Abstract
The development of targeted therapies increases treatment options for metastatic castration resistant prostate cancer (mCRPC) patients. There is a need for strong predictive and prognostic signatures to guide physicians in treating mCRPC patients. In this review we unravel the possible actionability in the AR pathway, PI3K AKT signaling, and DNA repair pathways. Additionally, we make recommendations on biomarker trial design, and the clinical use of this new type of data.
1. Introduction
Prostate cancer (PCa) is not a uniform disease as its outcome is highly associated with the corresponding clinical risk category. In general, PCa is strongly driven by androgen receptor (AR) regulated transcription [1]. However, PCa can become ‘castration resistant’ (CRPC) after escaping conventional androgen deprivation treatment (ADT, LHRH agonists, and antagonists). As prolonged and intense targeting of the AR in CRPC patients resulted in a survival benefit, ‘castration resistance’ often remains AR driven (Figure 1).
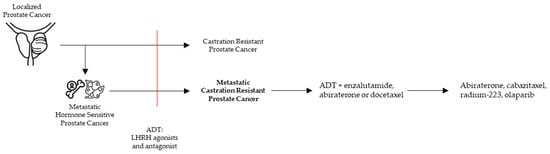
Figure 1.
Clinical pathway to metastatic castration resistant prostate cancer (mCRPC) with the current treatment options.
Metastatic castration resistant prostate cancer (mCRPC) results from many genomic and transcriptomic changes. The heterogeneous appearance of these changes underly the multifocal nature of PCa. It is thought that AR insensitive clones will survive the therapeutic selection pressure by AR targeting treatments. As mCRPC patients have a bad prognosis, these patients need to be rapidly treated with drugs likely to be effective. Identifying patients that would benefit from specific treatments will need to be based on specific markers, which could be detected as genomic and/or transcriptomic signatures.
In this review we will look at these genomic and transcriptomic alterations, with particular attention on those with prognostic and predictive potential.
2. mCRPC in the Clinic
2.1. Approved Treatments in Clinic
CRPC is diagnosed in PCa patients with castrate serum testosterone < 50 ng/dL and biochemical or radiological progression [2] (Figure 1). mCRPC is a noncurable, fatal disease with a median overall survival of 2.8 years [3,4]. In general, mCRPC patients will receive multiple lines of therapies. Accepted therapies available in the clinic can largely be classified in AR signaling inhibitors (ARSI), chemotherapeutics, and the alpha emitter radium-223. ARSI result in a strong inhibition of the AR pathway by drugs targeting the androgen synthesis (abiraterone) and antagonists of the AR (enzalutamide, darolutamide, apalutamide). Chemotherapeutics consist of tubulin binding docetaxel and the 2nd generation cabazitaxel (Figure 1).
Besides the standard of care, novel targeted therapies are entering clinic with PARP (poly-ADP ribose polymerase)-inhibitors being beneficial in PCa escaping from ARSI treatment on the condition that homologous recombination repair (HRR) alterations are present [5]. Other promising targeted therapies are still investigational and will be discussed throughout this review.
2.2. Treatment Allocation and Decision Making
It has been shown that the overall survival gain of consecutive treatments is not equal to the survival gain of each drug separately [3]. Combination treatments are thought to be superior over sequential treatments [6].
Most recent guidelines advise to base the choice of first-line treatment on different patient characteristics including performance status, symptoms, comorbidities, location and extent of disease, patient preference, and previous treatments [2]. The optimal treatment sequence, however, remains unclear. This contrasts the need for rapid and adequate treatment in mCRPC patients. Improvements in the understanding of PCa biology should help clinicians to decide on the optimal treatment sequence.
2.3. Role of Genetic Sequencing
In mCRPC, genetic sequencing on different time point can guide the management throughout the course of the disease [7,8]. Although sequencing of metastatic lesions reveals the main oncogenic drivers, obtaining this tissue can be practically challenging [9]. Sequencing circulating tumor cells or DNA isolated from liquid biopsies offers practical alternatives, as their mutations possibly reflect genomic alterations in the tissue [8].
TCGA, a molecular classification initiative, aimed to classify different tumors, including prostate cancer, by their genomic alterations. In mCRPC, large sequencing initiatives identified the genomic alterations and confirmed possible actionable targets to be present in most patients [10,11,12]. As discussed further, some alterations will predict a good response to drugs already available in the clinic, while others require the development of novel targeted therapies.
Studies confirmed a possible benefit of using genomic information to help treatment allocation [5,13,14]. However, as PCa is heterogenous, more clinical studies are needed to define the contribution of genomic information to the management of mCRPC patients.
3. PCa Biology and Markers
3.1. Genomic and Transcriptomic Pathways in PCa
3.1.1. AR Pathway
The AR signaling pathway is essential for prostate development and throughout different stages of PCa [1]. As ADT and/or ARSI are administered before patients develop mCRPC, AR pathways are altered in about 70% of the mCRPC cases [10]. These alterations can result in both AR dependent and independent disease [15].
The AR dependent resistance mechanisms cover AR overexpression, AR gene amplifications, mutations, splice variants, and alterations of coregulators, with the majority being amplifications. Amplifications result in an increased sensitivity to androgens due to an overexpression of AR [10]. In another study more than 70% of mCRPC patients had duplications of an upstream enhancer of AR, suggesting this to be a causative step in the development to CRPC [16].
AR mutations are most often found in the Ligand Binding Domain (LBD) [10]. These mutations are mostly situated within the ligand binding pocket, resulting in altered hormone binding and an altered response to AR targeting. Mutations L702H, T878A, and H875Y were associated with poor response to abiraterone, while enzalutamide and apalutamide act as agonists for the F877L and F876L mutated AR [17,18,19]. The agonistic effect of anti-androgens on the F877L mutation is found to be more apparent when comutated with the T878A mutation [20]. Darolutamide, a novel ARSI, maintains its antagonistic characteristics in F876L mutated PCa [21]. Taken together, clinical PCa treatment could be guided by the presence of AR mutations.
Although still strongly debated, expression of splice variants, e.g., AR-V7, Arv567es, may cause the PCa cells to become insensitive to ADT or ARSI [22]. AR-V7 has been proposed as clear prognostic factor, yet its detection by immunohistochemistry or qPCR needs optimization before routine application. Once this is available, it will be possible to assess the true value of functional AR-V7 status [22,23]. As these AR splice variants all change or lose the ligand binding domain of the AR, classic therapies targeting this region lose their effectiveness [24,25]. Yet, splice variants are always coexpressed with high levels of full-length AR. Additionally, these splice variants interact differently with the microtubular structure of the cell, rendering them more resistant to taxane chemotherapeutics [6,26]. The anti-helminthic drug niclosamide is under clinical investigation as it led to degradation of AR-V7, and resulted in a reappearance of sensitivity to anti-androgens in preclinical settings [6,27,28].
Different coregulators have been described to be involved in the AR axis, including: NCOA1, 2, 3, p300, transcription factor FOXA1, negative regulator NCOR1/2, and AR induced negative regulator ZBTB16 [10,29]. Preclinical p300 inhibitors and investigational inhibitory strategies for the coactivators are available and show benefit in preclinical settings [30]. Interestingly, these coregulators are used by different families of transcription factors, making their PCa specific targeting challenging. Possible clinical benefits of targeting the coregulators remain to be discovered.
SPOP is described as tumor suppressor gene, degrading oncogenic substrates like NCOA3/SRC3 (co-activator of AR), ERG, and DEK [31,32]. Besides this, SPOP is also involved in ubiquitination and degradation of the AR [33]. In PCa SPOP is often mutated leading to increased androgen signaling and tumor progression [34,35]. Consequently, these tumors are highly AR regulated and form a distinct cluster in the TCGA molecular classification [13]. In the localized setting SPOP mutations are the most frequent alteration (13%), but it is less frequently found in the metastatic cohort (around 8%) [10,36,37]. In the later stages of PCa, SPOP mutations have therapeutic implications, being more resistant to chemotherapeutics and more susceptible for AR targeting [38,39]. Interestingly SPOP mutations are mutually exclusive with changes in TP53, PTEN, or TMPRSS2-ERG [37].
Erythroblast Transformation Specific (ETS) gene fusions are the most common genetic alteration in cancer and it is found in approximately 50% of localized PCa as well as in mCRPC [10,40]. Fusions link the transcriptional control regions of a prostate-specific, AR regulated, TMPRSS2 gene to the protein coding part of members of the oncogenic ETS gene family (e.g., ERG). This renders the ETS oncogene family AR dependent and highly expressed. TMPRSS2-ERG fusions are more resistant to treatment with taxane-chemotherapeutics and, as they are AR regulated, these patients are more susceptible to ADT and ARSI [41,42,43].
A lot of attention has been attributed to glucocorticoid receptor (GR) upregulation as escape mechanism after ARSI treatment. As GR and AR have very similar mechanisms of action, the GR can take over part of the AR pathway [44]. Moreover, in neoadjuvant ARSI trials, GR expression and activity are correlated with higher residual tumor volume in different studies based on immunohistochemistry and transcriptome changes [45,46].
GR antagonists were found to be beneficial as adjuvant treatment to ARSI in preclinical models, as the expression of GR and AR seemed to be inversely correlated [47]. Androgens and glucocorticoids are known to affect each other’s signaling pathways, which suggests both pathways should be targeted in order to be effective [48]. In different clinical mCRPC treatment regiments (chemotherapeutics and abiraterone) glucocorticoids are coadministered to diminish side effects, possibly stimulating GR upregulated tumors to progress [47]. A phase II trial investigated the use of the GR-antagonist mifepristone monotherapy as an AR antagonist in 19 both non-metastatic and metastatic CRPC patients [49]. GR blockage resulted in upregulated circulating androgens due to a feedback via adrenocorticotropic hormone (ACTH) inducing adrenal androgens and their conversion to testosterone and DHT. This feedback likely masked the therapeutic value of mifepristone in CRPC patients. It will therefore be interesting to see the effect of combined treatment with ARSI and GR antagonists, currently under investigation in a phase I/II trial [50].
Castration and abiraterone both target the AR axis by depleting its ligands, but this inhibition is overcome by the activation of the tumoral steroid synthesis. Cancer cells of local and metastatic disease can synthesize DHT from adrenal precursors, resulting in a release of the inhibited AR [51]. For example, the 3β-hydroxysteroid dehydrogenase isoenzyme-1 (HSD3B1) can become expressed in these cells and will convert dehydroepiandrosterone (DHEA) to androstenedione and androstenediol to testosterone. ARSI treatments induce HSD3B1 levels, by decreasing proteasomal degradation. Interestingly, single nucleotide polymorphisms in the HSD3B1 gene also affect the expression levels. This results in higher concentrations of androstenedione, and therefore DHT [52,53]. Preclinical models are moreover suggestive that some adrenal steroidogenesis remains upon CYP17A inhibition, by proving that adrenalectomy has stronger effects than CYP17A inhibition [54]. Further translational studies should try to target these escape mechanisms, in order to exploit this new knowledge clinically.
In conclusion, as PCa is an androgen driven tumor, different escape mechanisms alter the AR pathway. Future efforts should not only be directed to the characterization of AR alterations and common other AR escape mechanisms, but also focus on targeting the steroid metabolism and the GR pathway.
3.1.2. PI3K–AKT–MAPK Pathway—PTEN Loss
As PTEN is a major regulator of the cell cycle and tumor suppressor gene, its loss is associated with poor clinical outcome and progression to mCRPC [55,56,57,58,59]. Deletion of PTEN is more often present in mCRPC (17% in localized and 40% of mCRPC cases), independent of metastatic load [10,36,60]. In mCRPC, PTEN loss is associated with ETS rearrangements (see above), enforcing their mutagenic capacities [61,62].
The exact mechanisms explaining how PTEN leads to castration resistance are still debated. The inhibition of the PI 3K pathway (PI3K, AKT, mTORC1/2), via AKT inhibition by PTEN, is considered an important contributor [63]. As AKT promotes cell survival and its activation is associated with multiple cancers, AKT inhibitors have been developed [63]. Preclinical evidence in PTEN deleted models showed lower AR activity after activation of the PI 3K pathway. As AR and PI 3K compensate for each other’s inhibition, a dual inhibition of both pathways, consisting of an AKT inhibitor and an ARSI, seems promising [64]. A phase III trial studying this dual inhibition (ipatasertib/abiraterone) is currently running in mCRPC patients with PTEN loss [65].
3.1.3. DNA Repair
Of all germline variants found in metastatic cancers, 75% were related to defects in DNA repair confirming the importance of aberrant DNA repair in carcinogenesis [66]. Although localized PCa has a low mutational burden, germline mutations in DNA repair genes are present in 12% of mCRPC patients [66,67]. During synthesis (S) or Gap 2 (G2) phase homologous recombination repair (HRR) accurately repairs double strand breaks. Whenever the HRR pathway is deficient, other repair pathways take over to repair DNA damage, albeit all with higher failure rates. Most common genetic mutations affecting HRR are BRCA2, CHEK2, ATM, and BRCA1 [5,68].
Mutations in HRR render patients sensitive to PARP inhibitors (PARPi), as PARP is involved in the repair of single strand DNA breaks. If unrepaired, these are transformed to double strand DNA breaks during replication. PARPi are therefore called synthetically lethal because these are only effective when repair mechanisms fail [5,68]. This explains why PARPi would preferentially target tumor cells, contributing to the favorable risk profile. The phase III PROfound trial studied PARPi in 387 progressive mCRPC patients, with confirmed prolonged progression free survival and good functional outcomes in patients with HRR defects [5].
Mismatch repair (MMR) genes will correct nucleotide allocation mistakes by an excision and reallocation of the base by verifying the genetic code on the adjacent strand. Different genes have been described to be involved in MMR, including: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. Tumors with MMR mutations typically accumulate large numbers of mutations and microsatellite instability. Such tumors can be relatively easily identified with immunohistochemistry (see below) and have been shown to respond well to both ADT and ARSI treatment and possibly immune checkpoint inhibitors [69,70].
Cyclin dependent kinase 12 (CDK12) is a member of the kinases, involved in the regulation of the cell cycle. Biallelic loss of CDK12 defines a distinct subtype of PCa and is present in 5% of mCRPC [10]. When present, most patients have no alterations in genes involved in other oncogenic pathways like ERG, SPOP, TP53, and PTEN [71]. Tumors with CDK12 loss show aberrant DNA replication during S-phase and have a high immune infiltration [72]. A retrospective multicenter study suggested CDK12 loss to form an aggressive PCa subtype with poor responses to hormonal and taxane treatment as well as PARPi. Surprisingly, good response of this subtype was seen after treatment with immune checkpoint inhibitors, possibly linked to their higher mutation burden [71]. Further prospective studies should be performed to investigate the underlying biology as well as the opportunities in clinical management of these patients.
3.1.4. Neuroendocrine Differentiation
In this review we focused on adenocarcinoma of the prostate. Although this is not the only histopathologic form of mCRPC, it covers more than 95% of all mCRPC cases [10]. Preclinical evidence suggests neuroendocrine features to be selected during AR targeting treatments or via an accrual of mutations [73,74]. Different gene sets have been developed to assess neuroendocrine transdifferentiation, yet these should be validated in the mCRPC setting [75].
In mCRPC patients it is unclear what percentage of tumors develop into a neuroendocrine state, and what percentage result from a neuroendocrine population being positively selected as a result of the treatments. The latter has enriched losses of p53 and Rb1 and should be treated early on [76,77].
An overview of the discussed pathways from this section is represented in Figure 2.
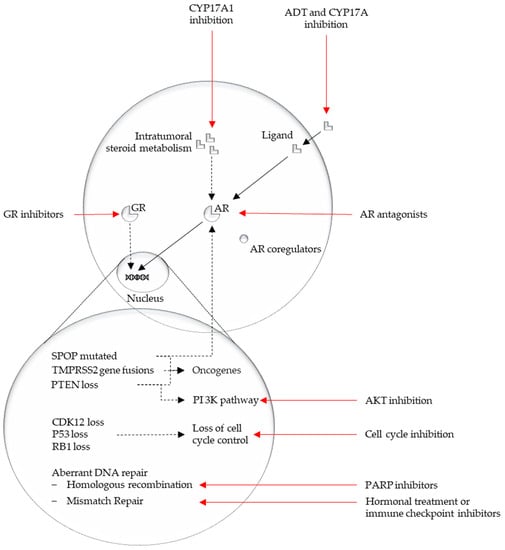
Figure 2.
Overview major affected pathways in mCRPC and their possible actionability.
3.2. Biomarkers
A good biomarker has reproducible predictive or prognostic characteristics in a well-defined disease state, and provides information that can impact the treatment allocation and the further care. As a biomarker should be reproducible in a certain disease state, different patients, and trial settings, a reliable predictive risk profile will most likely consist of different biomarkers. Here, we focus on the possible contribution of genomic biomarkers in future PCa care.
Although there are many reports suggesting new biomarkers for different specific PCa settings, limited clinical improvements have been made [78,79]. One approach using the adapted genomic PAM50 classifier from breast cancer can classify PCa into luminal and basal subtypes [80]. AR expression is higher in luminal than basal subtypes. Furthermore, luminal B subtype was proven to benefit from additional ADT after primary treatment [81]. Luminal A subtypes had the best survival and overall, the luminal subtypes were more sensitive than the basal subtype to docetaxel and less sensitive to platinum and etoposide chemotherapeutics [82].
Histopathology is an elegant way to detect protein-biomarkers. Firstly, although hematoxylin-eosin staining suffices to find malignant cells, some markers (like AR) can help to reduce doubt [83]. Second, some histological entities (e.g., cribriform PCa) are associated with molecular alterations (e.g., PTEN loss, SPOP, and ATM mutant) and can therefore reflect corresponding underlying biomarkers [84]. Lastly, histological markers reflect underlying transcriptomic or genomic alterations and can therefore often predict outcomes accordingly. A big advantage of immunohistochemistry to check for underlying biomarkers is that this methodology and corresponding antibodies are readily available and mostly rather cheap (e.g., AR targeted treatment: PTEN and ERG) [59].
3.3. Prospects
Most candidate biomarkers result from small, hypothesis generating, studies. Subsequent biomarker trials should therefore be run to validate them for specific indications. Besides treatment allocation, signatures could identify reasons for treatment intensification, drug selection, and duration of therapy. In further disease stages the role in defining the treatment-sequence becomes equally important [14]. Good examples of upcoming treatment allocation trials are the GUNS and the ProBio trial. The GUNS trial will investigate the guided treatment allocation using gene signatures from a prostate biopsy [85]. In mCRPC patients, the ProBio trial uses cfDNA in mCRPC patients to allocate them to treatment arms based upon genomic alterations [86].
Multiple attempts have already been done to categorize PCa based on its molecular characteristics, with the TCGA being the most promising [13,79,87]. However, in a study where each PCa tumor focus was sequenced, only 30% of patients could be classified in one of the proposed classes [88]. Furthermore, a preclinical actionable target is present in almost 90% of mCRPC patients, but this cannot be implemented into clinic due to the lack of specific inhibitors and high-quality clinical trials [10].
Besides genomic alterations, which were the topic of this review, there are multiple clinical biomarkers being studied. The overarching aim is that some of these factors will contribute to the identification of specific patient profiles that will allow clinicians to propose treatments likely to be effective (Figure 3).
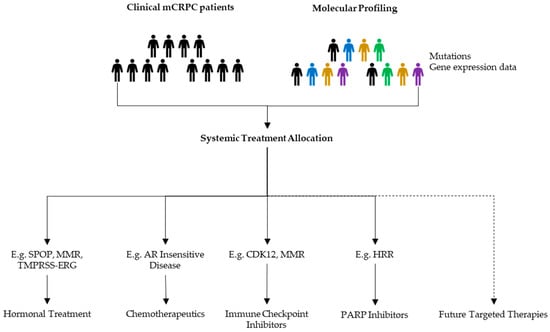
Figure 3.
Possible benefits of molecular profiling in mCRPC management. When progressive disease appears, discussions upon next line treatments should be based upon all available data to allocate a patient tailored follow-up treatment.
Additionally, by increasing knowledge on cancer biology throughout the following years, novel treatment options that are not included in this review will be suggested and investigated. These can be targeted towards mCRPC or act as an adjuvant treatment to block certain escape mechanisms.
4. Conclusions
To increase PCa survival, multidisciplinary oncological PCa boards should be able to use all available diagnostic data for their discussions and treatment decisions (Figure 3). These data should include clinical characteristics, imaging, and histology. Additional predictive or prognostic biomarker-profiles based on molecular and genomic information may help to guide the clinical team towards the optimal treatment for each mCRPC patient.
Defining better subclassifications of mCRPC populations that respond well to given treatments is therefore a high priority in mCRPC care. Predictive molecular biomarkers for the treatment of mCRPC are intensively investigated, but until now, only limited clinical implementation has been realized.
Author Contributions
Conceptualization, W.D., T.G., F.C.; writing—original draft preparation, W.D., T.G., F.C.; writing—review and editing, W.D., T.G., F.C., M.E., A.C., G.D., L.M., T.V.d.B., R.M., S.J.; supervision, F.C. and T.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. This research was supported by the ‘Jozef De Wever’ fund for prostate cancer prevention of the KU Leuven, Leuven, Belgium.
Acknowledgments
Steven Joniau is a Senior Clinical Investigators of the Research Foundation Flanders (FWO).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sehgal, P.D.; Bauman, T.M.; Nicholson, T.M.; Vellky, J.E.; Ricke, E.A.; Tang, W.; Xu, W.; Huang, W.; Ricke, W.A. Tissue-specific quantification and localization of androgen and estrogen receptors in prostate cancer. Hum. Pathol. 2019, 89, 99–108. [Google Scholar] [CrossRef] [PubMed]
- EAU Guidelines. EAU Annual Congress; EAU Guidelines Office: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Mottet, N.; Gillessen, S.; Mottet, N. EAU Guidelines: Prostate Cancer|Uroweb. 2020. Available online: https://uroweb.org/guideline/prostate-cancer/ (accessed on 7 August 2020).
- Francini, E.; Gray, K.P.; Shaw, G.K.; Evan, C.P.; Hamid, A.A.; Perry, C.E.; Kantoff, P.W.; Taplin, M.-E.; Sweeney, C.J. Impact of new systemic therapies on overall survival of patients with metastatic castration-resistant prostate cancer in a hospital-based registry. Prostate Cancer Prostatic Dis. 2019, 22, 420–427. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Carceles-Cordon, M.; Kelly, W.K.; Gomella, L.; Knudsen, K.E.; Rodriguez-Bravo, V.; Domingo-Domenech, J. Cellular rewiring in lethal prostate cancer: The architect of drug resistance. Nat. Rev. Urol. 2020, 17, 292–307. [Google Scholar] [CrossRef] [PubMed]
- Hovelson, D.H.; Tomlins, S.A. The Role of Next-Generation Sequencing in Castration-Resistant Prostate Cancer Treatment. Cancer J. 2016, 22, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Adalsteinsson, V.A.; Cibulskis, K.; Choudhury, A.D.; Rosenberg, M.; Cruz-Gordillo, P.; Francis, J.M.; Zhang, C.-Z.; Shalek, A.K.; Satija, R.; et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat. Biotechnol. 2014, 32, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Dawson, N.A.; Zibelman, M.; Lindsay, T.; Feldman, R.; Saul, M.; Gatalica, Z.; Korn, W.M.; Heath, E.I. An Emerging Landscape for Canonical and Actionable Molecular Alterations in Primary and Metastatic Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1373–1382. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nat. Cell Biol. 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Naing, A.; Li, Y.; Stephen, B.; Zinner, R.; Subbiah, V.; Fu, S.; Karp, D.; Falchook, G.S.; et al. Cancer Therapy Directed by Comprehensive Genomic Profiling: A Single Center Study. Cancer Res. 2016, 76, 3690–3701. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Helsen, C.; Prekovic, S.; Broeck, T.V.D.; Spans, L.; van Poppel, H.; Joniau, S. Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol. 2014, 11, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef]
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G.; Jenster, G.; Berrevoets, C.; Claassen, E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990, 173, 534–540. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Mizuno, K.; Yamasaki, T.; Miyazaki, Y.; Makino, Y.; Okasho, K.; Li, X.; Utsunomiya, N.; Goto, T.; Kobayashi, T.; et al. Clinical utility of androgen receptor gene aberrations in circulating cell-free DNA as a biomarker for treatment of castration-resistant prostate cancer. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2013, 2, e00499. [Google Scholar] [CrossRef]
- Prekovic, S.; van Royen, M.E.; Voet, A.R.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.; Broeck, T.V.D.; Smeets, E.; Spans, L.; et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712. [Google Scholar] [CrossRef]
- Moilanen, A.-M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykänen, P.S.; Törmäkangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, srep12007. [Google Scholar] [CrossRef]
- Handle, F.; Claessens, F. AR variants: Lost in translation to clinical practice? Nat. Rev. Urol. 2019, 16, 451–452. [Google Scholar] [CrossRef]
- Kaczorowski, A.; Chen, X.; Herpel, E.; Merseburger, A.S.; Kristiansen, G.; Bernemann, C.; Hohenfellner, M.; Cronauer, M.V.; Duensing, S. Antibody selection influences the detection of AR-V7 in primary prostate cancer. Cancer Treat. Res. Commun. 2020, 24, 100186. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen Receptor Splice Variants Activate Androgen Receptor Target Genes and Support Aberrant Prostate Cancer Cell Growth Independent of Canonical Androgen Receptor Nuclear Localization Signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Bernemann, C.; Schnoller, T.J.; Luedeke, M.; Steinestel, K.; Boegemann, M.; Schrader, A.J.; Steinestel, J. Expression of AR-V7 in Circulating Tumour Cells Does Not Preclude Response to Next Generation Androgen Deprivation Therapy in Patients with Castration Resistant Prostate Cancer. Eur. Urol. 2017, 71, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Thadani-Mulero, M.; Portella, L.; Sun, S.; Sung, M.; Matov, A.; Vessella, R.L.; Corey, E.; Nanus, D.M.; Plymate, S.R.; Giannakakou, P. Androgen Receptor Splice Variants Determine Taxane Sensitivity in Prostate Cancer. Cancer Res. 2014, 74, 2270–2282. [Google Scholar] [CrossRef] [PubMed]
- Abiraterone Acetate, Niclosamide, and Prednisone in Treating Patients with Hormone-Resistant Prostate Cancer—Full Text View—ClinicalTrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02807805 (accessed on 7 August 2020).
- Enzalutamide and Niclosamide in Treating Patients with Recurrent or Metastatic Castration-Resistant Prostate Cancer—Full Text View—ClinicalTrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03123978 (accessed on 7 August 2020).
- Alen, P.; Claessens, F.; Verhoeven, G.; Rombauts, W.; Peeters, B. The Androgen Receptor Amino-Terminal Domain Plays a Key Role in p160 Coactivator-Stimulated Gene Transcription. Mol. Cell. Biol. 1999, 19, 6085–6097. [Google Scholar] [CrossRef]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef]
- Ju, L.-G.; Zhu, Y.; Long, Q.-Y.; Li, X.-J.; Lin, X.; Tang, S.-B.; Yin, L.; Xiao, Y.; Wang, X.-H.; Li, L.; et al. SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 2019, 26, 1156–1168. [Google Scholar] [CrossRef]
- Mani, R.-S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef]
- Geng, C.; Rajapakshe, K.; Shah, S.S.; Shou, J.; Eedunuri, V.K.; Foley, C.; Fiskus, W.; Rajendran, M.; Chew, S.A.; Zimmermann, M.; et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014, 74, 5631–5643. [Google Scholar] [CrossRef]
- Geng, C.; He, B.; Xu, L.; Barbieri, C.E.; Eedunuri, V.K.; Chew, S.A.; Zimmermann, M.; Bond, R.; Shou, J.; Li, C.; et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc. Natl. Acad. Sci. USA 2013, 110, 6997–7002. [Google Scholar] [CrossRef]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Spans, L.; Clinckemalie, L.; Helsen, C.; Vanderschueren, D.; Boonen, S.; Lerut, E.; Joniau, S.; Claessens, F. The Genomic Landscape of Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 10822–10851. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhu, Y.; Ma, J.; Chang, K.; Ding, D.; Bai, Y.; Gao, K.; Zhang, P.; Mo, R.; Feng, K.; et al. Prostate Cancer-associated SPOP mutations enhance cancer cell survival and docetaxel resistance by upregulating Caprin1-dependent stress granule assembly. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Sumanasuriya, S.; Bianchini, D.; et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593. [Google Scholar] [CrossRef]
- Clark, J.; Cooper, C.S. ETS gene fusions in prostate cancer. Nat. Rev. Urol. 2009, 6, 429–439. [Google Scholar] [CrossRef]
- Reig, Ò.; Marín-Aguilera, M.; Carrera, G.; Jiménez, N.; Paré, L.; García-Recio, S.; Gaba, L.; Pereira, M.V.; Fernandez, P.; Prat, A.; et al. TMPRSS2-ERG in Blood and Docetaxel Resistance in Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 709–713. [Google Scholar] [CrossRef]
- Semaan, L.; Mander, N.; Cher, M.L.; Chinni, S.R. TMPRSS2-ERG fusions confer efficacy of enzalutamide in an in vivo bone tumor growth model. BMC Cancer 2019, 19, 972–1010. [Google Scholar] [CrossRef]
- Graff, R.E.; Pettersson, A.; Lis, R.T.; Dupre, N.C.; Jordahl, K.M.; Nuttall, E.; Rider, J.R.; Fiorentino, M.; Sesso, H.D.; Kenfield, S.A.; et al. The TMPRSS2:ERG fusion and response to androgen deprivation therapy for prostate cancer. Prostate 2015, 75, 897–906. [Google Scholar] [CrossRef]
- Claessens, F.; Joniau, S.; Helsen, C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell. Mol. Life Sci. 2017, 74, 2217–2228. [Google Scholar] [CrossRef]
- Efstathiou, E.; Davis, J.W.; Pisters, L.; Li, W.; Wen, S.; McMullin, R.P.; Gormley, M.; Ricci, D.; Titus, M.; Hoang, A.; et al. Clinical and Biological Characterisation of Localised High-risk Prostate Cancer: Results of a Randomised Preoperative Study of a Luteinising Hormone-releasing Hormone Agonist with or Without Abiraterone Acetate plus Prednisone. Eur. Urol. 2019, 76, 418–424. [Google Scholar] [CrossRef]
- Sowalsky, A.G.; Ye, H.; Bhasin, M.; van Allen, A.T.; Loda, M.; Lis, R.T.; Montaser-Kouhsari, L.; Calagua, C.; Ma, F.; Russo, J.W.; et al. Neoadjuvant-Intensive Androgen Deprivation Therapy Selects for Prostate Tumor Foci with Diverse Subclonal Oncogenic Alterations. Cancer Res. 2018, 78, 4716–4730. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.M.; Eder, I.E.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2017, 24, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Alyamani, M.; Li, J.; Patel, M.; Taylor, S.; Nakamura, F.; Berk, M.; Przybycin, C.; Posadas, E.; Madan, R.; Gulley, J.; et al. Deep androgen receptor suppression in prostate cancer exploits sexually dimorphic renal expression for systemic glucocorticoid exposure. Ann. Oncol. 2020, 31, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.-E.; Manola, J.; Oh, W.K.; Kantoff, P.W.; Bubley, G.J.; Smith, M.; Barb, D.; Mantzoros, C.; Gelmann, E.P.; Balk, S.P. A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 2008, 101, 1084–1089. [Google Scholar] [CrossRef]
- Enzalutamide and Mifepristone in Treating Patients with Metastatic Hormone Resistant Prostate Cancer—Full Text View—ClinicalTrials. Available online: https://clinicaltrials.gov/ct2/show/NCT02012296 (accessed on 30 June 2020).
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased Expression of Genes Converting Adrenal Androgens to Testosterone in Androgen-Independent Prostate Cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef]
- Hearn, J.W.; AbuAli, G.; Reichard, C.A.; Reddy, C.A.; Magi-Galluzzi, C.; Chang, K.-H.; Carlson, R.; Rangel, L.; Reagan, K.; Davis, B.J.; et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: A retrospective, multicohort study. Lancet Oncol. 2016, 17, 1435–1444. [Google Scholar] [CrossRef]
- Sabharwal, N.; Sharifi, N. HSD3B1 Genotypes Conferring Adrenal-Restrictive and Adrenal-Permissive Phenotypes in Prostate Cancer and Beyond. Endocrinology 2019, 160, 2180–2188. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Zhang, A.; Hernandez, S.; Marck, B.T.; Zhang, X.; Tamae, D.; Biehl, H.E.; Tretiakova, M.S.; Bartlett, J.; Burns, J.F.; et al. Contribution of Adrenal Glands to Intratumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2018, 25, 426–439. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Cunha, I.; Coudry, R.; Fonseca, F.; Torres, C.; Soares, F.; Squire, J. MP-17.17: FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Urology 2007, 70, 135. [Google Scholar] [CrossRef]
- Lotan, T.L.; Gurel, B.; Sutcliffe, S.; Esopi, D.; Liu, W.; Xu, J.; Hicks, J.L.; Park, B.H.; Humphreys, E.; Partin, A.W.; et al. PTEN Protein Loss by Immunostaining: Analytic Validation and Prognostic Indicator for a High Risk Surgical Cohort of Prostate Cancer Patients. Clin. Cancer Res. 2011, 17, 6563–6573. [Google Scholar] [CrossRef] [PubMed]
- Köksal, I.T.; Dirice, E.; Yasar, D.; Sanlioglu, A.D.; Ciftcioglu, A.; Gulkesen, K.H.; O Ozes, N.; Baykara, M.; Luleci, G.; Sanlioglu, S. The assessment of PTEN tumor suppressor gene in combination with Gleason scoring and serum PSA to evaluate progression of prostate carcinoma. Urol. Oncol. Semin. Orig. Investig. 2004, 22, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Chaux, A.; Peskoe, S.B.; Gonzalez-Roibon, N.; Schultz, L.; Albadine, R.; Hicks, J.; de Marzo, A.M.; Platz, E.A.; Netto, G. Loss of PTEN expression is associated with increased risk of recurrence after prostatectomy for clinically localized prostate cancer. Mod. Pathol. 2012, 25, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Ye, H.; Xie, W.; Lis, R.; Calagua, C.; Zhang, Z.; Trinh, Q.-D.; Chang, S.L.; Harshman, L.C.; Ross, A.E.; et al. Evaluation of Intense Androgen Deprivation Before Prostatectomy: A Randomized Phase II Trial of Enzalutamide and Leuprolide With or Without Abiraterone. J. Clin. Oncol. 2019, 37, 923–931. [Google Scholar] [CrossRef]
- Gilson, C.; Ingleby, F.; Gilbert, D.C.; Parry, M.A.; Atako, N.B.; Ali, A.; Hoyle, A.; Clarke, N.W.; Gannon, M.; Wanstall, C.; et al. Genomic Profiles of De Novo High- and Low-Volume Metastatic Prostate Cancer: Results From a 2-Stage Feasibility and Prevalence Study in the STAMPEDE Trial. JCO Precis. Oncol. 2020, 2020, 882–897. [Google Scholar] [CrossRef]
- Gevaert, T.; Montironi, R.; Lopez-Beltran, A.; van Leenders, G.; Allory, Y.; de Ridder, D.; Claessens, F.; Kockx, M.; Akand, M.; Joniau, S.; et al. Genito-urinary genomics and emerging biomarkers for immunomodulatory cancer treatment. Semin. Cancer Biol. 2018, 52, 216–227. [Google Scholar] [CrossRef]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Roche, H.-L. Ipatasertib Plus Abiraterone Plus Prednisone/Prednisolone, Relative to Placebo Plus Abiraterone Plus Prednisone/Prednisolone in Adult Male Patients with Metastatic Castrate-Resistant Prostate Cancer (IPATential150). Available online: https://clinicaltrials.gov/ct2/show/NCT03072238 (accessed on 7 August 2020).
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nat. Cell Biol. 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; de Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Shaukat, F.; Velho, P.I.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Velho, P.I.; Fu, W.; Wang, H.; Agarwal, N.; Santos, V.S.; Maughan, B.L.; Pili, R.; Adra, N.; Sternberg, C.N.; et al. CDK12-Altered Prostate Cancer: Clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors. JCO Precis. Oncol. 2020, 4, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782. [Google Scholar] [CrossRef]
- Ci, X.; Hao, J.; Dong, X.; Xue, H.; Wu, R.; Choi, S.Y.C.; Haegert, A.; Collins, C.C.; Liu, X.; Lin, D.; et al. Conditionally Reprogrammed Cells from Patient-Derived Xenograft to Model Neuroendocrine Prostate Cancer Development. Cells 2020, 9, 1398. [Google Scholar] [CrossRef]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell-derived eXplant model. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Ostano, P.; Mello-Grand, M.; Sesia, D.; Gregnanin, I.; Peraldo-Neia, C.; Guana, F.; Jachetti, E.; Farsetti, A.; Chiorino, G. Gene Expression Signature Predictive of Neuroendocrine Transformation in Prostate Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 1078. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Stoyanova, T. Biomarkers for Diagnosis and Prognosis of Prostate Cancer. Prostatectomy 2019. [Google Scholar] [CrossRef]
- Eggener, S.E.; Rumble, R.B.; Armstrong, A.J.; Morgan, T.M.; Crispino, T.; Cornford, P.; van der Kwast, T.; Grignon, D.J.; Rai, A.J.; Agarwal, N.; et al. Molecular Biomarkers in Localized Prostate Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1474–1494. [Google Scholar] [CrossRef] [PubMed]
- Mian, O.Y.; Tendulkar, R.D.; Abazeed, M.E. The evolving role of molecular profiling in prostate cancer: Basal and luminal subtyping transcends tissue of origin. Transl. Cancer Res. 2017, 6, S1441–S1445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef]
- Kim, W.; Small, E.J.; Aggarwal, R.R.; Den, R.B.; Lehrer, J.; Zhang, L.; Youngren, J.; Goldstein, T.C.; Alumkal, J.J.; Gleave, M.; et al. Luminal and basal subtyping of metastatic castration-resistant prostate cancer (mCRPC) and its clinical implications. J. Clin. Oncol. 2018, 36, 197. [Google Scholar] [CrossRef]
- Kehr, E.; Masry, P.; Lis, R.; Loda, M.; Taplin, M.-E.; Hirsch, M.S. Detecting metastatic prostate carcinoma in pelvic lymph nodes following neoadjuvant hormone therapy: The eyes have it! Histopathology 2015, 68, 303–307. [Google Scholar] [CrossRef]
- Elfandy, H.; Armenia, J.; Pederzoli, F.; Pullman, E.; Pértega-Gomes, N.; Schultz, N.; Viswanathan, K.; Vosoughi, A.; Blattner, M.; Stopsack, K.H.; et al. Genetic and Epigenetic Determinants of Aggressiveness in Cribriform Carcinoma of the Prostate. Mol. Cancer Res. 2018, 17, 446–456. [Google Scholar] [CrossRef]
- SUO 2019: Genomic Umbrella Neoadjuvant Study and other Biomarker Trials. 2020. Available online: https://www.urotoday.com/conference-highlights/suo-2019/suo-2019-prostate-cancer/117559-suo-2019-guns-and-other-biomarker-trials.html (accessed on 7 August 2020).
- Case Medical Research ProBio: A Biomarker Driven Study in Patients with Metastatic Castrate Resistant Prostate Cancer. Case Med Res. 2019. [CrossRef]
- Broeck, T.V.D.; Moris, L.; Gevaert, T.; Tosco, L.; Smeets, E.; Fishbane, N.; Liu, Y.; Helsen, C.; Margrave, J.; Buerki, C.; et al. Validation of the Decipher Test for Predicting Distant Metastatic Recurrence in Men with High-risk Nonmetastatic Prostate Cancer 10 Years After Surgery. Eur. Urol. Oncol. 2019, 2, 589–596. [Google Scholar] [CrossRef]
- Carm, K.T.; Hoff, A.M.; Bakken, A.C.; Axcrona, U.; Axcrona, K.; Lothe, R.A.; Skotheim, R.I.; Løvf, M. Interfocal heterogeneity challenges the clinical usefulness of molecular classification of primary prostate cancer. Sci. Rep. 2019, 9, 13579. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).