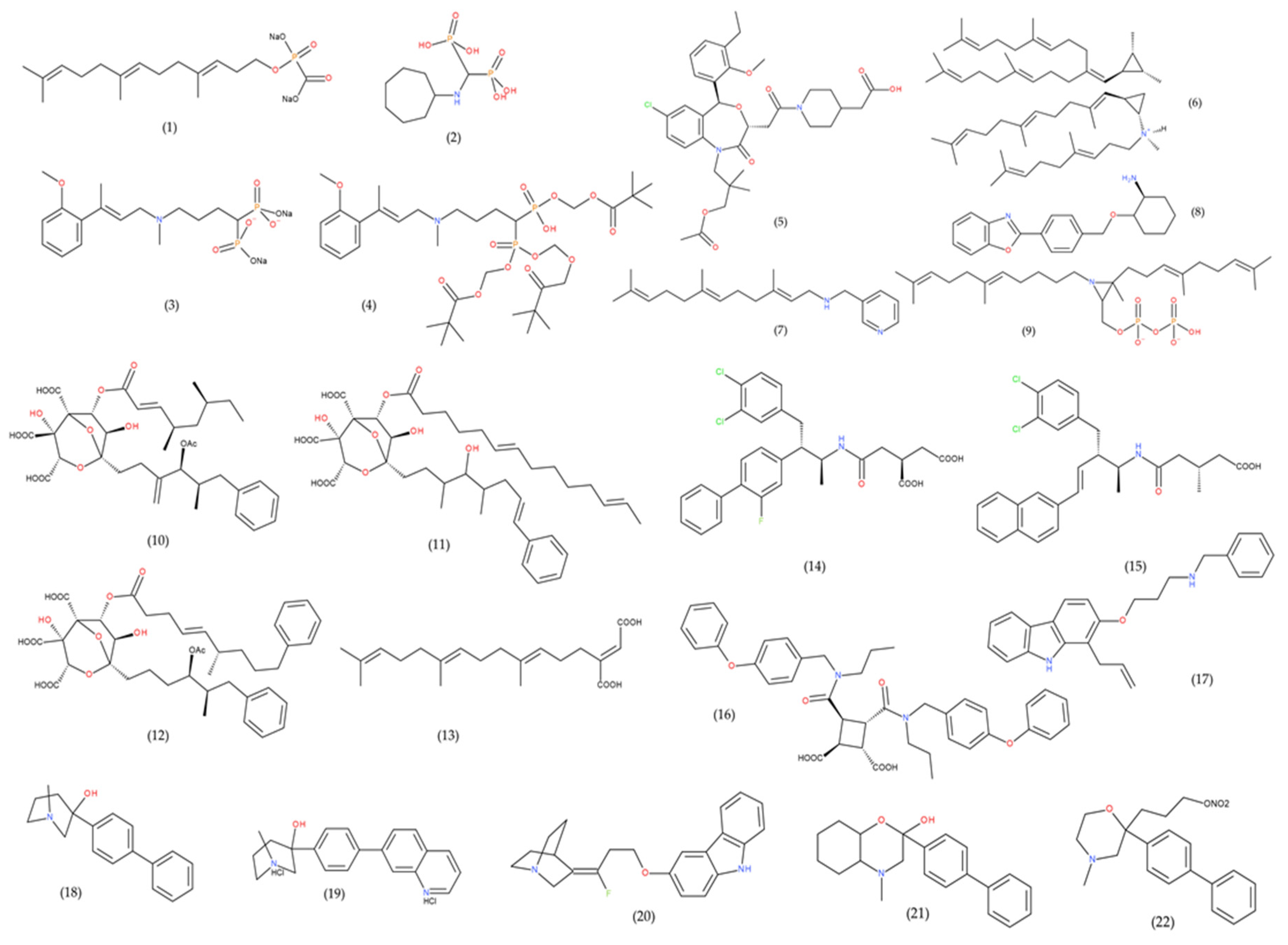
| Structure | Substances | IC50 (FDFT1 Inhibition) | Assay | Reference |
|---|---|---|---|---|
| SUBSTRATE (FPP) ANALOGUES | Isoprenyl Phosphinylformates (1) | 8.7–197 μM | Rat liver microsomes | [240] |
| YM175 (2) | 64 nM | Rat liver microsomes | [231] | |
| ER-28488 (3) | 3.6 nM | Rat liver microsomes | [230] | |
| ER-27856 (4) (prodrug of ER-28488) | 39 μM | Rat liver microsomes | [230] | |
| BENZOXAZEPINES | TAK-475 (Lapaquistat) (5) | 78 nM | HepG2 cells | [241] |
| TRANSITION- STATE ANALOGUES | Aza analogues (6) | 3–10 μM | Yeast microsomes (In the presence of PPi) | [242] |
| N-(arylalkyl) farnesylamine derivative (7) | 0.05 μM | Rat liver microsomes | [243] | |
| RPR 101821 (8) | 1 nM | Rat liver microsomes | [231] | |
| Aziridine diphosphate (9) | 1.2–1.9 μM | Yeast microsomes (In the presence of PPi) | [244] | |
| ZARAGOZIC ACIDS (SQUALESTATINS) | Zaragozic acid A (10) | 6 μM | HepG2 cells | [229] |
| Zaragozic acid B (11) | 0.6 μM | |||
| Zaragozic acid C (12) | 4 μM | |||
| DICARBOXYLIC ACID DERIVATIVES | Schizostatin (13) | 0.84 μM | Unknown | [233] |
| J-104,118 (14) | 0.52 nM | Hep G2 cells | [245] | |
| J-104,123 (15) | 2.5 nM | Hep G2 cells | [246] | |
| A-87049 (16) | 37 nM | Rat liver microsomes | [247] | |
| PROPYLAMINE DERIVATIVES | YM-75440 (17) | 63 nM | Hep G2 cells | [248] |
| QUINUCLIDINE DERIVATIVES | ZM-97480 (18) | 16 nM | Rat liver microsomes | [249] |
| RPR 107393 (19) | 0.6–0.9 nM | Rat liver microsomes | [231] | |
| YM-53601 (20) | 79 nM | Hep G2 cells | [190] | |
| MORPHOLINES | EP2306 (21) | 63 μM | Human liver microsomes | [250] |
| EP2302 (22) | 1 μM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, N.T.; Lee, C.H. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells 2020, 9, 2352. https://doi.org/10.3390/cells9112352
Ha NT, Lee CH. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells. 2020; 9(11):2352. https://doi.org/10.3390/cells9112352
Chicago/Turabian StyleHa, Nguyen Thi, and Chang Hoon Lee. 2020. "Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments" Cells 9, no. 11: 2352. https://doi.org/10.3390/cells9112352
APA StyleHa, N. T., & Lee, C. H. (2020). Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells, 9(11), 2352. https://doi.org/10.3390/cells9112352