An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Systemic and Islet Renin–Angiotensin–Aldosterone System (RAAS) Activation in MetS, T2DM and COVID-19
3. Islet Redox Stress in MetS, T2DM and COVID-19
4. Systemic and Islet Inflammation
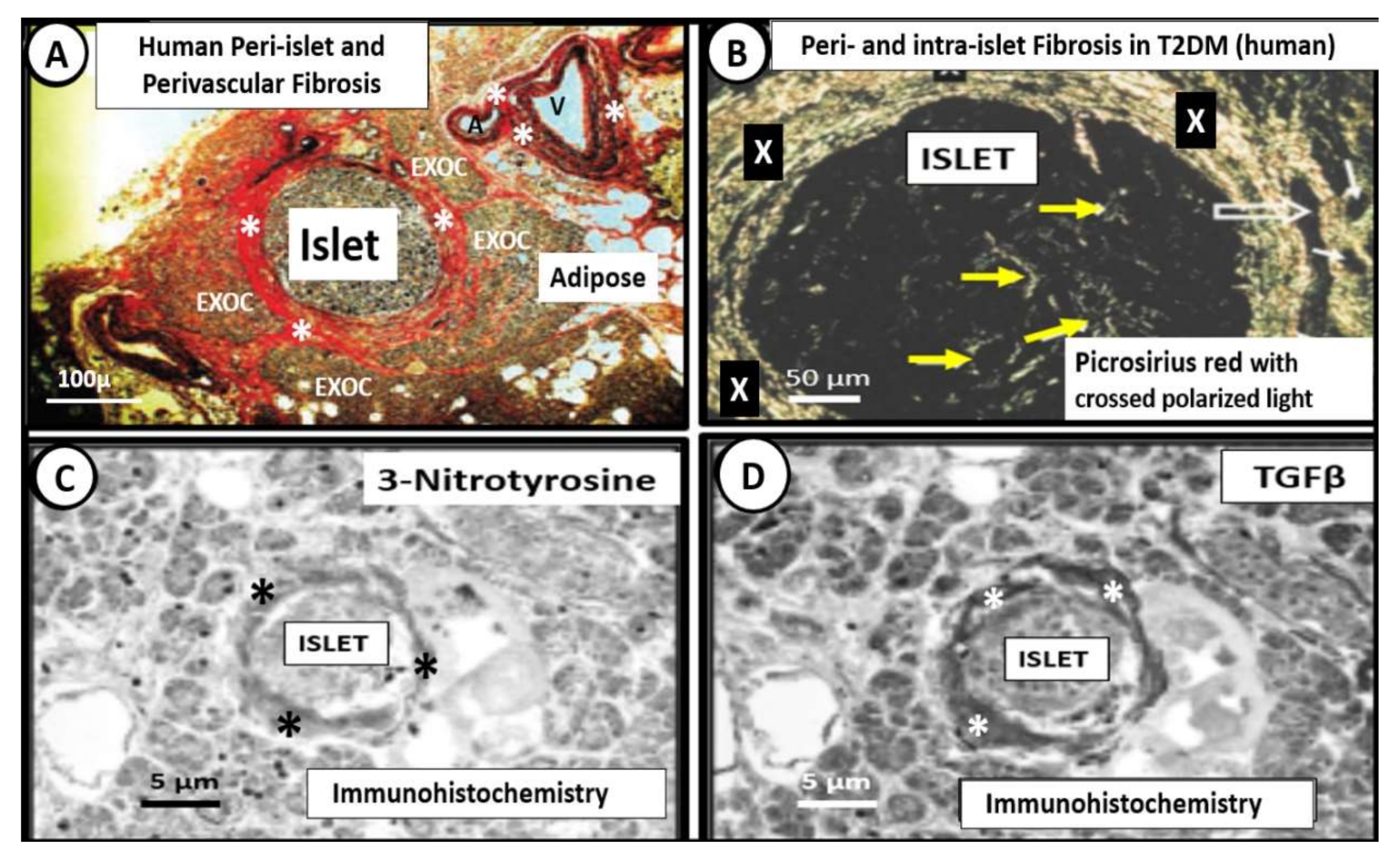
5. Islet Fibrosis
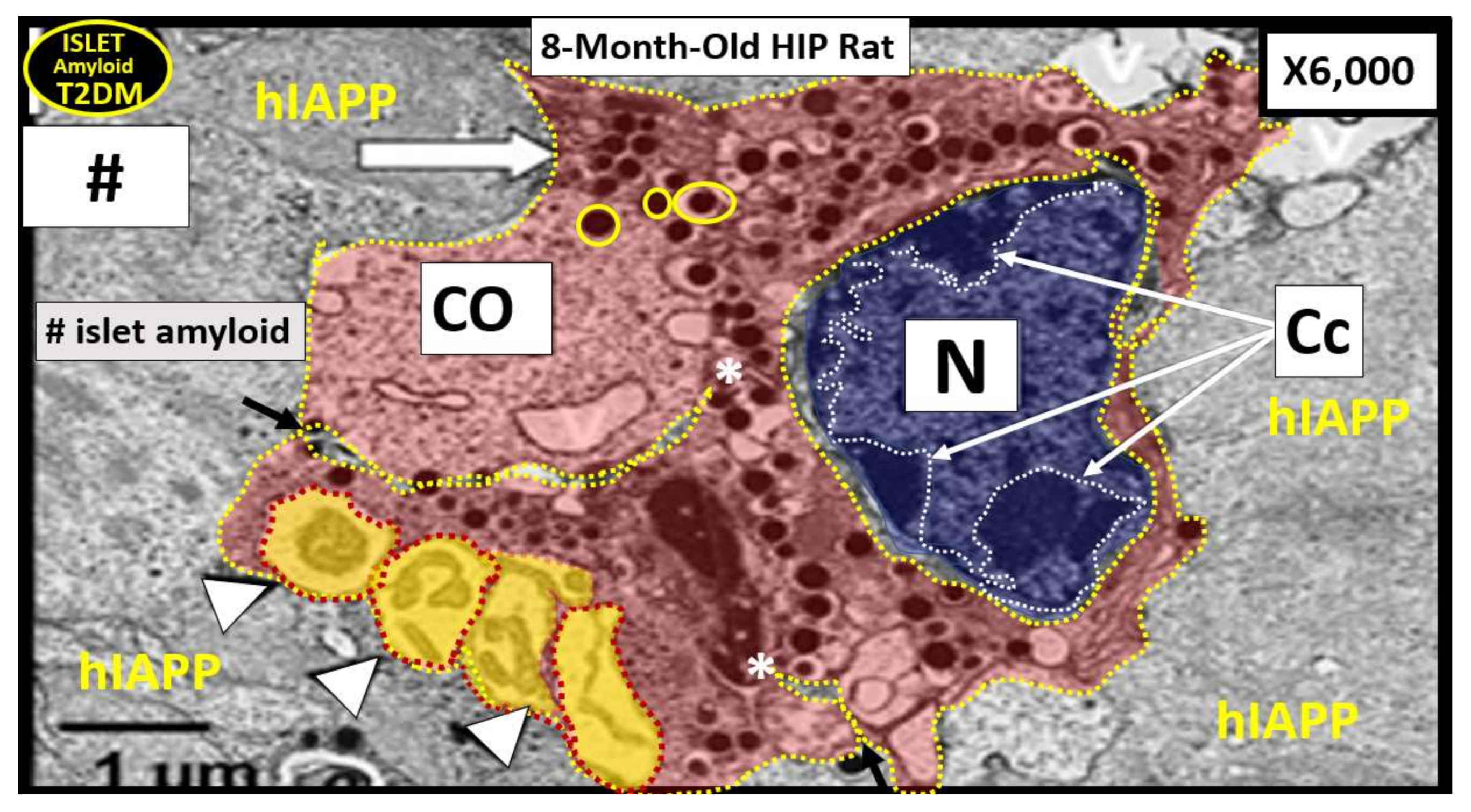
6. Islet Amyloid/Amylin/Islet Amyloid Polypeptide (IAPP)
7. Islet β-Cell Dysfunction and Failure Due to Loss (Apoptosis) and Capillary Rarefaction
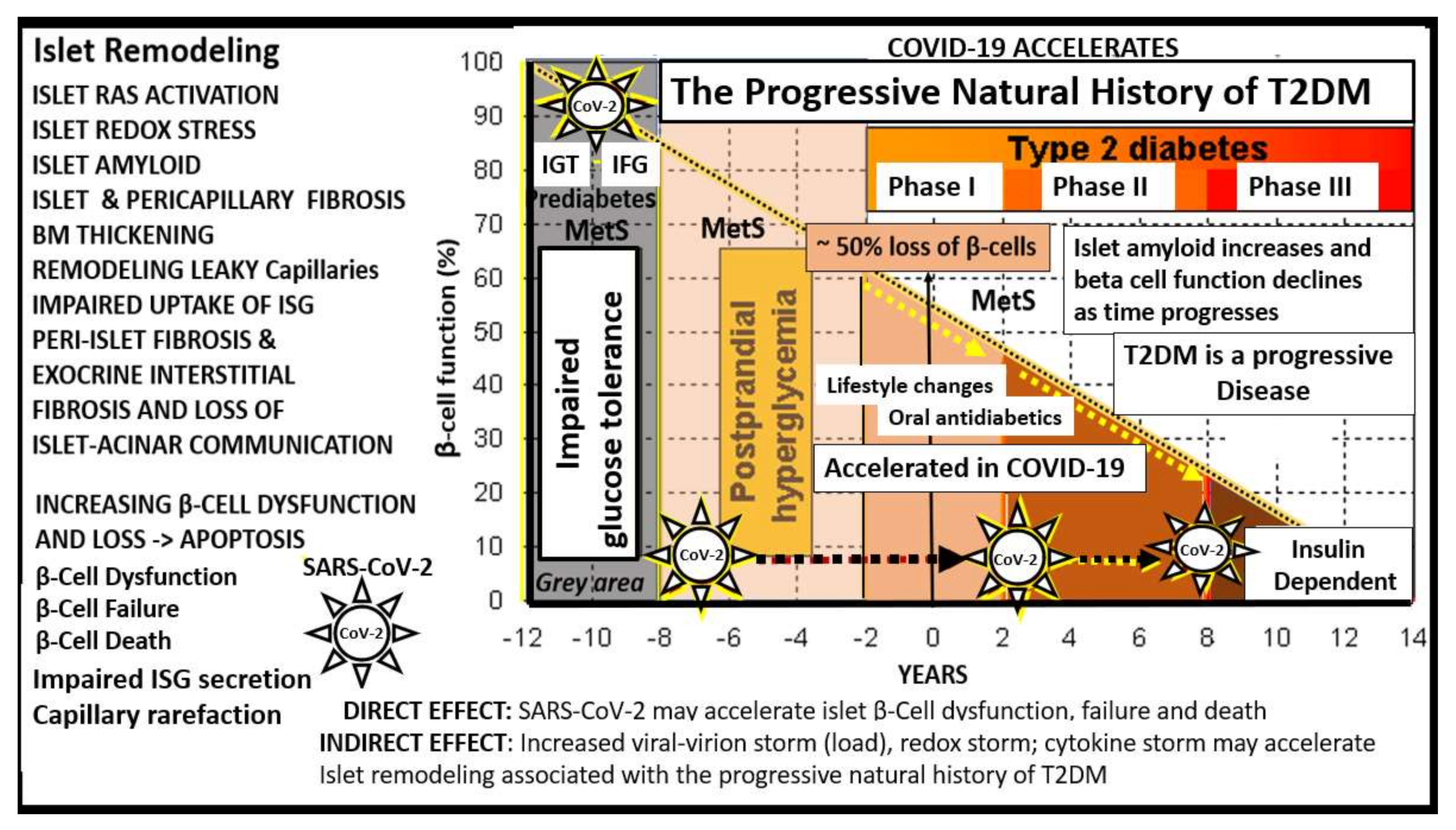
8. T2DM May Be Considered a Spectrum Disease
9. Glucotoxicity
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Ethics
References
- Cariou, B.; Hadjadj, S.; Wargny, M.; Pichelin, M.; Al-Salameh, A.; Allix, I.; Amadou, C.; Arnault, G.; Baudoux, F.; Bauduceau, B.; et al. Phenotypic characteristics and prognosis of inpatients with COVID-19 and diabetes: The CORONADO study. Diabetologia 2020, 63, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Gubbi, S.; Muniyappa, R. Metabolic Syndrome and COVID 19: Endocrine-Immune-Vascular Interactions Shapes Clinical Course. Endocrinology 2020, 161, bqaa112. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. The Role of Adipocytes and Adipocyte-Like Cells in the Severity of COVID-19 Infections. Obesity 2020, 28, 1187–1190. [Google Scholar] [CrossRef]
- Zhang, Y.; Somers, K.R.; Becari, C.; Polonis, K.; Pfeifer, M.A.; Allen, A.M.; Kellogg, T.A.; Covassin, N.; Singh, P. Comparative Expression of Renin-Angiotensin Pathway Proteins in Visceral Versus Subcutaneous Fat. Front. Physiol. 2018, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- Rubino, F.; Amiel, S.A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R.H.; Mingrone, G.; Boehm, B.; Cooper, M.E.; Chai, Z.; et al. New-Onset Diabetes in Covid-19. N. Engl. J. Med. 2020, 383, 789–790. [Google Scholar] [CrossRef]
- Hernandez, C.; Bruckner, A.L. Focus on “COVID Toes”. JAMA Dermatol. 2020, 156, 1003. [Google Scholar] [CrossRef]
- He, L.; Mae, M.A.; Sun, Y.; Muhl, L.; Nahar, K.; Liébanas, E.V.; Fagerlund, M.J.; Oldner, A.; Liu, J.; Genové, G.; et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2—Implications for microvascular inflammation and hypercoagulopathy in COVID-19 patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carlsson, P.O. The renin-angiotensin system in the endocrine pancreas. JOP. J. Pancreas 2001, 2, 26–32. [Google Scholar]
- Lau, T.; Carlsson, P.O.; Leung, P.S. Evidence for a local angiotensin system and dose-dependent inhibition of glucose-stimulated insulin release by angiotensin II in isolated pancreatic islets. Diabetologia 2004, 47, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, P.S. Pancreatic renin-angiotensin system: A novel target for the potential treatment of pancreatic diseases? JOP J. Pancreas 2003, 4, 89–91. [Google Scholar]
- Leung, P.S.; Carlsson, P.O. Tissue renin-angiotensin system: Its expression, localization, regulation and potential role in the pancreas. J. Mol. Endocrinol. 2001, 26, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, P.S. The physiology of a local renin–angiotensin system in the pancreas. J. Physiol. 2007, 580, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Chappell, M.C. A local pancreatic renin-angiotensin system: Endocrine and exocrine roles. Int. J. Biochem. Cell Biol. 2003, 35, 838–846. [Google Scholar] [CrossRef]
- Tahmasebi, M.; Inwang, E.R.; Vinson, G.P.; Puddefoot, J.R. The tissue renin-angiotensin system in human pancreas. J. Endocrinol. 1999, 161, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Tikellis, C.; Wookey, P.J.; Candido, R.; Andrikopoulos, S.; Thomas, M.C.; Cooper, M.E. Improved islet morphology after blockade of the renin-angiotensin system in the ZDF rat. Diabetes 2004, 53, 989–997. [Google Scholar] [CrossRef]
- Goossens, G.H. The Renin-Angiotensin System in the Pathophysiology of Type 2 Diabetes. Obes. Facts 2012, 5, 611–624. [Google Scholar] [CrossRef]
- Luther, J.M. Effects of aldosterone on insulin sensitivity and secretion. Steroids 2014, 91, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Sowers, K.M.; Pulakat, L.; Joginpally, T.; Krueger, B.; Whaley-Connell, A.; Sowers, J.R. Possible Mechanisms of Local Tissue Renin-Angiotensin System Activation in the Cardiorenal Metabolic Syndrome and Type 2 Diabetes Mellitus. Cardiorenal Med. 2011, 1, 193–210. [Google Scholar] [CrossRef] [Green Version]
- Prestes, T.R.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 Diabetes Mellitus: Implications of Islet RAS, Islet Fibrosis, Islet Amyloid, Remodeling, and Oxidative Stress. Antioxid. Redox Signal. 2007, 9, 891–910. [Google Scholar] [CrossRef]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Wasekar, C.; Lastra, G.; Manrique, C.; Stas, S.; Sowers, J.R. Ultrastructural islet study of early fibrosis in the Ren2 rat model of hypertension. Emerging role of the islet pancreatic pericyte-stellate cell. JOP J. Pancreas 2007, 8, 725–738. [Google Scholar]
- Hayden, M.R.; Sowers, J.R. Pancreatic Renin-Angiotensin-Aldosterone System in the Cardiometabolic Syndrome and Type 2 Diabetes Mellitus. J. Cardiometabolic Syndr. 2008, 3, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Aloysius, M.M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 presenting as acute pancreatitis. Pancreatology 2020, 20, 1026–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, H.; Fan, J.; Zhang, Y.; Wang, H.; Zhao, Q. Pancreatic Injury Patterns in Patients With Coronavirus Disease 19 Pneumonia. Gastroenterology 2020, 159, 367–370. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Redox Imbalance in Diabetes. Antioxid. Redox Signal. 2007, 9, 865–867. [Google Scholar] [CrossRef]
- Pina, A.F.; Patarrão, R.S.; Ribeiro, R.T.; Penha-Gonçalves, C.; Raposo, J.F.; Gardete-Correia, L.; Duarte, R.; Boavida, J.M.; Medina, J.L.; Henriques, R.; et al. Metabolic Footprint, Towards Understanding Type 2 Diabetes Beyond Glycemia. J. Clin. Med. 2020, 9, 2588. [Google Scholar] [CrossRef]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, M.; Yue, M.; Wang, P.; Yin, X.; Wang, L.; Yang, X.; Liu, H. Hyperinsulinemia precedes insulin resistance in offspring rats exposed to angiotensin II type 1 autoantibody in utero. Endocrine 2018, 62, 588–601. [Google Scholar] [CrossRef]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metabolism 2019, 93, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; Enden, M.V.D.; Kilo, C.; Tilton, R.G. Hyperglycemic Pseudohypoxia and Diabetic Complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef]
- Williamson, J.R.; Kilo, C.; Ido, Y. The role of cytosolic reductive stress in oxidant formation and diabetic complications. Diabetes Res. Clin. Pr. 1999, 45, 81–82. [Google Scholar] [CrossRef]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Roche, L.; Mesta, F. Oxidative Stress as Key Player in Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Cron, R.Q.; Behrens, E.M. Cytokine Storm Syndrome, 1st ed.; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G. Low-Grade Systemic Inflammation and the Development of Type 2 Diabetes: The Atherosclerosis Risk in Communities Study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Ehses, J.A. Islet Inflammation Impairs the Pancreatic β-Cell in Type 2 Diabetes. Physiology 2009, 24, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. Rev. 2019, 14, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böni-Schnetzler, M.; Meier, D.T. Islet inflammation in type 2 diabetes. Semin. Immunopathol. 2019, 41, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R. Empagliflozin ameliorates tunica adiposa expansion and vascular stiffening of the descending aorta in female db/db mice: An ultrastructure study. Adipobiology 2019, 10, 41–54. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Khaper, N.; Bryan, S.; Dhingra, S.; Singal, R.; Bajaj, A.; Pathak, C.M.; Singal, P.K. Targeting the Vicious Inflammation–Oxidative Stress Cycle for the Management of Heart Failure. Antioxid. Redox Signal. 2010, 13, 1033–1049. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the `Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Hayden, M.R.; Patel, K.; Habibi, J.; Gupta, D.; Tekwani, S.S.; Whaley-Connell, A.; Sowers, J.R. Attenuation of endocrine-exocrine pancreatic communication in type 2 diabetes: Pancreatic extracellular matrix ultrastructural abnormalities. J. Cardiometabolic Syndr. 2008, 3, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-W. Loss of beta-cells with fibrotic islet destruction in type 2 diabetes mellitus. Front. Biosci. 2008, 13, 6022–6033. [Google Scholar] [CrossRef] [Green Version]
- Habibi, J.; Whaley-Connell, A.; Hayden, M.R.; Demarco, V.G.; Schneider, R.; Sowers, S.D.; Karuparthi, P.; Ferrario, C.M.; Sowers, J.R. Renin Inhibition Attenuates Insulin Resistance, Oxidative Stress, and Pancreatic Remodeling in the Transgenic Ren2 Rat. Endocrinology 2008, 149, 5643–5653. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kim, J.-W.; Park, H.-S.; Lee, E.-Y.; Yoon, K.-H. Pancreatic stellate cells in the islets as a novel target to preserve the pancreatic β-cell mass and function. J. Diabetes Investig. 2020, 11, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Yang, Y.; Habibi, J.; Bagree, S.V.; Sowers, J.R. Pericytopathy: Oxidative Stress and Impaired Cellular Longevity in the Pancreas and Skeletal Muscle in Metabolic Syndrome and Type 2 Diabetes. Oxidative Med. Cell. Longev. 2010, 3, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R.; Sowers, J.R. Childhood-Adolescent Obesity in the Cardiorenal Syndrome: Lessons from Animal Models. Cardiorenal Med. 2011, 1, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R. Islet amyloid and fibrosis in the cardiometabolic syndrome and type 2 diabetes mellitus. J. Cardiometabolic Syndr. 2007, 2, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Opie, E.L. The relation of diabetes mellitus to lesions of the pancreas: Hyaline degeneration of the islands of Langerhans. J. Exp. Med. 1901, 5, 527–540. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. “A” is for amylin and amyloid in type 2 diabetes mellitus. JOP J. Pancreas 2001, 2, 124–139. [Google Scholar]
- Hayden, M.R.; Tyagi, S.C. Remodeling of the endocrine pancreas: The central role of amylin and insulin resistance. South. Med. J. 2000, 93, 24–28. [Google Scholar] [CrossRef]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2001, 1537, 179–203. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Tyagi, S.C. Islet redox stress: The manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP J. Pancreas 2002, 3, 86–108. [Google Scholar]
- Hayden, M.R.; Tyagi, S.C.; Kerklo, M.M.; Nicolls, M.R. Type 2 diabetes mellitus as a conformational disease. JOP J. Pancreas 2005, 6, 287–302. [Google Scholar]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.C.; Courtade, J.A.; Ehses, J.A.; Verchere, C.B. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2014, 58, 575–585. [Google Scholar] [CrossRef]
- D’Alessio, D.A.; Verchere, C.B.; Kahn, S.E.; Hoagland, V.; Baskin, D.G.; Palmiter, R.D.; Ensinck, J.W. Pancreatic Expression and Secretion of Human Islet Amyloid Polypeptide in a Transgenic Mouse. Diabetes 1994, 43, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, P.C.; Jang, J.; Gurlo, T.; Carty, M.D.; Soeller, W.C.; Butler, P.C. Diabetes Due to a Progressive Defect in Cell Mass in Rats Transgenic for Human Islet Amyloid Polypeptide (HIP Rat): A New Model for Type 2 Diabetes. Diabetes 2004, 53, 1509–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R.; Karuparthi, P.R.; Manrique, C.M.; Lastra, G.; Habibi, J.; Sowers, J.R. Longitudinal ultrastructure study of islet amyloid in the HIP rat model of type 2 diabetes mellitus. Exp. Biol. Med. 2007, 232, 772–779. [Google Scholar]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Lastra, G.; Patel, K.; Wasekar, C.; Manrique, C.M.; Ozerdem, U.; Stas, S.; Sowers, J.R. Ultrastructure of islet microcirculation, pericytes and the islet exocrine interface in the HIP rat model of diabetes. Exp. Biol. Med. 2008, 233, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Shah, A. Novel Coronavirus-Induced NLRP3 Inflammasome Activation: A Potential Drug Target in the Treatment of COVID-19. Front. Immunol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Rivera, J.F.; Gurlo, T.; Daval, M.; Huang, C.J.; Matveyenko, A.V.; Butler, P.C.; Costes, S. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: Protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2010, 18, 415–426. [Google Scholar] [CrossRef]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-cell assassin: IAPP cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef]
- Coate, K.C.; Cha, J.; Shrestha, S.; Wang, W.; Fasolino, M.; Morgan, A.; Dai, C.; Saunders, D.C.; Aramandla, R.; Jenkins, R.; et al. SARS-CoV-2 Cell Entry Factors ACE2 and TMPRSS2 are Expressed in the Pancreas but Not in Islet Endocrine Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kusmartseva, I.; Wu, W.; Syed, F.; Van Der Heide, V.; Jorgensen, M.; Joseph, P.; Tang, X.; Candelario-Jalil, E.; Yang, C.; Nick, H.; et al. ACE2 and SARS-CoV-2 Expression in the Normal and COVID-19 Pancreas. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 2020, 27, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Fignani, D.; Licata, G.; Brusco, N.; Nigi, L.; Grieco, G.E.; Marselli, L.; Overbergh, L.; Gysemans, C.; Colli, M.L.; Marchetti, P.; et al. SARS-CoV-2 receptor Angiotensin I-Converting Enzyme type 2 is expressed in human pancreatic islet β-cells and in pancreas microvasculature. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef] [Green Version]
- Tangvarasittichai, S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J. Diabetes 2015, 6, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I.H.; Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance andβ-Cell Dysfunction. Oxidative Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef] [Green Version]
- Walker, N.I.; Harmon, B.V.; Gobé, G.; Kerr, J.F. Patterns of cell death. Methods Achiev. Exp. Pathol. 1988, 13, 18–54. [Google Scholar]
- Lastra, G.; Manrique, C.M.; Hayden, M.R. The Role of Beta-Cell Dysfunction in the Cardiometabolic Syndrome. J. Cardiometabolic Syndr. 2006, 1, 41–46. [Google Scholar] [CrossRef]
- Kaiser, N.; Leibowitz, G.; Nesher, R. Glucotoxicity and Beta-Cell Failure in Type 2 Diabetes Mellitus. J. Pediatr. Endocrinol. Metab. 2003, 16, 5–22. [Google Scholar] [CrossRef]
- Wang, S.; Ma, P.; Zhang, S.; Song, S.; Wang, Z.; Ma, Y.; Xu, J.; Wu, F.; Duan, L.; Yin, Z.; et al. Fasting blood glucose at admission is an independent predictor for 28-day mortality in patients with COVID-19 without previous diagnosis of diabetes: A multi-centre retrospective study. Diabetologia 2020, 63, 2102–2111. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, X.; Liu, M.; Li, Y.; Zhang, J.; Li, A.; Zhang, H.; Xiu, R. Insulin protects against type 1 diabetes mellitus-induced ultrastructural abnormalities of pancreatic islet microcirculation. Microscopy 2020, dfaa036. [Google Scholar] [CrossRef]
- Tomita, T. Apoptosis in pancreatic β-islet cells in Type 2 diabetes. Bosn. J. Basic Med. Sci. 2016, 16, 162–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.A.; Mantzoros, C.; Sowers, J.R. Commentary: COVID-19 in patients with diabetes. Metabolism 2020, 107, 154217. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Gillies, C.; Singh, R.; Singh, A.; Chudasama, Y.; Coles, B.; Seidu, S.; Zaccardi, F.; Davies, M.J.; Khunti, K. Prevalence of co-morbidities and their association with mortality in patients with COVID-19: A systematic review and meta-analysis. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Li, C.; Sattar, N. Metabolic Syndrome and Incident Diabetes: Current state of the evidence. Diabetes Care 2008, 31, 1898–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unanue, E.R.; Wan, X. The Immunoreactive Platform of the Pancreatic Islets Influences the Development of Autoreactivity. Diabetes 2019, 68, 1544–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wali, J.A.; Masters, S.L.; Thomas, H.E. Linking Metabolic Abnormalities to Apoptotic Pathways in Beta Cells in Type 2 Diabetes. Cells 2013, 2, 266–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippi, G.; Lavie, C.J.; Henry, B.M.; Sanchis-Gomar, F. Do genetic polymorphisms in angiotensin converting enzyme 2 (ACE2) gene play a role in coronavirus disease 2019 (COVID-19)? Clin. Chem. Lab. Med. 2020, 58, 1415–1422. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R. An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms. Cells 2020, 9, 2475. https://doi.org/10.3390/cells9112475
Hayden MR. An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms. Cells. 2020; 9(11):2475. https://doi.org/10.3390/cells9112475
Chicago/Turabian StyleHayden, Melvin R. 2020. "An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms" Cells 9, no. 11: 2475. https://doi.org/10.3390/cells9112475
APA StyleHayden, M. R. (2020). An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms. Cells, 9(11), 2475. https://doi.org/10.3390/cells9112475