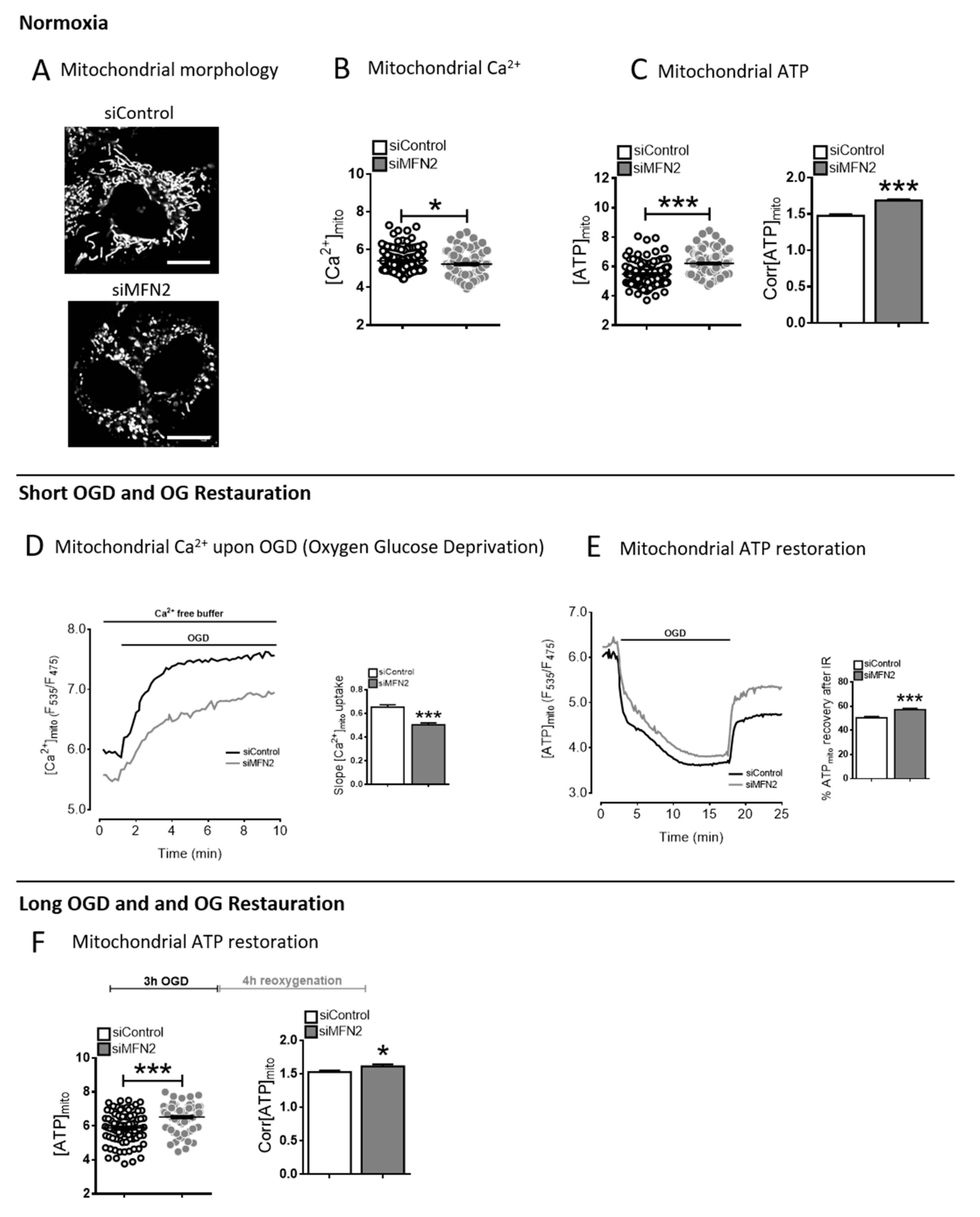
Figure 1.
The Calcium/ATP paradox in mitofusin 2 (MFN2)-KD cells. (
A) Confocal images of H9C2-sv40 cells expressing the mitochondrial Ateam sensor in siControl and MFN2-KD conditions (Scale bars, 10 μm). (
B) Dot plot of steady-state mitochondrial Ca
2+ level in siControl (black) and siMFN2 (grey) H9C2-sv40 cells (F
535/F
475).
n = 125, 116 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. *
p < 0.05. (
C) (left inset) Dot plot of steady-state mitochondrial ATP level in control (black) and MFN2-KD (grey) H9C2-sv40 cells (F
535/F
475). (right inset) Corrected steady-state mitochondrial ATP level (Corr[ATP]
mito) calculated as ratio of Ateam values before/after 2-Deoxyglucose and oligomycin A for each cell (see
Figure S2).
n = 126, 139 cells respectively. Data shown represent the mean ± SEM of 4 independent experiments. (
D) Mitochondrial calcium measurement during oxygen glucose deprivation (OGD) in siControl (black) and siMFN2 (grey) H9C2-sv40 cells. Maximal slope analysis of mitochondrial Ca
2+ entry upon OGD in H9C2-sv40 cells.
n = 125, 116 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. ***
p < 0.001. These experiments have been performed in absence of external calcium. (
E) Measurement of the mitochondrial ATP level during a 15 min-oxygen glucose deprivation (OGD) and a 5 min-reoxygenation with glucose (reox + Glucose), in siControl (black) and siMFN2 (grey) H9C2-sv40 cells. Inset: Percentage of mitochondrial ATP recovery calculated as Ateam ratioat reperfusion divided by its steady-state value in siControl (white) and siMFN2 (grey) H9C2-sv40 cells.
n = 85, 164 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. (
F) Measurements of steady-state mitochondrial ATP level after a 3 h-OGD and a 4 h-reoxygenation with glucose (reox + Glucose). (left inset) Dot-plot shows the steady-state mitochondrial ATP level ([ATP]
mito) in each cell (F
535/F
475). (right inset) Corrected steady-state mitochondrial ATP level (Corr[ATP]
mito).
n = 75 cells for siControl and siMFN2 H9C2-sv40 cells. Data shown represent the mean ± SEM of 3 independent experiments, *
p < 0.05; ***
p < 0.001.
Figure 1.
The Calcium/ATP paradox in mitofusin 2 (MFN2)-KD cells. (
A) Confocal images of H9C2-sv40 cells expressing the mitochondrial Ateam sensor in siControl and MFN2-KD conditions (Scale bars, 10 μm). (
B) Dot plot of steady-state mitochondrial Ca
2+ level in siControl (black) and siMFN2 (grey) H9C2-sv40 cells (F
535/F
475).
n = 125, 116 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. *
p < 0.05. (
C) (left inset) Dot plot of steady-state mitochondrial ATP level in control (black) and MFN2-KD (grey) H9C2-sv40 cells (F
535/F
475). (right inset) Corrected steady-state mitochondrial ATP level (Corr[ATP]
mito) calculated as ratio of Ateam values before/after 2-Deoxyglucose and oligomycin A for each cell (see
Figure S2).
n = 126, 139 cells respectively. Data shown represent the mean ± SEM of 4 independent experiments. (
D) Mitochondrial calcium measurement during oxygen glucose deprivation (OGD) in siControl (black) and siMFN2 (grey) H9C2-sv40 cells. Maximal slope analysis of mitochondrial Ca
2+ entry upon OGD in H9C2-sv40 cells.
n = 125, 116 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. ***
p < 0.001. These experiments have been performed in absence of external calcium. (
E) Measurement of the mitochondrial ATP level during a 15 min-oxygen glucose deprivation (OGD) and a 5 min-reoxygenation with glucose (reox + Glucose), in siControl (black) and siMFN2 (grey) H9C2-sv40 cells. Inset: Percentage of mitochondrial ATP recovery calculated as Ateam ratioat reperfusion divided by its steady-state value in siControl (white) and siMFN2 (grey) H9C2-sv40 cells.
n = 85, 164 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 4 independent experiments. (
F) Measurements of steady-state mitochondrial ATP level after a 3 h-OGD and a 4 h-reoxygenation with glucose (reox + Glucose). (left inset) Dot-plot shows the steady-state mitochondrial ATP level ([ATP]
mito) in each cell (F
535/F
475). (right inset) Corrected steady-state mitochondrial ATP level (Corr[ATP]
mito).
n = 75 cells for siControl and siMFN2 H9C2-sv40 cells. Data shown represent the mean ± SEM of 3 independent experiments, *
p < 0.05; ***
p < 0.001.
![Cells 09 02542 g001]()
Figure 2.
Decreased MFN2 expression induces a glycolytic phenotype. (A) Measurements of mitochondrial ATP level before and after a 1 µM rotenone treatment was performed in order to figure out the proportion of complex I-dependent ATP production. n = 40, 46 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments (See method details for the formula). (B) same as (A) with a 500 nM antimycin A treatment to calculate the proportion of complex III-dependent ATP level in the mitochondria. n = 20, 21 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. (C) NADH/NAD+ ratio measurements obtained in control (black) and MFN2-KD (grey) H9C2-sv40 cells. A 10mM Pyruvate treatment, in absence of glucose, is achieved to determine the minimal NADH/NAD+ ratio value corresponding to the NADH insensitivity threshold of the probe (free NADH is consumed by the lactate dehydrogenase). (inset) Value represent mean ± SEM variation of NADH/NAD+ ratio between time 0 and post-pyruvate treatment (Normalized NADH/NAD+ ratio) obtained from 4 independent experiments. n = 81, 115 cells for siControl and siMFN2 H9C2-sv40 cells, respectively, ** p < 0.01, *** p < 0.001.
Figure 2.
Decreased MFN2 expression induces a glycolytic phenotype. (A) Measurements of mitochondrial ATP level before and after a 1 µM rotenone treatment was performed in order to figure out the proportion of complex I-dependent ATP production. n = 40, 46 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments (See method details for the formula). (B) same as (A) with a 500 nM antimycin A treatment to calculate the proportion of complex III-dependent ATP level in the mitochondria. n = 20, 21 cells for siControl and siMFN2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. (C) NADH/NAD+ ratio measurements obtained in control (black) and MFN2-KD (grey) H9C2-sv40 cells. A 10mM Pyruvate treatment, in absence of glucose, is achieved to determine the minimal NADH/NAD+ ratio value corresponding to the NADH insensitivity threshold of the probe (free NADH is consumed by the lactate dehydrogenase). (inset) Value represent mean ± SEM variation of NADH/NAD+ ratio between time 0 and post-pyruvate treatment (Normalized NADH/NAD+ ratio) obtained from 4 independent experiments. n = 81, 115 cells for siControl and siMFN2 H9C2-sv40 cells, respectively, ** p < 0.01, *** p < 0.001.
![Cells 09 02542 g002]()
Figure 3.
MFN2 loss of function induces an ANT2-dependent ATP import into mitochondria. (A) Estimation of the proportion of mitochondrial ATP originated from the cytosolic anaerobic glycolysis determined by mitochondrial ATP measurements in basal condition and after incubation with oxamate (20 mM) in control (white), siANT2 (stripped) MFN2-KD (grey) and siMFN2 + siANT2 (dashed) H9C2-sv40 cells. n = 125, 113, 122, 103, 106, 97, 104, 117 cells for siControl, siMFN2 and siMFN2 + siANT2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. (B) Estimation of the proportion of cytosolic ATP originated from the cytosolic anaerobic glycolysis determined by cytosolic ATP measurements in basal condition and after incubation with oxamate (20 mM) in the cytosol of control (white), siANT2 (stripped), MFN2-KD (grey) and siMFN2 + siANT2 (dashed). n = 150, 195, 157, 121, 154, 131, 188, 134 cells for siControl, siMFN2 and siMFN2 + siANT2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. ** p < 0.01; *** p < 0.001. (C) Western-blot showing ANT1 protein level in siControl (white) and siMFN2 (grey) H9C2-sv40 cells. ANT1 expression was normalized by the one of Tom20. *** p < 0.001 (see S4A for blot). (D) Western-blot of ANT2 protein normalized to Tom20 protein in siControl (white) and siMFN2 (grey) H9C2-sv40 cells. Data shown represent the mean ± SEM of 3 independent experiments (see S4B for blot). (E) Measurement of steady-state mitochondrial ATP in siControl, siANT2, siMFN2 and siMFN2 + siANT2 transfected cells. n = 226, 226, 188, and 217. Data shown represent the mean ± SEM of 3 independent experiments (F) Decay of the mitochondrial ATP content was measured by mean of mito-Ateam during oxygen glucose deprivation (OGD), in siControl (black), siANT2 (dotted), siMFN2 (grey) and siMFN2 + siANT2 (dashed) H9C2-sv40 cells. (inset) Mean lifetime of an exponential decay fit for mitochondrial ATP evoked by OGD in siControl, siANT2, siMFN2 and siMFN2 + siANT2. n = 153, 19, 231, and 89 cells. Data shown represent the mean ± SEM of 3–4 independent experiments. ** p < 0.01; *** p < 0.001.
Figure 3.
MFN2 loss of function induces an ANT2-dependent ATP import into mitochondria. (A) Estimation of the proportion of mitochondrial ATP originated from the cytosolic anaerobic glycolysis determined by mitochondrial ATP measurements in basal condition and after incubation with oxamate (20 mM) in control (white), siANT2 (stripped) MFN2-KD (grey) and siMFN2 + siANT2 (dashed) H9C2-sv40 cells. n = 125, 113, 122, 103, 106, 97, 104, 117 cells for siControl, siMFN2 and siMFN2 + siANT2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. (B) Estimation of the proportion of cytosolic ATP originated from the cytosolic anaerobic glycolysis determined by cytosolic ATP measurements in basal condition and after incubation with oxamate (20 mM) in the cytosol of control (white), siANT2 (stripped), MFN2-KD (grey) and siMFN2 + siANT2 (dashed). n = 150, 195, 157, 121, 154, 131, 188, 134 cells for siControl, siMFN2 and siMFN2 + siANT2 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments. ** p < 0.01; *** p < 0.001. (C) Western-blot showing ANT1 protein level in siControl (white) and siMFN2 (grey) H9C2-sv40 cells. ANT1 expression was normalized by the one of Tom20. *** p < 0.001 (see S4A for blot). (D) Western-blot of ANT2 protein normalized to Tom20 protein in siControl (white) and siMFN2 (grey) H9C2-sv40 cells. Data shown represent the mean ± SEM of 3 independent experiments (see S4B for blot). (E) Measurement of steady-state mitochondrial ATP in siControl, siANT2, siMFN2 and siMFN2 + siANT2 transfected cells. n = 226, 226, 188, and 217. Data shown represent the mean ± SEM of 3 independent experiments (F) Decay of the mitochondrial ATP content was measured by mean of mito-Ateam during oxygen glucose deprivation (OGD), in siControl (black), siANT2 (dotted), siMFN2 (grey) and siMFN2 + siANT2 (dashed) H9C2-sv40 cells. (inset) Mean lifetime of an exponential decay fit for mitochondrial ATP evoked by OGD in siControl, siANT2, siMFN2 and siMFN2 + siANT2. n = 153, 19, 231, and 89 cells. Data shown represent the mean ± SEM of 3–4 independent experiments. ** p < 0.01; *** p < 0.001.
![Cells 09 02542 g003]()
Figure 4.
MFN2 loss upregulates ATPase inhibitory factor 1. (
A) Estimation of F
1F
0 ATP hydrolase activity calculated as the difference in mitochondrial ATP decay during OGD with or without the F
1F
0 ATP hydrolase inhibitor BMS-199264 (10 µM) in siControl, siANT2, siMFN2, and siMFN2+siANT2 transfected cells.
n = 48, 20, 62, and 48 cells. Mean lifetime of an exponential decay fit is used to calculate the mitochondrial ATP decay during OGD. δ1 = Mean lifetime in control condition and δ2 = Mean lifetime in presence of BMS. Data shown represent the mean ± SEM of 4 independent experiments. (
B) Histogram showing the contribution of other ATP-catabolic processes during OGD in presence of F
1F
0ATP hydrolase inhibitor BMS-199264in H9C2-sv40 cells. Activity of these other ATP catabolic processes was quantified as the averaged mitochondrial ATP decay figured out by the mean lifetime of an exponential decay fitting the Ateam sensor fluorescence ratio over time.
n = 65 (control), 73 (siMFN2), 48 (siMFN2 + siANT2) and 20 (siANT2) cells. Data shown represent the mean ± SEM of 3–4 independent experiments *
p < 0.05. (
C) Western-blot (WB) of ATP5a and IF1 protein normalized to Tubulin protein in siControl (white), siMFN2 (grey) and siMFN2+siANT2 (grey dashed line) in H9C2-sv40 cells. (
D) Fold change of ATP5a WB density normalized to Tubulin protein represents the variation in ATP5a protein expression. (
E) Fold change of IF1 WB density normalized to ATP5a protein represents the variation in negative regulatory IF-1 protein expression as compared to F
1F
0ATP synthase/hydrolase activity. Data shown represent the mean ± SEM of 4 independent experiments. *
p < 0.05. (
F) ATP hydrolase activity (calculated as explain in the
Figure 4A) in siControl, siIF1, siMFN2, siMFN2 + siIF1, and siIF1 transfected cells.
n = 48, 30, 62, and 48 cells. δ1 = Mean lifetime in control condition and δ2 = Mean lifetime in presence of BMS. Data shown represent the mean ± SEM of 4 independent experiments. (
G) Estimation of the proportion of mitochondrial ATP originated from the cytosolic anaerobic glycolysis. Mitochondrial ATP measurements were achieved with mito-Ateam sensor expressed in basal condition and after incubation with oxamate (20 mM) in control, IF1, MFN2-KD and siMFN2 + siIF1 transfected H9C2-sv40 cells.
n = 125, 113, 100, 106, 106, 97, 96, 84 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments *
p < 0.05, **
p < 0.01; ***
p < 0.001.
Figure 4.
MFN2 loss upregulates ATPase inhibitory factor 1. (
A) Estimation of F
1F
0 ATP hydrolase activity calculated as the difference in mitochondrial ATP decay during OGD with or without the F
1F
0 ATP hydrolase inhibitor BMS-199264 (10 µM) in siControl, siANT2, siMFN2, and siMFN2+siANT2 transfected cells.
n = 48, 20, 62, and 48 cells. Mean lifetime of an exponential decay fit is used to calculate the mitochondrial ATP decay during OGD. δ1 = Mean lifetime in control condition and δ2 = Mean lifetime in presence of BMS. Data shown represent the mean ± SEM of 4 independent experiments. (
B) Histogram showing the contribution of other ATP-catabolic processes during OGD in presence of F
1F
0ATP hydrolase inhibitor BMS-199264in H9C2-sv40 cells. Activity of these other ATP catabolic processes was quantified as the averaged mitochondrial ATP decay figured out by the mean lifetime of an exponential decay fitting the Ateam sensor fluorescence ratio over time.
n = 65 (control), 73 (siMFN2), 48 (siMFN2 + siANT2) and 20 (siANT2) cells. Data shown represent the mean ± SEM of 3–4 independent experiments *
p < 0.05. (
C) Western-blot (WB) of ATP5a and IF1 protein normalized to Tubulin protein in siControl (white), siMFN2 (grey) and siMFN2+siANT2 (grey dashed line) in H9C2-sv40 cells. (
D) Fold change of ATP5a WB density normalized to Tubulin protein represents the variation in ATP5a protein expression. (
E) Fold change of IF1 WB density normalized to ATP5a protein represents the variation in negative regulatory IF-1 protein expression as compared to F
1F
0ATP synthase/hydrolase activity. Data shown represent the mean ± SEM of 4 independent experiments. *
p < 0.05. (
F) ATP hydrolase activity (calculated as explain in the
Figure 4A) in siControl, siIF1, siMFN2, siMFN2 + siIF1, and siIF1 transfected cells.
n = 48, 30, 62, and 48 cells. δ1 = Mean lifetime in control condition and δ2 = Mean lifetime in presence of BMS. Data shown represent the mean ± SEM of 4 independent experiments. (
G) Estimation of the proportion of mitochondrial ATP originated from the cytosolic anaerobic glycolysis. Mitochondrial ATP measurements were achieved with mito-Ateam sensor expressed in basal condition and after incubation with oxamate (20 mM) in control, IF1, MFN2-KD and siMFN2 + siIF1 transfected H9C2-sv40 cells.
n = 125, 113, 100, 106, 106, 97, 96, 84 cells, respectively. Data shown represent the mean ± SEM of 3 independent experiments *
p < 0.05, **
p < 0.01; ***
p < 0.001.
![Cells 09 02542 g004]()
Figure 5.
MFN2 loss upregulates ATPase inhibitory factor 1 preserving mitochondrial membrane potential. (A) Wide-field images of siControl H9C2-sv40 cells loaded with TMRM dye before and after a 10 min OGD (Scale bars, 20 μm). (B) Wide-field images of MFN2-KD H9C2-sv40 cells loaded with TMRM dye before and after an 10 min OGD (Scale bars, 20 μm). (C) Dissipation of mitochondrial membrane potential, Δψm, during OGD as measured by the variation in TMRM fluorescence in mitochondria of siControl (black), siMFN2 (grey), siMFN2 + siANT2 (dashed) and siMFN2 + siIF1 (dotted) transfected H9C2-sv40 cells (representative curves). (D) Δψm is characterized by the mean lifetime of an exponential decay fit on TMRM fluorescent time-lapse recordings. n = 276 (siControl), 383 (siANT2), 352 (siIF1), 289 (siMFN2), 192 (siMFN2 + siANT2), and 184 (siMFN2 + siIF1) cells. Mean lifetime of each condition has been normalized with the control’s mean lifetime of the experiment day. Data shown represent the mean ± SEM of 6 independent experiments. (E) Restoration of Δψm upon 60 min-reoxygenation with glucose after a 40 min-OGD as measured by the variation in TMRM fluorescence in mitochondria of H9C2-sv40 cells. (F) Values show mean ± SEMΔψm recovery after OGD and reoxygenation (reox + Glucose). (Δ2/Δ1) in4 independent experiments. H9C2-sv40 cells. n = 232 (siControl), 172 (siANT2), 284 (siIF1), 221 (siMFN2), 242 (siMFN2 + siANT2), 152 (siMFN2 + siIF1) cells. Δψm recovery of each condition has been normalized with the control’s Δψm recovery of the experiment day. *** p < 0.001.
Figure 5.
MFN2 loss upregulates ATPase inhibitory factor 1 preserving mitochondrial membrane potential. (A) Wide-field images of siControl H9C2-sv40 cells loaded with TMRM dye before and after a 10 min OGD (Scale bars, 20 μm). (B) Wide-field images of MFN2-KD H9C2-sv40 cells loaded with TMRM dye before and after an 10 min OGD (Scale bars, 20 μm). (C) Dissipation of mitochondrial membrane potential, Δψm, during OGD as measured by the variation in TMRM fluorescence in mitochondria of siControl (black), siMFN2 (grey), siMFN2 + siANT2 (dashed) and siMFN2 + siIF1 (dotted) transfected H9C2-sv40 cells (representative curves). (D) Δψm is characterized by the mean lifetime of an exponential decay fit on TMRM fluorescent time-lapse recordings. n = 276 (siControl), 383 (siANT2), 352 (siIF1), 289 (siMFN2), 192 (siMFN2 + siANT2), and 184 (siMFN2 + siIF1) cells. Mean lifetime of each condition has been normalized with the control’s mean lifetime of the experiment day. Data shown represent the mean ± SEM of 6 independent experiments. (E) Restoration of Δψm upon 60 min-reoxygenation with glucose after a 40 min-OGD as measured by the variation in TMRM fluorescence in mitochondria of H9C2-sv40 cells. (F) Values show mean ± SEMΔψm recovery after OGD and reoxygenation (reox + Glucose). (Δ2/Δ1) in4 independent experiments. H9C2-sv40 cells. n = 232 (siControl), 172 (siANT2), 284 (siIF1), 221 (siMFN2), 242 (siMFN2 + siANT2), 152 (siMFN2 + siIF1) cells. Δψm recovery of each condition has been normalized with the control’s Δψm recovery of the experiment day. *** p < 0.001.
![Cells 09 02542 g005]()
Figure 6.
ANT2 and inhibitory factor 1 (IF1) mediate the modulation of mitochondria bioenergetics. Scheme summarizing the organization of cytosolic ATP import in mitochondria and IF-1 activity on the biochemical reaction of ATP hydrolysis and maintenance of Δψm in mitochondria of MFN2-KD cells. In the inset, curves represent a rough estimate of the relation between inner mitochondrial membrane (IMM) potential (ψm) and ATP concentration in mitochondria ([ATP]mito) in 3 cell populations transfected either with siCTL, siMFN2, or siANT2. This graph schematizes the effects of MFN2 KD shifting mitochondrial ATP concentration towards higher values and decreasing, in parallel, the maximal consumption rate of F1F0 ATP hydrolase via an increase in IF-1 expression. Cells knocked down for ANT2 show impaired ATP import which result in a shift of ATP concentration in mitochondria towards lower values.
Figure 6.
ANT2 and inhibitory factor 1 (IF1) mediate the modulation of mitochondria bioenergetics. Scheme summarizing the organization of cytosolic ATP import in mitochondria and IF-1 activity on the biochemical reaction of ATP hydrolysis and maintenance of Δψm in mitochondria of MFN2-KD cells. In the inset, curves represent a rough estimate of the relation between inner mitochondrial membrane (IMM) potential (ψm) and ATP concentration in mitochondria ([ATP]mito) in 3 cell populations transfected either with siCTL, siMFN2, or siANT2. This graph schematizes the effects of MFN2 KD shifting mitochondrial ATP concentration towards higher values and decreasing, in parallel, the maximal consumption rate of F1F0 ATP hydrolase via an increase in IF-1 expression. Cells knocked down for ANT2 show impaired ATP import which result in a shift of ATP concentration in mitochondria towards lower values.
Figure 7.
MFN2 loss triggers a metabolic reprogramming which confers resistance to hypoxic injury. (A) Calcein/cobalt experiment assesses the kinetic of PTP opening after treatment with ionomycin in siControl, siANT2, siIF1, siMFN2, and siMFN2 + siANT2 H9C2-sv40 cells. (left inset) Decay time of the calcein fluorescence decreases upon ionomycin treatment (right inset) n = 756, 110, 362,219, 282 and 365cells for siControl, siANT2, siIF1, siMFN2, siMFN2 + siANT2, and siMFN2 + siIF1 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 5 independent experiments. (B) Percentage of propidium iodide positive cells measured by FACS after 4 h OGD and 2 h reoxygenation in siControl, siANT2, siIF1, siMFN2, siMFN2 + siANT2, and siMFN2 + siIF1 H9C2-sv40 cells. Data shown represent the mean ± SEM of 9 independent experiments * p < 0.05; *** p < 0.001. (C) Principal component analysis shows the first and second principle components of the multi-variate scattering of 4 experimental conditions: Control (siCTL), MFN2 KD cells (siMFN2), ANT2 KD cells (siA) and MFN2/ANT2 KD cells (siAM) by the variance of mean values of 10 experimental variables: Steady-state cytosolic ATP concentration ([ATP]c), steady-state mitochondrial ATP concentration ([ATP]m), anaerobic glycolysis-dependent ATP level in cytosol ([ATP]c-LDH), mitochondrial ATP originated from cytosolic anaerobic glycolysis ([ATP]m-LDH), mPTP opening, cell death, ATP hydrolase activity during OGD (F1F0 ATP hydrolase), lifetime of the drop in mitochondrial ATP concentration during OGD ([ATP]m decay), lifetime of the drop in IMM potential during OGD (ψm decay) and the recovery of IMM potential after OGD-reoxygenation (ψm recovery). This shows that, regarding the 10 experimental variables, the phenotypes of cells knock-down for both ANT2 and MFN2 are similar to control cells. Conversely to control cells, single knocked-down cells either for MFN2 or for ANT2 display a dispersion along PC1 and PC2, respectively, demonstrating that they do not share an identical phenotype.
Figure 7.
MFN2 loss triggers a metabolic reprogramming which confers resistance to hypoxic injury. (A) Calcein/cobalt experiment assesses the kinetic of PTP opening after treatment with ionomycin in siControl, siANT2, siIF1, siMFN2, and siMFN2 + siANT2 H9C2-sv40 cells. (left inset) Decay time of the calcein fluorescence decreases upon ionomycin treatment (right inset) n = 756, 110, 362,219, 282 and 365cells for siControl, siANT2, siIF1, siMFN2, siMFN2 + siANT2, and siMFN2 + siIF1 H9C2-sv40 cells, respectively. Data shown represent the mean ± SEM of 5 independent experiments. (B) Percentage of propidium iodide positive cells measured by FACS after 4 h OGD and 2 h reoxygenation in siControl, siANT2, siIF1, siMFN2, siMFN2 + siANT2, and siMFN2 + siIF1 H9C2-sv40 cells. Data shown represent the mean ± SEM of 9 independent experiments * p < 0.05; *** p < 0.001. (C) Principal component analysis shows the first and second principle components of the multi-variate scattering of 4 experimental conditions: Control (siCTL), MFN2 KD cells (siMFN2), ANT2 KD cells (siA) and MFN2/ANT2 KD cells (siAM) by the variance of mean values of 10 experimental variables: Steady-state cytosolic ATP concentration ([ATP]c), steady-state mitochondrial ATP concentration ([ATP]m), anaerobic glycolysis-dependent ATP level in cytosol ([ATP]c-LDH), mitochondrial ATP originated from cytosolic anaerobic glycolysis ([ATP]m-LDH), mPTP opening, cell death, ATP hydrolase activity during OGD (F1F0 ATP hydrolase), lifetime of the drop in mitochondrial ATP concentration during OGD ([ATP]m decay), lifetime of the drop in IMM potential during OGD (ψm decay) and the recovery of IMM potential after OGD-reoxygenation (ψm recovery). This shows that, regarding the 10 experimental variables, the phenotypes of cells knock-down for both ANT2 and MFN2 are similar to control cells. Conversely to control cells, single knocked-down cells either for MFN2 or for ANT2 display a dispersion along PC1 and PC2, respectively, demonstrating that they do not share an identical phenotype.
![Cells 09 02542 g007]()
Figure 8.
ANT2-mediated ATP import into mitochondria protects against cardiac hypoxia lethal injury. Ischemia-reperfusion injury triggers an endoplasmic reticulum (ER) Ca2+ transfer to mitochondria (Mito), leading to membrane potential imbalance and opening of the permeability transition pore (PTP). Loss of MFN2 disrupts ER-mitochondria contact sites, lessened ER-to-mitochondria Ca2+ transfer and caused a metabolic reprogramming (Warburg shift) of the cell bioenergetics. The increased import of cytosolic ATP into mitochondria via ANT2 and the fine-tuning of F1F0ATP hydrolase by IF1 overexpression allowed a better tolerance to hypoxia by mean of the prevention of the collapse of the mitochondrial membrane potential associated to a delayed opening of the permeability transition pore and a higher steady-state ATP content during hypoxia associated with a greater cell survival.
Figure 8.
ANT2-mediated ATP import into mitochondria protects against cardiac hypoxia lethal injury. Ischemia-reperfusion injury triggers an endoplasmic reticulum (ER) Ca2+ transfer to mitochondria (Mito), leading to membrane potential imbalance and opening of the permeability transition pore (PTP). Loss of MFN2 disrupts ER-mitochondria contact sites, lessened ER-to-mitochondria Ca2+ transfer and caused a metabolic reprogramming (Warburg shift) of the cell bioenergetics. The increased import of cytosolic ATP into mitochondria via ANT2 and the fine-tuning of F1F0ATP hydrolase by IF1 overexpression allowed a better tolerance to hypoxia by mean of the prevention of the collapse of the mitochondrial membrane potential associated to a delayed opening of the permeability transition pore and a higher steady-state ATP content during hypoxia associated with a greater cell survival.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}