Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Models
2.2. Morphometric Analysis of Aorta
2.3. Histological Analysis of Cardiac Slices
2.4. RNA Isolation, cDNA Synthesis and Quantitative Real Time PCR
2.5. Statistical Analysis
3. Results
3.1. Enhanced AngII-Induced Perivascular Fibrosis in Aortae of NO-GC1 KOs
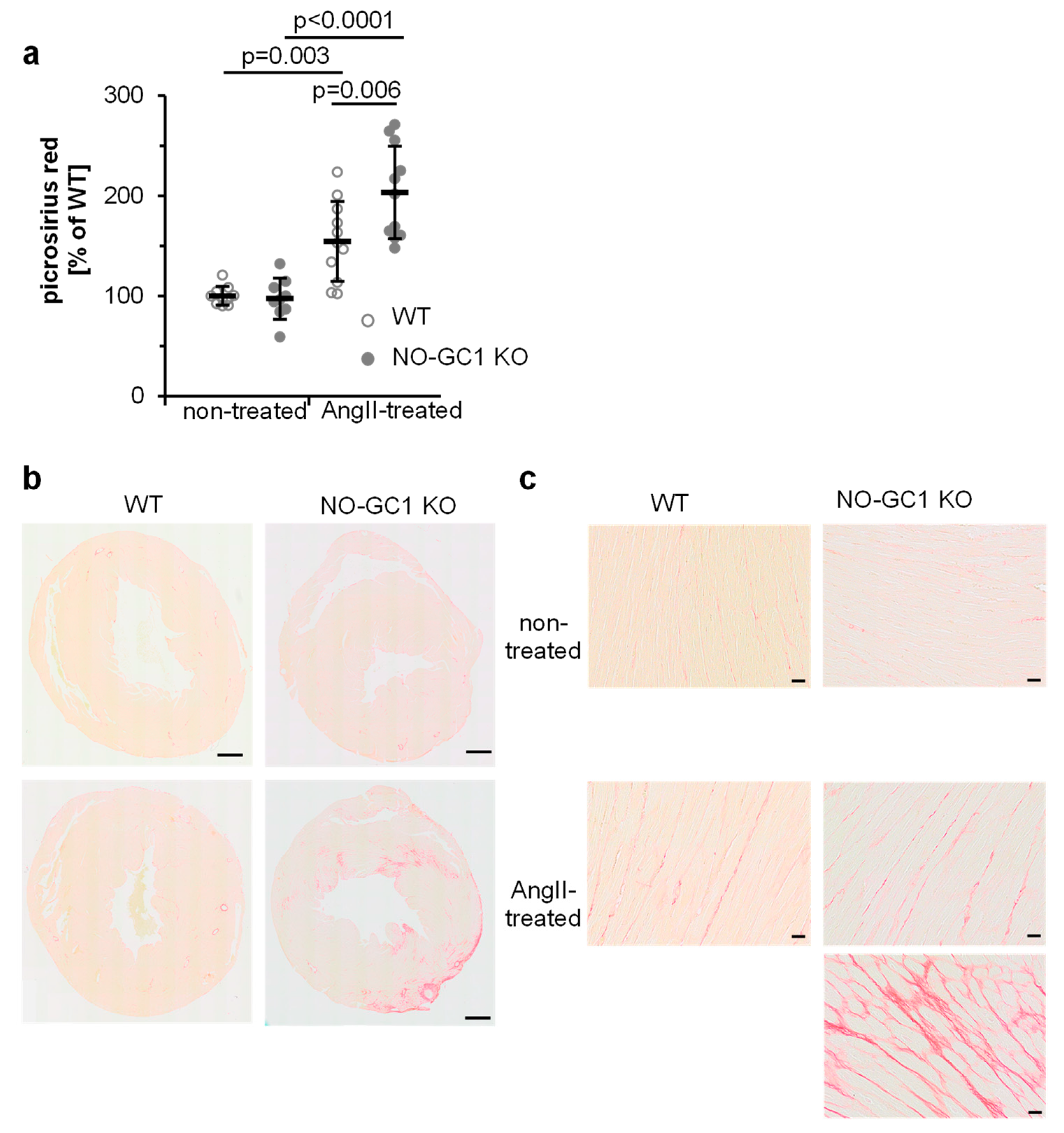
3.2. Enhanced AngII-Induced Cardiac Perivascular and Interstitial Fibrosis in NO-GC1 KOs
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hofmann, F. A concise discussion of the regulatory role of cGMP kinase I in cardiac physiology and pathology. Basic Res. Cardiol. 2018, 113, 31. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir. Med. 2017, 122, S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Preedy, M.E.J.; Baliga, R.S.; Hobbs, A.J. Multiplicity of Nitric Oxide and Natriuretic Peptide Signaling in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Russwurm, M.; Koesling, D. NO activation of guanylyl cyclase. EMBO J. 2004, 23, 4443–4450. [Google Scholar] [CrossRef]
- Russwurm, M.; Koesling, D. Isoforms of NO-sensitive guanylyl cyclase. Mol. Cell. Biochem. 2002, 230, 159–164. [Google Scholar] [CrossRef]
- Russwurm, M.; Behrends, S.; Harteneck, C.; Koesling, D. Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem. J. 1998, 335, 125–130. [Google Scholar] [CrossRef]
- Russwurm, M.; Wittau, N.; Koesling, D. Guanylyl cyclase/PSD-95 interaction: Targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. J. Biol. Chem. 2001, 276, 44647–44652. [Google Scholar] [CrossRef]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Invest. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Menges, L.; Krawutschke, C.; Füchtbauer, E.-M.; Füchtbauer, A.; Sandner, P.; Koesling, D.; Russwurm, M. Mind the gap (junction): CGMP induced by nitric oxide in cardiac myocytes originates from cardiac fibroblasts. Br. J. Pharmacol. 2019, 176, 4696–4707. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Touyz, R.M. From bedside to bench to bedside: Role of renin-angiotensin-aldosterone system in remodeling of resistance arteries in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H435–H446. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Broekmans, K.; Stegbauer, J.; Potthoff, S.A.; Russwurm, M.; Koesling, D.; Mergia, E. Angiotensin II-Induced Hypertension Is Attenuated by Reduction of Sympathetic Output in NO-Sensitive Guanylyl Cyclase 1 Knockout Mice. J. Pharmacol. Exp. Ther. 2016, 356, 191–199. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ; National Institutes of Health: Bethesda, MD, USA. Available online: http://imagej.nih.gov/ij/ (accessed on 29 November 2017).
- Kazakov, A.; Hall, R.; Jagoda, P.; Bachelier, K.; Müller-Best, P.; Semenov, A.; Lammert, F.; Böhm, M.; Laufs, U. Inhibition of endothelial nitric oxide synthase induces and enhances myocardial fibrosis. Cardiovasc. Res. 2013, 100, 211–221. [Google Scholar] [CrossRef]
- Masuyama, H.; Tsuruda, T.; Kato, J.; Imamura, T.; Asada, Y.; Stasch, J.-P.; Kitamura, K.; Eto, T. Soluble guanylate cyclase stimulation on cardiovascular remodeling in angiotensin II-induced hypertensive rats. Hypertension 2006, 48, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Tsuruda, T.; Sekita, Y.; Hatakeyama, K.; Imamura, T.; Kato, J.; Asada, Y.; Stasch, J.-P.; Kitamura, K. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens. Res. 2009, 32, 597–603. [Google Scholar] [CrossRef]
- Patrucco, E.; Domes, K.; Sbroggió, M.; Blaich, A.; Schlossmann, J.; Desch, M.; Rybalkin, S.D.; Beavo, J.A.; Lukowski, R.; Hofmann, F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Kapoun, A.M.; Liang, F.; O’Young, G.; Damm, D.L.; Quon, D.; White, R.T.; Munson, K.; Lam, A.; Schreiner, G.F.; Protter, A.A. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: Fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ. Res. 2004, 94, 453–461. [Google Scholar] [CrossRef]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.-F. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef]
- Burnett, J.C. Vericiguat—Another Victory for Targeting Cyclic GMP in Heart Failure. N. Engl. J. Med. 2020, 382, 1952–1953. [Google Scholar] [CrossRef]
- Castro, L.R.V.; Verde, I.; Cooper, D.M.F.; Fischmeister, R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar] [CrossRef]
- Castro, L.R.V.; Schittl, J.; Fischmeister, R. Feedback control through cGMP-dependent protein kinase contributes to differential regulation and compartmentation of cGMP in rat cardiac myocytes. Circ. Res. 2010, 107, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef]
- Götz, K.R.; Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Kuhn, M.; Gorelik, J.; Balligand, J.-L.; Nikolaev, V.O. Transgenic mice for real-time visualization of cGMP in intact adult cardiomyocytes. Circ. Res. 2014, 114, 1235–1245. [Google Scholar] [CrossRef]
- Takimoto, E.; Champion, H.C.; Belardi, D.; Moslehi, J.; Mongillo, M.; Mergia, E.; Montrose, D.C.; Isoda, T.; Aufiero, K.; Zaccolo, M.; et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ. Res. 2005, 96, 100–109. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broekmans, K.; Giesen, J.; Menges, L.; Koesling, D.; Russwurm, M. Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1. Cells 2020, 9, 2436. https://doi.org/10.3390/cells9112436
Broekmans K, Giesen J, Menges L, Koesling D, Russwurm M. Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1. Cells. 2020; 9(11):2436. https://doi.org/10.3390/cells9112436
Chicago/Turabian StyleBroekmans, Kathrin, Jan Giesen, Lukas Menges, Doris Koesling, and Michael Russwurm. 2020. "Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1" Cells 9, no. 11: 2436. https://doi.org/10.3390/cells9112436
APA StyleBroekmans, K., Giesen, J., Menges, L., Koesling, D., & Russwurm, M. (2020). Angiotensin II-Induced Cardiovascular Fibrosis Is Attenuated by NO-Sensitive Guanylyl Cyclase1. Cells, 9(11), 2436. https://doi.org/10.3390/cells9112436