Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence

 , and
, and
Abstract
:1. Introduction
2. Can Patients with NAFL Progress to a More Advanced Disease Stage?
2.1. Evidence in Favor of the Good Prognosis of NAFL Patients
2.2. Evidence of Progression from Steatosis to Fibrosing-Steatohepatitis and Mortality in NAFL
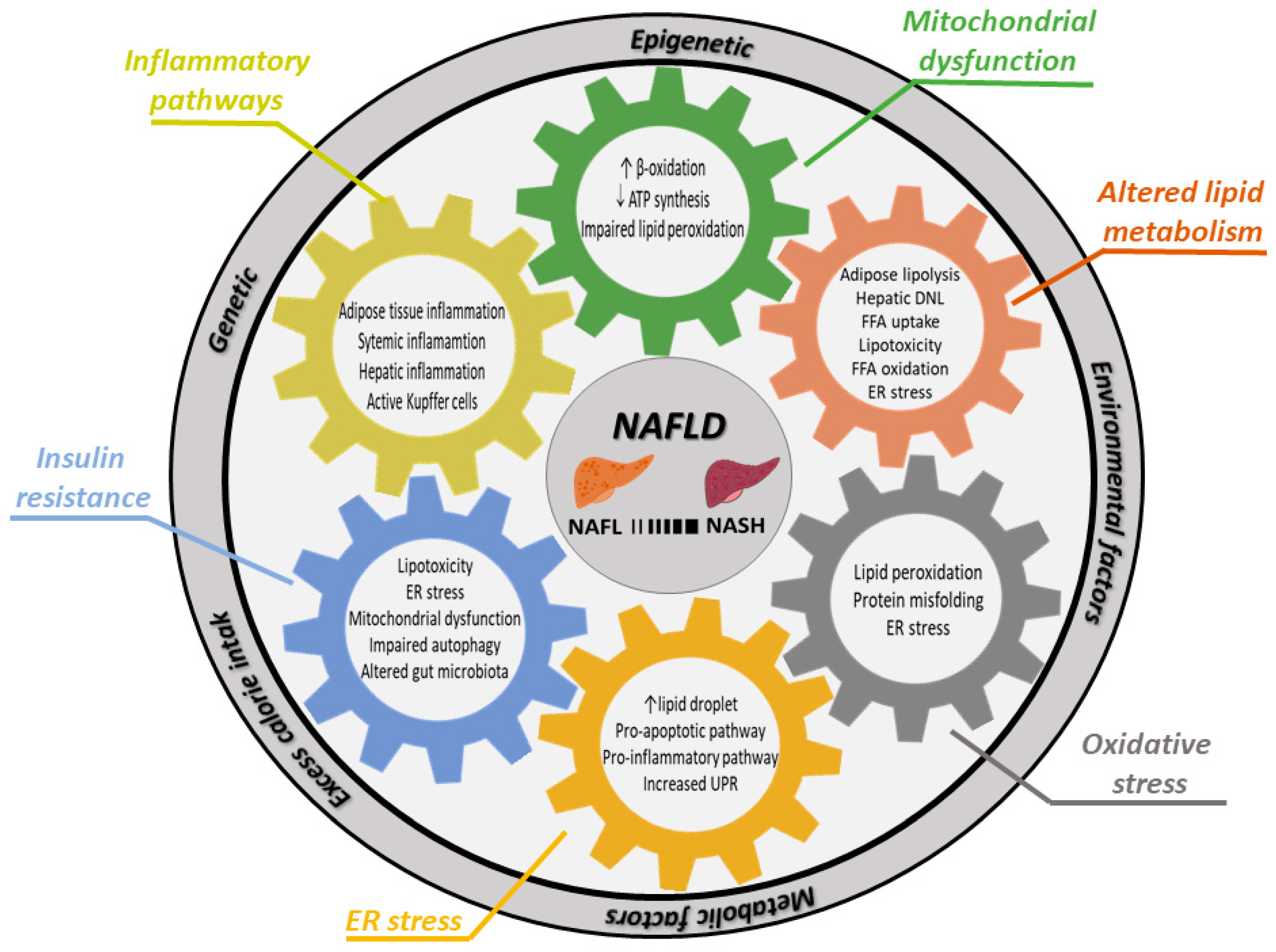
3. Pathophysiological and Molecular Mechanisms of Non-Alcoholic Fatty Liver Disease
3.1. Genetic Factors Involved in NAFLD
3.2. Insulin Resistance
3.3. Lipid Metabolism and Lipotoxicity
3.4. ER Stress
3.5. Mitochondrial Dysfunction
4. NAFLD Patients Are More Susceptible to Liver Injury Generated by Drugs and Alcohol
5. COVID-19 and NAFLD
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatol. Baltim. Md. 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.J. Pathophysiology of nonalcoholic steatohepatitis. J. Clin. Gastroenterol. 2006, 40 (Suppl. 1), S17–S29. [Google Scholar] [PubMed]
- Serfaty, L.; Lemoine, M. Definition and natural history of metabolic steatosis: Clinical aspects of NAFLD, NASH and cirrhosis. Diabetes Metab. 2008, 34, 634–637. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Kuwajima, V.; Nadelson, J.; Atiq, O.; Sanyal, A.J. Drug-induced fatty liver disease: An overview of pathogenesis and management. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatol. Baltim. Md. 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Tampi, R.; Priyadarshini, M.; Nader, F.; Younossi, I.M.; Racila, A. Burden of Illness and Economic Model for Patients With Nonalcoholic Steatohepatitis in the United States. Hepatol. Baltim. Md. 2019, 69, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatol. Baltim. Md. 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatol. Baltim. Md. 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Y. Review article: Is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment. Pharmacol. Ther. 2012, 36, 815–823. [Google Scholar] [CrossRef]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V.; LIDO Study Group. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Heurtier, A.; Gombert, S.; Giral, P.; Bruckert, E.; Grimaldi, A.; Capron, F.; Poynard, T. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 2005, 128, 1898–1906. [Google Scholar] [CrossRef]
- Rockey, D.C.; Caldwell, S.H.; Goodman, Z.D.; Nelson, R.C.; Smith, A.D. American Association for the Study of Liver Diseases Liver biopsy. Hepatol. Baltim. Md. 2009, 49, 1017–1044. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatol. Baltim. Md. 1999, 29, 664–669. [Google Scholar] [CrossRef]
- Poonawala, A.; Nair, S.P.; Thuluvath, P.J. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: A case-control study. Hepatol. Baltim. Md. 2000, 32, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Hannah, W.N.; Torres, D.M.; Harrison, S.A. Nonalcoholic Steatohepatitis and Endpoints in Clinical Trials. Gastroenterol. Hepatol. 2016, 12, 756–763. [Google Scholar]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.P. Natural history of NAFLD: Remarkably benign in the absence of cirrhosis. Gastroenterology 2005, 129, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.S.; Caldwell, S.H. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 371–379. [Google Scholar] [CrossRef]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatol. Baltim. Md 2006, 44, 865–873. [Google Scholar] [CrossRef]
- Dam-Larsen, S.; Becker, U.; Franzmann, M.-B.; Larsen, K.; Christoffersen, P.; Bendtsen, F. Final results of a long-term, clinical follow-up in fatty liver patients. Scand. J. Gastroenterol. 2009, 44, 1236–1243. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Wong, G.L.-H.; Choi, P.C.-L.; Chan, A.W.-H.; Li, M.K.-P.; Chan, H.-Y.; Chim, A.M.-L.; Yu, J.; Sung, J.J.-Y.; Chan, H.L.-Y. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Stender, S.; Pietrelli, A.; Mancina, R.M.; Cespiati, A.; Petta, S.; Pelusi, S.; Pingitore, P.; Badiali, S.; Maggioni, M.; et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J. Intern. Med. 2018, 283, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholam, P.M.; Flancbaum, L.; Machan, J.T.; Charney, D.A.; Kotler, D.P. Nonalcoholic fatty liver disease in severely obese subjects. Am. J. Gastroenterol. 2007, 102, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatol. Baltim. Md. 2008, 48, 792–798. [Google Scholar] [CrossRef]
- Kälsch, J.; Keskin, H.; Schütte, A.; Baars, T.; Baba, H.A.; Bechmann, L.P.; Canbay, A.; Sowa, J.P. Patients with ultrasound diagnosis of hepatic steatosis are at high metabolic risk. Z. Gastroenterol. 2016, 54, 1312–1319. [Google Scholar]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Teli, M.R.; James, O.F.; Burt, A.D.; Bennett, M.K.; Day, C.P. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatol. Baltim. Md. 1995, 22, 1714–1719. [Google Scholar] [CrossRef]
- Dam-Larsen, S.; Franzmann, M.; Andersen, I.B.; Christoffersen, P.; Jensen, L.B.; Sørensen, T.I.A.; Becker, U.; Bendtsen, F. Long term prognosis of fatty liver: Risk of chronic liver disease and death. Gut 2004, 53, 750–755. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef]
- Simon, T.G.; Roelstraete, B.; Khalili, H.; Hagström, H.; Ludvigsson, J.F. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: Results from a nationwide cohort. Gut 2020. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic criteria for nonalcoholic steatohepatitis: Interprotocol agreement and ability to predict liver-related mortality. Hepatol. Baltim. Md. 2011, 53, 1874–1882. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Unalp, A.; Behling, C.E.; Lavine, J.E.; Neuschwander-Tetri, B.A.; NASH Clinical Research Network. A list of members of the Nonalcoholic Steatohepatitis Clinical Research Network can be found in the Appendix Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): A histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatol. Baltim. Md. 2009, 49, 809–820. [Google Scholar]
- Kälsch, J.; Bechmann, L.P.; Kälsch, H.; Schlattjan, M.; Erhard, J.; Gerken, G.; Canbay, A. Evaluation of Biomarkers of NAFLD in a Cohort of Morbidly Obese Patients. J. Nutr. Metab. 2011, 2011, 369168. [Google Scholar] [CrossRef] [PubMed]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatol. Baltim. Md. 2003, 37, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Ratziu, V. Non-alcoholic fatty liver-perhaps not so benign. J. Hepatol. 2015, 62, 1002–1004. [Google Scholar] [CrossRef] [Green Version]
- Takyar, V.; Nath, A.; Beri, A.; Gharib, A.M.; Rotman, Y. How healthy are the “healthy volunteers”? Penetrance of NAFLD in the biomedical research volunteer pool. Hepatol. Baltim. Md. 2017, 66, 825–833. [Google Scholar] [CrossRef]
- Canbay, A.; Kachru, N.; Haas, J.S.; Sowa, J.-P.; Meise, D.; Ozbay, A.B. Patterns and predictors of mortality and disease progression among patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2020, 52, 1185–1194. [Google Scholar]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C.P. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver Dis. 2015, 35, 270–290. [Google Scholar] [PubMed] [Green Version]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oniki, K.; Saruwatari, J.; Izuka, T.; Kajiwara, A.; Morita, K.; Sakata, M.; Otake, K.; Ogata, Y.; Nakagawa, K. Influence of the PNPLA3 rs738409 Polymorphism on Non-Alcoholic Fatty Liver Disease and Renal Function among Normal Weight Subjects. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wong, G.L.-H.; Chan, H.L.-Y.; Chan, R.S.-M.; Chan, H.-Y.; Chu, W.C.-W.; Cheung, B.H.-K.; Yeung, D.K.-W.; Li, L.S.; Sea, M.M.-M.; et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2014, 30, 139–146. [Google Scholar] [CrossRef]
- Salameh, H.; Hanayneh, M.A.; Masadeh, M.; Naseemuddin, M.; Matin, T.; Erwin, A.; Singal, A.K. PNPLA3 as a Genetic Determinant of Risk for and Severity of Non-alcoholic Fatty Liver Disease Spectrum. J. Clin. Transl. Hepatol. 2016, 4, 175–191. [Google Scholar]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatol. Baltim. Md. 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Pirola, C.J.; Sookoian, S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatol. Baltim. Md. 2015, 62, 1742–1756. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Llauradó, G.; Orešič, M.; Hyötyläinen, T.; Orho-Melander, M.; Yki-Järvinen, H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. J. Hepatol. 2015, 62, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology-divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Moon, Y.-A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, C.J.; Barve, S.; Deaciuc, I. Good fat/bad fat. Hepatol. Baltim. Md. 2007, 45, 1343–1346. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Birkenfeld, A.L.; Jurczak, M.J.; Kanda, S.; Guigni, B.A.; Jiang, D.C.; Zhang, D.; Lee, H.-Y.; Samuel, V.T.; Shulman, G.I. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc. Natl. Acad. Sci. USA 2011, 108, 5748–5752. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.-P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, G.; Sowa, J.-P.; Canbay, A. Cell death mechanisms in human chronic liver diseases: A far cry from clinical applicability. Clin. Sci. Lond. Engl. 1979 2016, 130, 2121–2138. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.D.; Lee, Y.-H.; Guo, G.L. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Aspects Med. 2017, 56, 34–44. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Fujii, J.; Homma, T.; Kobayashi, S.; Seo, H.G. Mutual interaction between oxidative stress and endoplasmic reticulum stress in the pathogenesis of diseases specifically focusing on non-alcoholic fatty liver disease. World J. Biol. Chem. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Transcriptional mediators of lipid homeostasis. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.S.; Laybutt, D.R. Lipid-induced endoplasmic reticulum stress in liver cells results in two distinct outcomes: Adaptation with enhanced insulin signaling or insulin resistance. Endocrinology 2012, 153, 2164–2177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of mitochondria in nonalcoholic fatty liver disease--from origin to propagation. Clin. Biochem. 2012, 45, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Einer, C.; Hohenester, S.; Wimmer, R.; Wottke, L.; Artmann, R.; Schulz, S.; Gosmann, C.; Simmons, A.; Leitzinger, C.; Eberhagen, C.; et al. Data on chow, liver tissue and mitochondrial fatty acid compositions as well as mitochondrial proteome changes after feeding mice a western diet for 6-24 weeks. Data Brief 2017, 15, 163–169. [Google Scholar] [CrossRef]
- Lee, J.; Homma, T.; Fujii, J. Mice in the early stage of liver steatosis caused by a high fat diet are resistant to thioacetamide-induced hepatotoxicity and oxidative stress. Toxicol. Lett. 2017, 277, 92–103. [Google Scholar] [CrossRef]
- Willy, J.A.; Young, S.K.; Stevens, J.L.; Masuoka, H.C.; Wek, R.C. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol. Biol. Cell 2015, 26, 2190–2204. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatol. Baltim. Md. 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Investig. J. Tech. Methods Pathol. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canbay, A.; Chen, S.-Y.; Gieseler, R.K.; Malago, M.; Karliova, M.; Gerken, G.; Broelsch, C.E.; Treichel, U. Overweight patients are more susceptible for acute liver failure. Hepatogastroenterology 2005, 52, 1516–1520. [Google Scholar] [PubMed]
- Fromenty, B. Fat accretion in a subpopulation of hepatocytes as a strategy to protect the whole liver against oxidative stress and lipotoxicity. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Michaut, A.; Moreau, C.; Robin, M.-A.; Fromenty, B. Acetaminophen-induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int. Off. J. Int. Assoc. Study Liver 2014, 34, e171–e179. [Google Scholar] [CrossRef]
- Regev, A.; Palmer, M.; Avigan, M.I.; Dimick-Santos, L.; Treem, W.R.; Marcinak, J.F.; Seekins, D.; Krishna, G.; Anania, F.A.; Freston, J.W.; et al. Consensus: Guidelines: Best practices for detection, assessment and management of suspected acute drug-induced liver injury during clinical trials in patients with nonalcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2019, 49, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Bonkovsky, H.L.; Fontana, R.; Lee, W.; Stolz, A.; Talwalkar, J.; Reddy, K.R.; Watkins, P.B.; Navarro, V.; Barnhart, H.; et al. Features and Outcomes of 899 Patients with Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015, 148, 1340–1352. [Google Scholar] [CrossRef] [Green Version]
- Tarantino, G.; Conca, P.; Basile, V.; Gentile, A.; Capone, D.; Polichetti, G.; Leo, E. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2007, 37, 410–415. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef]
- Lammert, C.; Imler, T.; Teal, E.; Chalasani, N. Patients with Chronic Liver Disease Suggestive of Nonalcoholic Fatty Liver Disease May Be at Higher Risk for Drug-Induced Liver Injury. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 2814–2815. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, E.A.; Trogdon, J.G.; Cohen, J.W.; Dietz, W. Annual medical spending attributable to obesity: Payer-and service-specific estimates. Health Aff. Proj. Hope 2009, 28, w822–w831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.H. Drug-induced liver disease. Curr. Opin. Gastroenterol. 2002, 18, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.W.; Watkins, P.B. Are patients with elevated liver tests at increased risk of drug-induced liver injury? Gastroenterology 2004, 126, 1477–1480. [Google Scholar] [CrossRef]
- Chalasani, N.; Björnsson, E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology 2010, 138, 2246–2259. [Google Scholar] [CrossRef] [Green Version]
- Massart, J.; Begriche, K.; Moreau, C.; Fromenty, B. Role of nonalcoholic fatty liver disease as risk factor for drug-induced hepatotoxicity. J. Clin. Transl. Res. 2017, 3, 212–232. [Google Scholar]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Weltman, M.D.; Farrell, G.C.; Hall, P.; Ingelman-Sundberg, M.; Liddle, C. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatol. Baltim. Md. 1998, 27, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Kolwankar, D.; Vuppalanchi, R.; Ethell, B.; Jones, D.R.; Wrighton, S.A.; Hall, S.D.; Chalasani, N. Association between nonalcoholic hepatic steatosis and hepatic cytochrome P-450 3A activity. Clin. Gastroenterol. Hepatol. 2007, 5, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight risk factor for alcoholic liver disease. Hepatol. Baltim. Md. 1997, 25, 108–111. [Google Scholar] [CrossRef]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Davey Smith, G. Effect of body mass index and alcohol consumption on liver disease: Analysis of data from two prospective cohort studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lai, K.K.Y.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamoto, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everitt, H.; Hu, M.; Ajmo, J.M.; Rogers, C.Q.; Liang, X.; Zhang, R.; Yin, H.; Choi, A.; Bennett, E.S.; You, M. Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G38–G47. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.H.; Crawford, D.H.G.; Jaskowski, L.A.; Subramaniam, V.N.; Clouston, A.D.; Crane, D.I.; Bridle, K.R.; Anderson, G.J.; Fletcher, L.M. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 1295–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. Severe acute respiratory syndrome-related coronavirus: The species and its viruses—A statement of the Coronavirus Study Group. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Ji, D.; Qin, E.; Xu, J.; Zhang, D.; Cheng, G.; Wang, Y.; Lau, G. Implication of non-alcoholic fatty liver diseases (NAFLD) in patients with COVID-19: A preliminary analysis. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Russell, T.W.; Diamond, C.; Liu, Y.; Edmunds, J.; Funk, S.; Eggo, R.M.; Centre for Mathematical Modelling of Infectious Diseases COVID-19 working group. Early dynamics of transmission and control of COVID-19: A mathematical modelling study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Boettler, T.; Newsome, P.N.; Mondelli, M.U.; Maticic, M.; Cordero, E.; Cornberg, M.; Berg, T. Care of patients with liver disease during the COVID-19 pandemic: EASL-ESCMID position paper. JHEP Rep. Innov. Hepatol. 2020, 2, 100113. [Google Scholar] [CrossRef]


{kind=link}
| Paired Liver Biopsy | # of Patients | Endpoints | Type of Study | Observations | Reference # |
|---|---|---|---|---|---|
| yes | 40 | natural history | retrospective | no progression to cirrhosis or liver-related complications; low number of patients | [24] |
| no | 132 | cirrhosis outcome, overall mortality, liver-related mortality | retrospective | poor outcome only in patients with ballooning, Mallory hyaline or fibrosis | [12] |
| no | 209 | liver-related mortality | retrospective | only fibrosis as independent factor of liver-related mortality | [25] |
| no | 646 | liver-related mortality, overall survival | retrospective | only fibrosis associated with liver-related mortality and overall survival | [26] |
| yes | 221 | natural history of fibrosis progression; predictors of progression to F3 fibrosis | retrospective systematic review | age and inflammation are predictors of progression to advanced fibrosis | [27] |
| Yes (n = 60) | 129 | survival and cause of death | retrospective | survival is lower in NASH but not in simple steatosis; low number of patients; heterogeneity of patient population; | [28] |
| no | 170 NAFLD 246 ALD | risk of cirrhosis development; risk of death | retrospective | patients with simple steatosis have similar survival to Danish population | [29] |
| no | 547 | potential risk factors (index biopsy) for survival and cirrhosis development | retrospective | long-term follow-up study of ref. #31 | [30] |
| No | 619 | long-term prognostic relevance of histologic features | retrospective | only fibrosis showed decreased overall survival, low number of patients with NASH and without fibrosis | [13] |
| Paired Liver Biopsy | # of Patients | Endpoints | Type of Study | Observations | Reference # |
|---|---|---|---|---|---|
| yes | 52 | disease progression | prospective longitudinal study | 20–30% of patients with simple steatosis had fibrosis progression; patients received lifestyle advice and metabolic monitoring | [31] |
| yes | 108 | factors predicting progression on liver biopsy | progression to NASH in 44% of patients with baseline NAFL | [32] | |
| yes | 70 | progression of simple steatosis and mild inflammation to NASH and fibrosis | retrospective | ballooning in 16 of the 25 patients with simple steatosis, and bridging fibrosis in 6 | [16] |
| no | 1515 (liver biopsy cohort) | hepatic fat accumulation has a causal role in determining liver damage and insulin resistance | mendelian randomization approach | long-term hepatic fat accumulation plays a causal role in the development of chronic liver disease | [33] |
| yes | 411 NAFLD - 150 NAFL - 261 NASH | clinical risk factors associated with progression | systematic review and meta-analysis | Liver fibrosis progresses in NAFL and NASH | [34] |
| yes | 103 | histological course of patients with sequential liver biopsies | retrospective | 2 out of 3 patients with steatosis develop NASH; 4 out of 4 patients with steatosis and mild inflammation develop NASH; low number of patients | [35] |
| no | 10,568 | mortality in NAFLD | retrospective matched cohort study | increased risk of mortality for all histological stages 1.71-times increased risk in NAFL | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzolini, G.; Sowa, J.-P.; Atorrasagasti, C.; Kücükoglu, Ö.; Syn, W.-K.; Canbay, A. Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence. Cells 2020, 9, 2458. https://doi.org/10.3390/cells9112458
Mazzolini G, Sowa J-P, Atorrasagasti C, Kücükoglu Ö, Syn W-K, Canbay A. Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence. Cells. 2020; 9(11):2458. https://doi.org/10.3390/cells9112458
Chicago/Turabian StyleMazzolini, Guillermo, Jan-Peter Sowa, Catalina Atorrasagasti, Özlem Kücükoglu, Wing-Kin Syn, and Ali Canbay. 2020. "Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence" Cells 9, no. 11: 2458. https://doi.org/10.3390/cells9112458
APA StyleMazzolini, G., Sowa, J.-P., Atorrasagasti, C., Kücükoglu, Ö., Syn, W.-K., & Canbay, A. (2020). Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence. Cells, 9(11), 2458. https://doi.org/10.3390/cells9112458