Regulatory T Cell Stability and Plasticity in Atherosclerosis



Abstract
:1. Introduction
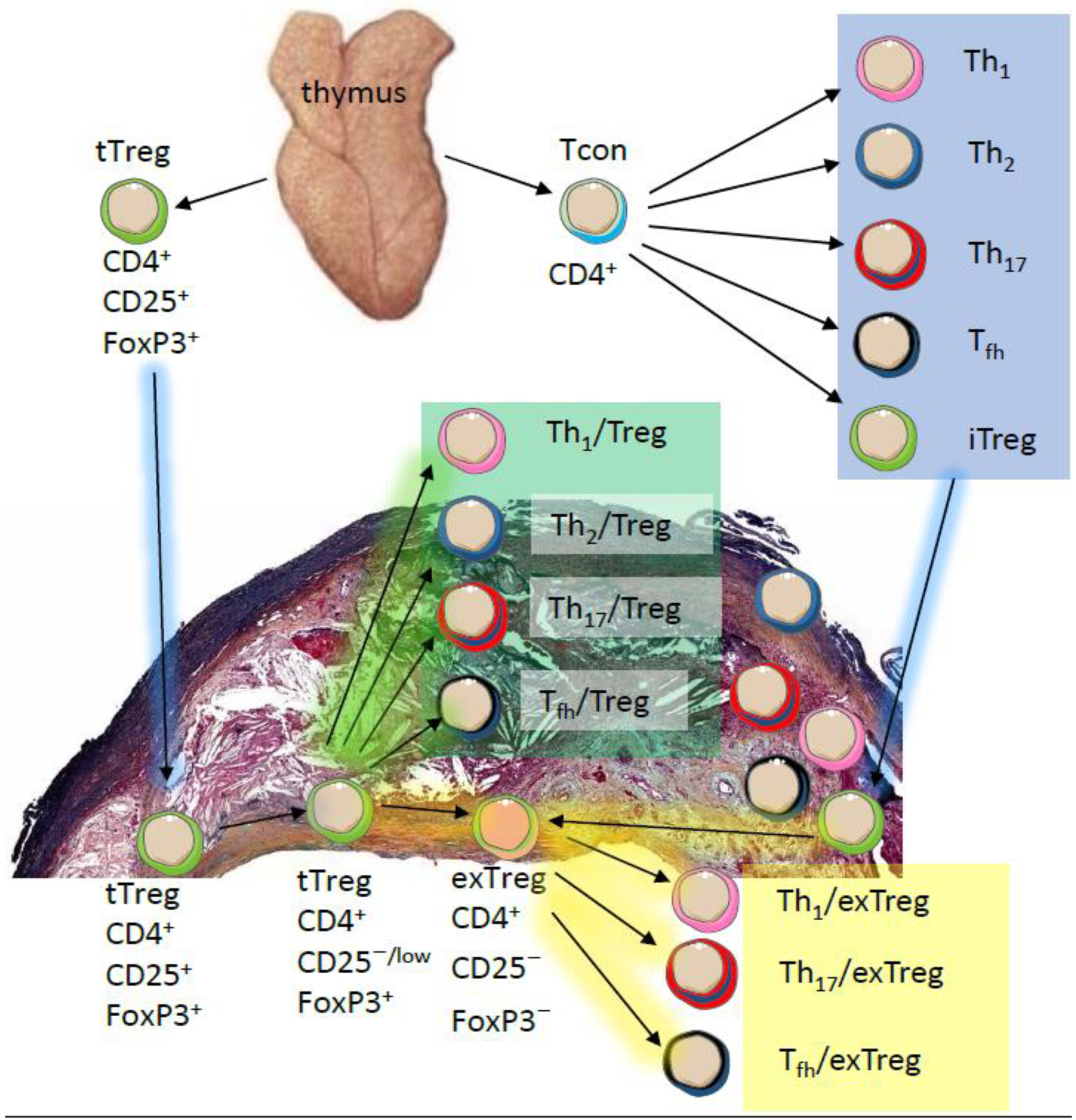
2. Phenotypic and Functional Adaptability of Tregs

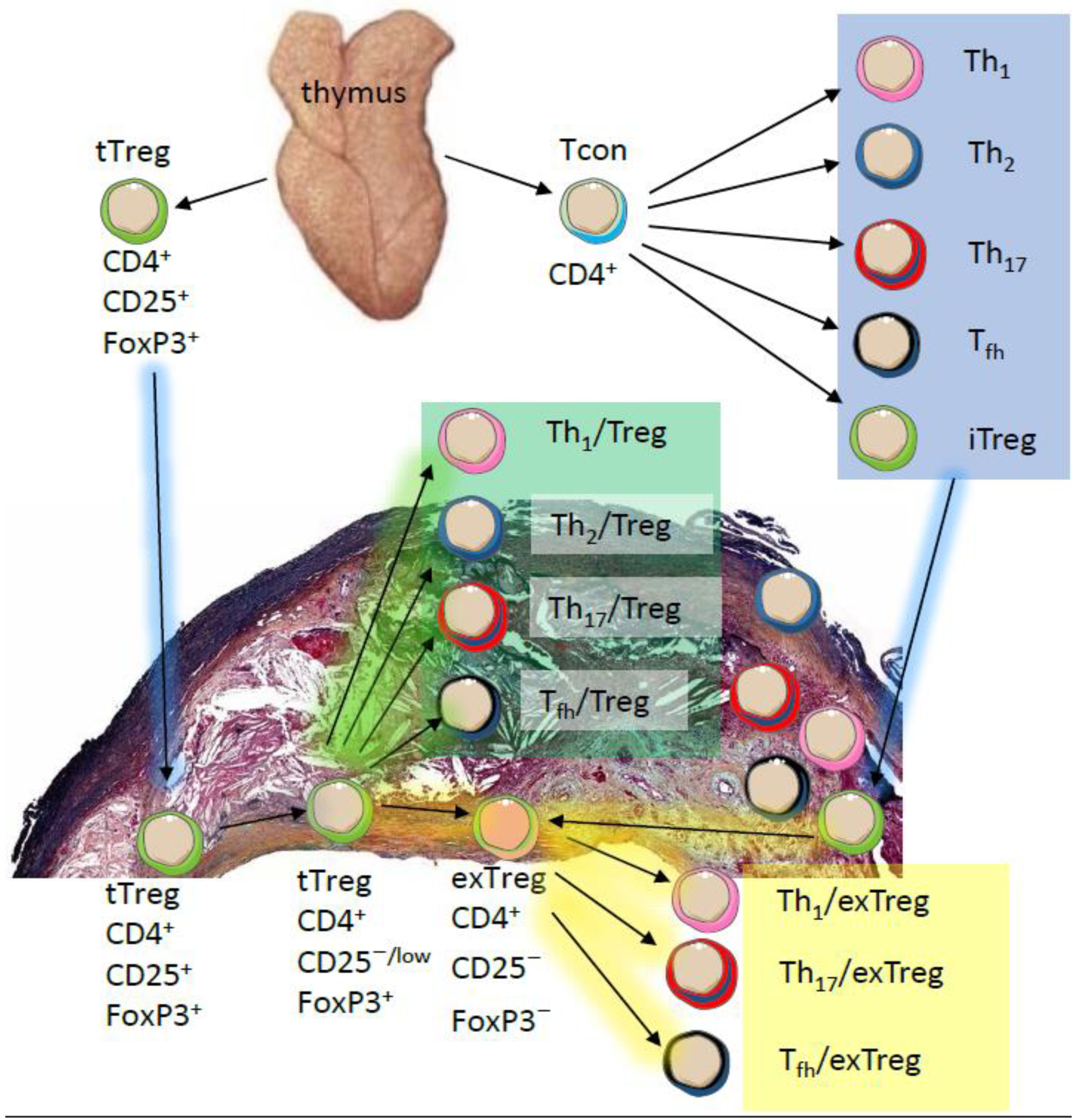{kind=link}
| Foxp3 Allele | IRES for Reporter/Cre | Allele Type | ROSA-Locus | Inducible | Advantages | Limitations | References |
|---|---|---|---|---|---|---|---|
| Foxp3-EGFP-Cre | No | Transgenic (BAC), Recombinase-expressing, Reporter | Rosa26-loxP-Stop-loxP-YFP | No | Natural FoxP3 locus intact | May overestimate the switched population. BAC transgene inactivation in some Foxp3+ cells has been reported. Promoter may be incomplete. Enhancers may be missing. | [40] |
| Foxp3-IRES-GFP-iCre | Yes | Targeted (knock-in), Recombinase-expressing, Reporter | Rosa26-loxP-Stop-loxP-RFP | No | Reporter expressed under the natural FoxP3 promoter with enhancers intact. | May overestimate the switched population. Foxp3 activation can occur early during development in non-Treg cells. In this mouse, Cre activity was reported in CD8 T cells, B cells, and myeloid cells. | [49] |
| Foxp3-IRES-EGFP-Cre-ERT2 | Yes | Targeted (knock-in), Recombinase-expressing, Reporter, Inducible | Rosa26-loxP-Stop-loxP-YFP | Yes, with Tamoxifen | Reporter expressed under the natural FoxP3 promoter with enhancers intact. | May underestimate the switched population. This mouse tracks only the Tregs that were labeled during Tamoxifen injection. If the switched subset developed after tamoxifen injection, it will not be visible. | [44] |
| Foxp3-dCNS1-hCre-2A-eqFP650-2A-Thy1.1 | No | Transgenic (BAC), Recombinase-expressing, Reporter | Rosa26-loxP-Stop-loxP-YFP | No | This mouse detects the fate of tTregs but not pTregs. In YFP+ cells Thy1.1 is lost before Foxp3, making Thy1.1 a marker for cells that are about to lose Foxp3 expression. | In this model, to define exTregs, researchers need to stain for intracellular Foxp3. | [61] |
3. Possible Mechanisms of Treg Instability
4. Treg Adaptability in Atherosclerosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gistera, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. Role of the adaptive immune system in atherosclerosis. Biochem. Soc. Trans. 2020, 48, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Ou, H.X.; Guo, B.B.; Liu, Q.; Li, Y.K.; Yang, Z.; Feng, W.J.; Mo, Z.C. Regulatory T cells as a new therapeutic target for atherosclerosis. Acta Pharm. Sin. 2018, 39, 1249–1258. [Google Scholar] [CrossRef]
- Jordan, M.S.; Boesteanu, A.; Reed, A.J.; Petrone, A.L.; Holenbeck, A.E.; Lerman, M.A.; Naji, A.; Caton, A.J. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001, 2, 301–306. [Google Scholar] [CrossRef]
- Wei, S.; Kryczek, I.; Zou, W. Regulatory T-cell compartmentalization and trafficking. Blood 2006, 108, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Wang, J.H.; Gray, J.D.; Soucier, H.; Horwitz, D.A. Natural and induced CD4+CD25+ cells educate CD4+CD25− cells to develop suppressive activity: The role of IL-2, TGF-beta, and IL-10. J. Immunol. 2004, 172, 5213–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagiwa, S.; Gray, J.D.; Hashimoto, S.; Horwitz, D.A. A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J. Immunol. 2001, 166, 7282–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 is essential for TGF-beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Yuan, J.; Kuchroo, V.K.; Weiner, H.L. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGF-beta T cell-transgenic mice. J. Immunol. 2007, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Seddiki, N.; Santner-Nanan, B.; Martinson, J.; Zaunders, J.; Sasson, S.; Landay, A.; Solomon, M.; Selby, W.; Alexander, S.I.; Nanan, R.; et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 2006, 203, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. Pillars Article: An Essential Role for Scurfin in CD4+CD25+ T Regulatory Cells. J. Immunol. 2017, 198, 993–998. [Google Scholar]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Pillars Article: Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. J. Immunol. 2017, 198, 986–992. [Google Scholar]
- Wildin, R.S.; Smyk-Pearson, S.; Filipovich, A.H. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J. Med. Genet. 2002, 39, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, N.; Oida, T.; Hirota, K.; Nakamura, K.; Nomura, T.; Uchiyama, T.; Sakaguchi, S. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int. Immunol. 2006, 18, 1197–1209. [Google Scholar] [CrossRef] [Green Version]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavin, M.A.; Rasmussen, J.P.; Fontenot, J.D.; Vasta, V.; Manganiello, V.C.; Beavo, J.A.; Rudensky, A.Y. Foxp3-dependent programme of regulatory T-cell differentiation. Nature 2007, 445, 771–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.A.; Feuerer, M.; Tash, K.; Haxhinasto, S.; Perez, J.; Melamed, R.; Mathis, D.; Benoist, C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 2007, 27, 786–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Josefowicz, S.Z.; Kas, A.; Chu, T.T.; Gavin, M.A.; Rudensky, A.Y. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature 2007, 445, 936–940. [Google Scholar] [CrossRef]
- Williams, L.M.; Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007, 8, 277–284. [Google Scholar] [CrossRef]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 2010, 463, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Trifari, S.; Aijo, T.; Tsagaratou, A.; Pastor, W.A.; Zepeda-Martinez, J.A.; Lio, C.W.; Li, X.; Huang, Y.; Vijayanand, P.; et al. Control of Foxp3 stability through modulation of TET activity. J. Exp. Med. 2016, 213, 377–397. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liang, Y.; LeBlanc, M.; Benner, C.; Zheng, Y. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 2014, 158, 734–748. [Google Scholar] [CrossRef] [Green Version]
- Soper, D.M.; Kasprowicz, D.J.; Ziegler, S.F. IL-2Rbeta links IL-2R signaling with Foxp3 expression. Eur. J. Immunol. 2007, 37, 1817–1826. [Google Scholar] [CrossRef]
- Burchill, M.A.; Yang, J.; Vogtenhuber, C.; Blazar, B.R.; Farrar, M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007, 178, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.D.; Bopp, T.; Schmitt, E.; et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007, 5, e38. [Google Scholar] [CrossRef]
- Baron, U.; Floess, S.; Wieczorek, G.; Baumann, K.; Grutzkau, A.; Dong, J.; Thiel, A.; Boeld, T.J.; Hoffmann, P.; Edinger, M.; et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur. J. Immunol. 2007, 37, 2378–2389. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Sakaguchi, S. Transcriptional and epigenetic basis of Treg cell development and function: Its genetic anomalies or variations in autoimmune diseases. Cell Res. 2020, 30, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Helmin, K.A.; Morales-Nebreda, L.; Torres Acosta, M.A.; Anekalla, K.R.; Chen, S.Y.; Abdala-Valencia, H.; Politanska, Y.; Cheresh, P.; Akbarpour, M.; Steinert, E.M.; et al. Maintenance DNA methylation is essential for regulatory T cell development and stability of suppressive function. J. Clin. Investig. 2020, 130, 6571–6587. [Google Scholar] [CrossRef] [PubMed]
- Mikami, N.; Kawakami, R.; Chen, K.Y.; Sugimoto, A.; Ohkura, N.; Sakaguchi, S. Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation. Proc. Natl. Acad. Sci. USA 2020, 117, 12258–12268. [Google Scholar] [CrossRef]
- Duarte, J.H.; Zelenay, S.; Bergman, M.L.; Martins, A.C.; Demengeot, J. Natural Treg cells spontaneously differentiate into pathogenic helper cells in lymphopenic conditions. Eur. J. Immunol. 2009, 39, 948–955. [Google Scholar] [CrossRef]
- Kastner, L.; Dwyer, D.; Qin, F.X. Synergistic effect of IL-6 and IL-4 in driving fate revision of natural Foxp3+ regulatory T cells. J. Immunol. 2010, 185, 5778–5786. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martinez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Filipowicz, A.R.; Waseem, T.C.; McGary, C.M.; Crow, K.J.; Magilnick, N.; Boldin, M.; Lundberg, P.S.; Galkina, E.V. Atherosclerosis-Driven Treg Plasticity Results in Formation of a Dysfunctional Subset of Plastic IFNgamma+ Th1/Tregs. Circ. Res. 2016, 119, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenhove, G.; Bouladoux, N.; Wohlfert, E.A.; Hall, J.A.; Chou, D.; Dos Santos, L.; O’Brien, S.; Blank, R.; Lamb, E.; Natarajan, S.; et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 2009, 31, 772–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubtsov, Y.P.; Niec, R.E.; Josefowicz, S.; Li, L.; Darce, J.; Mathis, D.; Benoist, C.; Rudensky, A.Y. Stability of the regulatory T cell lineage in vivo. Science 2010, 329, 1667–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Chi, H. Metabolic Control of Treg Cell Stability, Plasticity, and Tissue-Specific Heterogeneity. Front. Immunol. 2019, 10, 2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Li, J.; McArdle, S.; Gholami, A.; Kimura, T.; Wolf, D.; Gerhardt, T.; Miller, J.; Weber, C.; Ley, K. CCR5+T-bet+FoxP3+ Effector CD4 T Cells Drive Atherosclerosis. Circ. Res. 2016, 118, 1540–1552. [Google Scholar] [CrossRef] [Green Version]
- Miyao, T.; Floess, S.; Setoguchi, R.; Luche, H.; Fehling, H.J.; Waldmann, H.; Huehn, J.; Hori, S. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 2012, 36, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Mariotti-Ferrandiz, M.E.; Wang, Y.; Malissen, B.; Waldmann, H.; Hori, S. Heterogeneity of natural Foxp3+ T cells: A committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA 2009, 106, 1903–1908. [Google Scholar] [CrossRef] [Green Version]
- Tan, T.G.; Mathis, D.; Benoist, C. Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc. Natl. Acad. Sci. USA 2016, 113, 14103–14108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovenschen, H.J.; van de Kerkhof, P.C.; van Erp, P.E.; Woestenenk, R.; Joosten, I.; Koenen, H.J. Foxp3+ regulatory T cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J. Investig. Derm. 2011, 131, 1853–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, R.; Zhou, L.; Ma, Y.; Zhou, L.; Liang, T.; Shi, L.; Long, J.; Yuan, D. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 58, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Chaudhry, A.; Kas, A.; deRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.O.; Beiting, D.P.; Tato, C.; John, B.; Oldenhove, G.; Lombana, C.G.; Pritchard, G.H.; Silver, J.S.; Bouladoux, N.; Stumhofer, J.S.; et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 2012, 37, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Wagner, J.; Metzger, D.; Chambon, P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 1997, 237, 752–757. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Niec, R.E.; Kim, H.Y.; Treuting, P.; Chinen, T.; Zheng, Y.; Umetsu, D.T.; Rudensky, A.Y. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 2012, 482, 395–399. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Guo, J.; Gu, Q.; Zhu, X.; Zhou, X. Activation and Functional Specialization of Regulatory T Cells Lead to the Generation of Foxp3 Instability. J. Immunol. 2017, 198, 2612–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbi, J.; Pardoll, D.; Pan, F. Treg functional stability and its responsiveness to the microenvironment. Immunol. Rev. 2014, 259, 115–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xun, L.; Zhang, R.; Hu, F.; Luan, J.; Lao, K.; Wang, X.; Gou, X. Stability and inhibitory function of Treg cells under inflammatory conditions in vitro. Exp. Ther. Med. 2019, 18, 2443–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, J.; Perlman, S. Differential effects of IL-12 on Tregs and non-Treg T cells: Roles of IFN-gamma, IL-2 and IL-2R. PLoS ONE 2012, 7, e46241. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, K.; Misaki, Y.; Yamauchi, M.; Tsunekawa, S.; Setoguchi, K.; Miyazaki, J.; Yamamoto, K. Generation of CD4+CD25+ regulatory T cells from autoreactive T cells simultaneously with their negative selection in the thymus and from nonautoreactive T cells by endogenous TCR expression. J. Immunol. 2002, 168, 4399–4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrysova, L.; Christensen, J.R.; Wu, X.; Kissenpfennig, A.; Malissen, B.; O’Garra, A. Integrated T-cell receptor and costimulatory signals determine TGF-beta-dependent differentiation and maintenance of Foxp3+ regulatory T cells. Eur. J. Immunol. 2011, 41, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802. [Google Scholar] [CrossRef] [Green Version]
- Haxhinasto, S.; Mathis, D.; Benoist, C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 2008, 205, 565–574. [Google Scholar] [CrossRef]
- Hoffmann, P.; Boeld, T.J.; Eder, R.; Huehn, J.; Floess, S.; Wieczorek, G.; Olek, S.; Dietmaier, W.; Andreesen, R.; Edinger, M. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur. J. Immunol. 2009, 39, 1088–1097. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X. Foxp3 Instability Helps tTregs Distinguish Self and Non-self. Front. Immunol. 2019, 10, 2226. [Google Scholar] [CrossRef] [Green Version]
- Smigiel, K.S.; Richards, E.; Srivastava, S.; Thomas, K.R.; Dudda, J.C.; Klonowski, K.D.; Campbell, D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014, 211, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Zemmour, D.; Zilionis, R.; Kiner, E.; Klein, A.M.; Mathis, D.; Benoist, C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 2018, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, M.L.; Scavuzzo, M.A.; Blum, S.; Shevchenko, I.; Lee, T.; Makedonas, G.; Borowiak, M.; Bettini, M.L.; Bettini, M. High self-reactivity drives T-bet and potentiates Treg function in tissue-specific autoimmunity. JCI Insight 2018, 3, e97322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.G.; Wang, J.; Horwitz, D.A. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J. Immunol. 2008, 180, 7112–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massoud, A.H.; Charbonnier, L.M.; Lopez, D.; Pellegrini, M.; Phipatanakul, W.; Chatila, T.A. An asthma-associated IL4R variant exacerbates airway inflammation by promoting conversion of regulatory T cells to TH17-like cells. Nat. Med. 2016, 22, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Tarique, M.; Saini, C.; Naqvi, R.A.; Khanna, N.; Sharma, A.; Rao, D.N. IL-12 and IL-23 modulate plasticity of FoxP3+ regulatory T cells in human Leprosy. Mol. Immunol. 2017, 83, 72–81. [Google Scholar] [CrossRef]
- Tang, Q.; Adams, J.Y.; Penaranda, C.; Melli, K.; Piaggio, E.; Sgouroudis, E.; Piccirillo, C.A.; Salomon, B.L.; Bluestone, J.A. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008, 28, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Bailey-Bucktrout, S.L.; Martinez-Llordella, M.; Zhou, X.; Anthony, B.; Rosenthal, W.; Luche, H.; Fehling, H.J.; Bluestone, J.A. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 2013, 39, 949–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Brand, D.; Zheng, S.G. Targeting IL-2: An unexpected effect in treating immunological diseases. Signal. Transduct Target. Ther. 2018, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.S.; Suwannasaen, D.; Mann, E.H.; Urry, Z.; Richards, D.F.; Lertmemongkolchai, G.; Hawrylowicz, C.M. 1alpha,25-dihydroxyvitamin D3 in combination with transforming growth factor-beta increases the frequency of Foxp3+ regulatory T cells through preferential expansion and usage of interleukin-2. Immunology 2014, 143, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Urry, Z.; Chambers, E.S.; Xystrakis, E.; Dimeloe, S.; Richards, D.F.; Gabrysova, L.; Christensen, J.; Gupta, A.; Saglani, S.; Bush, A.; et al. The role of 1alpha, 25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur. J. Immunol. 2012, 42, 2697–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Lan, Q.; Li, Z.; Zhou, X.; Gu, J.; Li, Q.; Wang, J.; Chen, M.; Liu, Y.; Shen, Y.; et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc. Natl. Acad. Sci. USA 2014, 111, E3432–E3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puccetti, P.; Fallarino, F. Generation of T cell regulatory activity by plasmacytoid dendritic cells and tryptophan catabolism. Blood Cells Mol. Dis. 2008, 40, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F. The P2X7 Receptor as Regulator of T Cell Development and Function. Front. Immunol. 2020, 11, 1179. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martinez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Zeng, H.; Nguyen, T.M.; Wang, Y.; Vogel, P.; Dhungana, Y.; Liu, X.; Neale, G.; Locasale, J.W.; Chi, H. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun. 2018, 9, 2095. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Luo, Y.; Guo, W.; Niu, Q.; Xue, T.; Yang, F.; Sun, X.; Chen, S.; Liu, Y.; Liu, J.; et al. Lkb1 maintains Treg cell lineage identity. Nat. Commun. 2017, 8, 15876. [Google Scholar] [CrossRef]
- Timilshina, M.; You, Z.; Lacher, S.M.; Acharya, S.; Jiang, L.; Kang, Y.; Kim, J.A.; Chang, H.W.; Kim, K.J.; Park, B.; et al. Activation of Mevalonate Pathway via LKB1 Is Essential for Stability of Treg Cells. Cell Rep. 2019, 27, 2948–2961.e7. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Vallion, R.; Divoux, J.; Glauzy, S.; Ronin, E.; Lombardi, Y.; Lubrano di Ricco, M.; Gregoire, S.; Nemazanyy, I.; Durand, A.; Fradin, D.; et al. Regulatory T Cell Stability and Migration Are Dependent on mTOR. J. Immunol. 2020, 205, 1799–1809. [Google Scholar] [CrossRef]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitz, A.; de Marcken, M.; Gautron, A.S.; Mitrovic, M.; Hafler, D.A.; Dominguez-Villar, M. AKT isoforms modulate Th1-like Treg generation and function in human autoimmune disease. EMBO Rep. 2016, 17, 1169–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerdiles, Y.M.; Stone, E.L.; Beisner, D.R.; McGargill, M.A.; Ch’en, I.L.; Stockmann, C.; Katayama, C.D.; Hedrick, S.M. Foxo transcription factors control regulatory T cell development and function. Immunity 2010, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, W.; Beckett, O.; Ma, Q.; Paik, J.H.; DePinho, R.A.; Li, M.O. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 618–627. [Google Scholar] [CrossRef]
- Harada, Y.; Harada, Y.; Elly, C.; Ying, G.; Paik, J.H.; DePinho, R.A.; Liu, Y.C. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J. Exp. Med. 2010, 207, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Gaddis, D.E.; Padgett, L.E.; Wu, R.; McSkimming, C.; Romines, V.; Taylor, A.M.; McNamara, C.A.; Kronenberg, M.; Crotty, S.; Thomas, M.J.; et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun. 2018, 9, 1095. [Google Scholar] [CrossRef]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Anto Michel, N.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves from Initially Protective ApoB-Reactive CD4+ T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Qiu, G.; Lal, G.; Wu, Z.; Levy, D.E.; Ochando, J.C.; Bromberg, J.S.; Ding, Y. c-Maf regulates IL-10 expression during Th17 polarization. J. Immunol. 2009, 182, 6226–6236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.; Blume, J.; Roy, U.; Teh, P.P.; Vasanthakumar, A.; Beller, A.; Liao, Y.; Heinrich, F.; Arenzana, T.L.; Hackney, J.A.; et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol. 2019, 20, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Planer, D.; Luboshits, G.; Afek, A.; Metzger, S.; Chajek-Shaul, T.; Keren, G.; George, J. Role of naturally occurring CD4+ CD25+ regulatory T cells in experimental atherosclerosis. Arter. Thromb. Vasc. Biol. 2007, 27, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, A.; Luboshits, G.; Planer, D.; Keren, G.; George, J. Altered status of CD4+CD25+ regulatory T cells in patients with acute coronary syndromes. Eur. Heart J. 2006, 27, 2530–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.W.; Gistera, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. Hypercholesterolemia Induces Differentiation of Regulatory T Cells in the Liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef] [PubMed]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1alpha to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: Potential role in the regulation of aging process. Cell Mol. Life Sci. 2015, 72, 3897–3914. [Google Scholar] [CrossRef]
- Kimura, T.; Kobiyama, K.; Winkels, H.; Tse, K.; Miller, J.; Vassallo, M.; Wolf, D.; Ryden, C.; Orecchioni, M.; Dileepan, T.; et al. Regulatory CD4+ T Cells Recognize MHC-II-Restricted Peptide Epitopes of Apolipoprotein B. Circulation 2018, 138, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Tse, K.; McArdle, S.; Gerhardt, T.; Miller, J.; Mikulski, Z.; Sidney, J.; Sette, A.; Wolf, D.; Ley, K. Atheroprotective vaccination with MHC-II-restricted ApoB peptides induces peritoneal IL-10-producing CD4 T cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H781–H790. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.J.; Makings, J.; Ley, K. Regulatory T Cell Stability and Plasticity in Atherosclerosis. Cells 2020, 9, 2665. https://doi.org/10.3390/cells9122665
Ali AJ, Makings J, Ley K. Regulatory T Cell Stability and Plasticity in Atherosclerosis. Cells. 2020; 9(12):2665. https://doi.org/10.3390/cells9122665
Chicago/Turabian StyleAli, Amal J., Jeffrey Makings, and Klaus Ley. 2020. "Regulatory T Cell Stability and Plasticity in Atherosclerosis" Cells 9, no. 12: 2665. https://doi.org/10.3390/cells9122665
APA StyleAli, A. J., Makings, J., & Ley, K. (2020). Regulatory T Cell Stability and Plasticity in Atherosclerosis. Cells, 9(12), 2665. https://doi.org/10.3390/cells9122665