Adaptive Immunity and Pathogenesis of Diabetes: Insights Provided by the α4–Integrin Deficient NOD Mouse

 , ,
, , 
Abstract
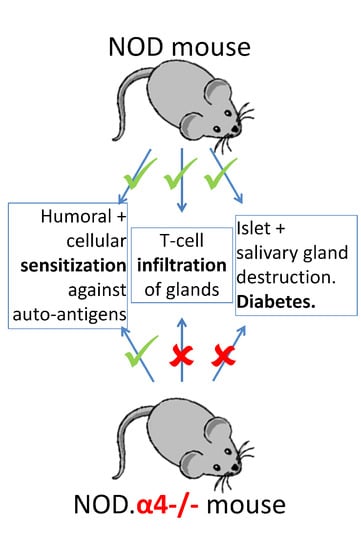
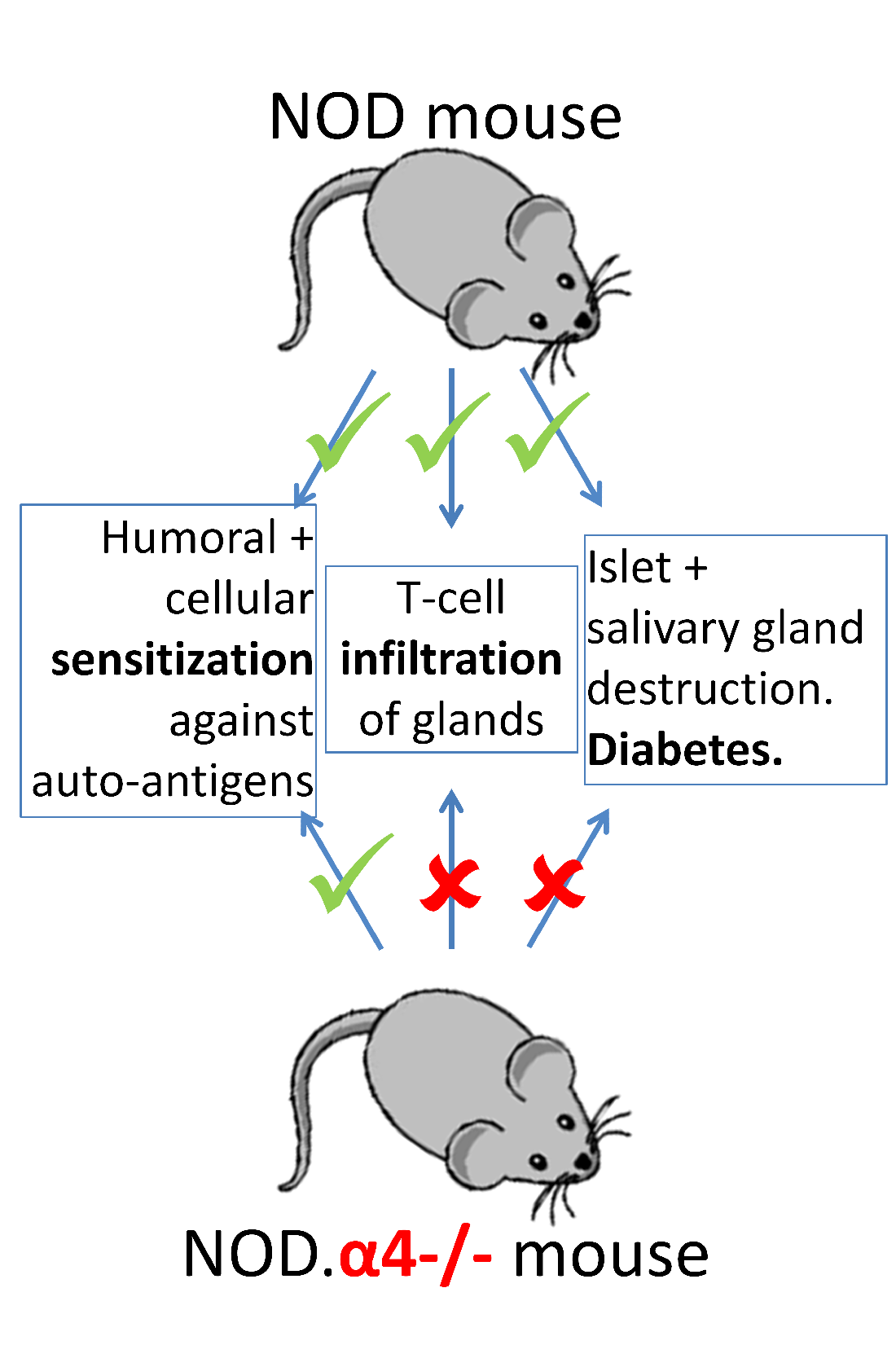{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Generation of NOD.α4-/- Mice
3.2. NOD.α4-/- Mice Are Protected from Autoimmune Diabetes
3.3. Adaptive Cellular and Humoral Immune Responses of NOD.α4-/- Mice against Islet Cell Antigens
3.4. Adoptive Transfer of a Mix of Diabetogenic α4+ Helper and Cytotoxic T-Cells, but Not α4+ Helper T-Cells Alone, Induce Diabetes in NOD.α4-/- Mice
3.5. α4 Ablation Protects Pre-Diabetic NOD Mice from Diabetes
3.6. Pre-Diabetic NOD Mice Have Normal Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gale, E.A. The rise of childhood type 1 diabetes in the 20th century. Diabetes 2002, 51, 3353–3361. [Google Scholar] [CrossRef]
- Patterson, C.C.; Dahlquist, G.G.; Gyurus, E.; Green, A.; Soltesz, G. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: A multicentre prospective registration study. Lancet 2009, 373, 2027–2033. [Google Scholar] [CrossRef]
- Steffes, M.W.; Sibley, S.; Jackson, M.; Thomas, W. Beta-cell function and the development of diabetes-related complications in the diabetes control and complications trial. Diabetes Care 2003, 26, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Streisand, R.; Monaghan, M. Young children with type 1 diabetes: Challenges, research, and future directions. Curr. Diab. Rep. 2014, 14, 520. [Google Scholar] [CrossRef] [PubMed]
- Keenan, H.A.; Sun, J.K.; Levine, J.; Doria, A.; Aiello, L.P.; Eisenbarth, G.; Bonner-Weir, S.; King, G.L. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010, 59, 2846–2853. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Smukler, S.R.; Arntfield, M.E.; Razavi, R.; Bikopoulos, G.; Karpowicz, P.; Seaberg, R.; Dai, F.; Lee, S.; Ahrens, R.; Fraser, P.E.; et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell 2011, 8, 281–293. [Google Scholar] [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef]
- Shoda, L.K.; Young, D.L.; Ramanujan, S.; Whiting, C.C.; Atkinson, M.A.; Bluestone, J.A.; Eisenbarth, G.S.; Mathis, D.; Rossini, A.A.; Campbell, S.E.; et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity 2005, 23, 115–126. [Google Scholar] [CrossRef]
- Bonifacio, E.; Ziegler, A.G. Advances in the prediction and natural history of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 513–525. [Google Scholar] [CrossRef]
- Podolsky, D.K.; Lobb, R.; King, N.; Benjamin, C.D.; Pepinsky, B.; Sehgal, P.; Debeaumont, M. Attenuation of colitis in the cotton-top tamarin by anti-alpha 4 integrin monoclonal antibody. J. Clin. Investig. 1993, 92, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, E.R.; Jiang, Y.; Henderson, W.R., Jr.; Latchman, Y.; Papayannopoulou, T. Absence of alpha 4 but not beta 2 integrins restrains development of chronic allergic asthma using mouse genetic models. Exp. Hematol. 2009, 37, 715–727. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mulligan, M.S.; Johnson, K.J.; Todd, R.F.; Issekutz, T.B.; Miyasaka, M.; Tamatani, T.; Smith, C.W.; Anderson, D.C.; Ward, A.P. Requirements for leukocyte adhesion molecules in nephrotoxic nephritis. J. Clin. Investig. 1993, 91, 577–587. [Google Scholar] [CrossRef]
- Baron, J.L.; Reich, E.P.; Visintin, I.; Janeway, C.A., Jr. The pathogenesis of adoptive murine autoimmune diabetes requires an interaction between alpha 4-integrins and vascular cell adhesion molecule-1. J. Clin. Investig. 1994, 93, 1700–1708. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.B.; Colombel, J.-F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.-J.; Danese, S.; et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef]
- Yang, X.D.; Karin, N.; Tisch, R.; Steinman, L.; McDevitt, H.O. Inhibition of insulitis and prevention of diabetes in nonobese diabetic mice by blocking L-selectin and very late antigen 4 adhesion receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 10494–10498. [Google Scholar] [CrossRef]
- Yang, X.D.; Michie, S.A.; Tisch, R.; Karin, N.; Steinman, L.; McDevitt, H.O. A predominant role of integrin alpha 4 in the spontaneous development of autoimmune diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA 1994, 91, 12604–12608. [Google Scholar] [CrossRef]
- Kommajosyula, S.; Reddy, S.; Nitschke, K.; Kanwar, J.R.; Karanam, M.; Krissansen, G.W. Leukocytes infiltrating the pancreatic islets of nonobese diabetic mice are transformed into inactive exiles by combinational anti-cell adhesion therapy. J. Leukoc. Biol. 2001, 70, 510–517. [Google Scholar] [PubMed]
- Yang, X.D.; Michie, S.A.; Tisch, R.; Karin, N.; Steinman, L.; McDevitt, H.O. Cell adhesion molecules: A selective therapeutic target for alleviation of IDDM. J. Autoimmun. 1994, 7, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Sytwu, H.K.; McDevitt, H.O.; Michie, S.A. Involvement of beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in the development of diabetes in obese diabetic mice. Diabetes 1997, 46, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Priestley, G.V.; Papayannopoulou, T. Deletion of alpha4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol. Cell Biol. 2003, 23, 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Koni, P.A.; Joshi, S.K.; Temann, U.A.; Olson, D.; Burkly, L.; Flavell, R.A. Conditional vascular cell adhesion molecule 1 deletion in mice: Impaired lymphocyte migration to bone marrow. J. Exp. Med. 2001, 193, 741–754. [Google Scholar] [CrossRef]
- Chudziak, D.; Spohn, G.; Karpova, D.; Dauber, K.; Wiercinska, E.; Miettinen, J.A.; Papayannopoulou, T.; Bonig, H. Functional consequences of perturbed CXCL12 signal processing: Analyses of immature hematopoiesis in GRK6-deficient mice. Stem Cells Dev. 2015, 24, 737–746. [Google Scholar] [CrossRef]
- Barclay, A.N.; Brown, M.H. Heterogeneity of interactions mediated by membrane glycoproteins of lymphocytes. Biochem. Soc. Trans. 1997, 25, 224–228. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr. The T cell receptor as a multicomponent signalling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annu. Rev. Immunol. 1992, 10, 645–674. [Google Scholar] [CrossRef]
- Walzer, T.; Chiossone, L.; Chaix, J.; Calver, A.R.; Carozzo, C.; Garrigue-Antar, L.; Jacques, Y.; Baratin, M.; Tomasello, E.; Vivier, E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 2007, 8, 1337–1344. [Google Scholar] [CrossRef]
- Chee, J.; Ko, H.-J.; Skowera, A.; Jhala, G.; Catterall, T.; Graham, K.L.; Sutherland, R.M.; Thomas, H.E.; Lew, A.M.; Peakman, M.; et al. Effector-Memory T Cells Develop in Islets and Report Islet Pathology in Type 1 Diabetes. J. Immunol. 2014, 192, 572–580. [Google Scholar] [CrossRef]
- Allman, D.; Lindsley, R.C.; DeMuth, W.; Rudd, K.; Shinton, S.A.; Hardy, R.R. Resolution of Three Nonproliferative Immature Splenic B Cell Subsets Reveals Multiple Selection Points During Peripheral B Cell Maturation. J. Immunol. 2001, 167, 6834–6840. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Carmack, C.E.; Shinton, S.A.; Kemp, J.D.; Hayakawa, K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Immunol. 1991, 189, 3271–3283. [Google Scholar] [CrossRef] [PubMed]
- Li, D.S.; Yuan, Y.H.; Tu, H.J.; Liang, Q.L.; Dai, L.J. A protocol for islet isolation from mouse pancreas. Nat. Protoc. 2009, 4, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.H.; Yurkovetskiy, L.A.; Chervonsky, A.V. Cutting Edge: Commensal Microbiota Has Disparate Effects on Manifestations of Polyglandular Autoimmune Inflammation. J. Immunol. 2016, 197, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Ulyanova, T.; Priestley, G.V.; Banerjee, E.R.; Papayannopoulou, T. Unique and redundant roles of alpha4 and beta2 integrins in kinetics of recruitment of lymphoid vs myeloid cell subsets to the inflamed peritoneum revealed by studies of genetically deficient mice. Exp. Hematol. 2007, 35, 1256–1265. [Google Scholar] [CrossRef]
- Banerjee, E.R.; Jiang, Y.; Henderson, W.R., Jr.; Scott, L.M.; Papayannopoulou, T. Alpha4 and beta2 integrins have nonredundant roles for asthma development, but for optimal allergen sensitization only alpha4 is critical. Exp. Hematol. 2007, 35, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Steptoe, R.J.; JRitchie, M.; Harrison, L.C. Transfer of hematopoietic stem cells encoding autoantigen prevents autoimmune diabetes. J. Clin. Investig. 2003, 111, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef]
- de Goffau, M.C.; Luopajarvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Harkonen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef]
- Ulyanova, T.; Georgolopoulos, G.; Papayannopoulou, T. Reappraising the role of alpha5 integrin and the microenvironmental support in stress erythropoiesis. Exp. Hematol. 2020, 81, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Traub, J.W.; Pellkofer, H.L.; Grondey, K.; Seeger, I.; Rowold, C.; Brück, W.; Husseini, L.; Häusser-Kinzel, S.; Weber, M.S. Natalizumab promotes activation and pro-inflammatory differentiation of peripheral B cells in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 228. [Google Scholar] [CrossRef]
- Wong, F.S.; Janeway, C.A., Jr. The role of CD4 vs. CD8 T cells in IDDM. J. Autoimmun. 1999, 13, 290–295. [Google Scholar] [CrossRef]
- Christianson, S.W.; Shultz, L.D.; Leiter, E.H. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 1993, 42, 44–55. [Google Scholar] [CrossRef]
- Bendelac, A.; Carnaud, C.; Boitard, C.; Bach, J.F. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J. Exp. Med. 1987, 166, 823–832. [Google Scholar] [CrossRef]
- Miller, B.J.; Appel, M.C.; O’Neil, J.J.; Wicker, L.S. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J. Immunol. 1988, 140, 52–58. [Google Scholar] [PubMed]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L.; et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Honkanen, J. Modulation of Type 1 Diabetes Risk by the Intestinal Microbiome. Curr. Diab. Rep. 2017, 17, 105. [Google Scholar] [CrossRef]
- King, C.; Sarvetnick, N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PLoS ONE 2011, 6, e17049. [Google Scholar] [CrossRef]
- Krych, L.; Nielsen, D.S.; Hansen, A.K.; Hansen, C.H. Gut microbial markers are associated with diabetes onset, regulatory imbalance, and IFN-gamma level in NOD mice. Gut Microbes 2015, 6, 101–109. [Google Scholar] [CrossRef]
- Lin, H.; An, Y.; Hao, F.; Wang, Y.; Tang, H. Correlations of Fecal Metabonomic and Microbiomic Changes Induced by High-fat Diet in the Pre-Obesity State. Sci. Rep. 2016, 6, 21618. [Google Scholar] [CrossRef]
- Leiter, E.H. The NOD mouse: A model for analysing the interplay between heredity and environment in development of autoimmune disease. ILAR News 1993, 35, 4. [Google Scholar] [CrossRef]
- Leijon, K.; Hammarstrom, B.; Holmberg, D. Non-obese diabetic (NOD) mice display enhanced immune responses and prolonged survival of lymphoid cells. Int. Immunol. 1994, 6, 339. [Google Scholar] [CrossRef]
- Serreze, D.V.; Leiter, E.H. Defective activation of T suppressor cell function in nonobese diabetic mice. Potential relation to cytokine deficiencies. J. Immunol. 1988, 140, 3801. [Google Scholar]
- Baxter, A.G.; Cooke, A. Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes 1993, 42, 1574. [Google Scholar] [CrossRef]
- Markees, T.G.; Serreze, D.V.; Phillips, N.E.; Sorli, C.H.; Gordon, E.J.; Shultz, L.D.; Noelle, R.J.; Greiner, B.A.W.D.L.; Mordes, J.P.; Rossini, A.A. NOD mice have a generalized defect in their response to transplantation tolerance induction. Diabetes 1999, 48, 967. [Google Scholar] [CrossRef]
- Nakhooda, A.F.; Like, A.A.; Chappel, C.J.; Murray, F.T.; Marliss, E.B. The spontaneously diabetic Wistar rat: Metabolic and morphologic studies. Diabetes 1977, 26, 100. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oulghazi, S.; Wegner, S.K.; Spohn, G.; Müller, N.; Harenkamp, S.; Stenzinger, A.; Papayannopoulou, T.; Bonig, H. Adaptive Immunity and Pathogenesis of Diabetes: Insights Provided by the α4–Integrin Deficient NOD Mouse. Cells 2020, 9, 2597. https://doi.org/10.3390/cells9122597
Oulghazi S, Wegner SK, Spohn G, Müller N, Harenkamp S, Stenzinger A, Papayannopoulou T, Bonig H. Adaptive Immunity and Pathogenesis of Diabetes: Insights Provided by the α4–Integrin Deficient NOD Mouse. Cells. 2020; 9(12):2597. https://doi.org/10.3390/cells9122597
Chicago/Turabian StyleOulghazi, Salim, Sarah K. Wegner, Gabriele Spohn, Nina Müller, Sabine Harenkamp, Albrecht Stenzinger, Thalia Papayannopoulou, and Halvard Bonig. 2020. "Adaptive Immunity and Pathogenesis of Diabetes: Insights Provided by the α4–Integrin Deficient NOD Mouse" Cells 9, no. 12: 2597. https://doi.org/10.3390/cells9122597
APA StyleOulghazi, S., Wegner, S. K., Spohn, G., Müller, N., Harenkamp, S., Stenzinger, A., Papayannopoulou, T., & Bonig, H. (2020). Adaptive Immunity and Pathogenesis of Diabetes: Insights Provided by the α4–Integrin Deficient NOD Mouse. Cells, 9(12), 2597. https://doi.org/10.3390/cells9122597