Abstract
The treatment of tumors requires the induction of cell death. Radiotherapy, chemotherapy, and immunotherapy are administered to kill cancer cells; however, some cancer cells are resistant to these therapies. Therefore, effective treatments require various strategies for the induction of cell death. Regulated cell death (RCD) is systematically controlled by intracellular signaling proteins. Apoptosis and autophagy are types of RCD that are morphologically different from necrosis, while necroptosis, pyroptosis, and ferroptosis are morphologically similar to necrosis. Unlike necrosis, regulated necrotic cell death (RNCD) is caused by disruption of the plasma membrane under the control of specific proteins and induces tissue inflammation. Various types of RNCD, such as necroptosis, pyroptosis, and ferroptosis, have been used as therapeutic strategies against various tumor types. In this review, the mechanisms of necroptosis, pyroptosis, and ferroptosis are described in detail, and a potential effective treatment strategy to increase the anticancer effects on apoptosis- or autophagy-resistant tumor types through the induction of RNCD is suggested.
Keywords:
apoptosis; autophagy; necrosis; necroptosis; pyroptosis; ferroptosis; therapy-resistant tumors 1. Introduction
Cancer is one of the major causes of death worldwide, and it leads to approximately 1 in 6 deaths globally [1]. In 2018, 9.6 million people died from various types of cancer such as lung (1.76 million), colorectal (862,000), stomach (783,000), liver (782,000), and breast (627,000) cancer [2]. In general, cancer is induced by not only genetic factors but also physical, chemical, and biological carcinogens including radiation, asbestos, and viruses [3]. However, if these harmful factors are avoided and early screening for cancer prevention is performed thoroughly, 30~50% of tumors can be prevented, thus reducing the cancer burden [4]. Cancer patients are treated with surgery, chemotherapy, immunotherapy, and radiotherapy to relieve pain or symptoms [5]. The ultimate goal of these therapies is to kill cancer cells in the patients [6]. However, it is difficult to completely kill all cancer cells, since some cells are resistant to cancer therapies through various survival mechanisms [7].
Various types of cell death can be induced (Figure 1). Traditionally, the goal of anticancer therapy has been to induce apoptosis in cancer cells; however, cancer cells can avoid this type of death by inhibiting the activation of apoptotic signals [8]. In various types of cancer cells, the basal expression level of anti-apoptotic proteins such as survivin, PED, Bcl-xL, Bcl-2, and cFLIP, is high, and the expression of pro-apoptotic proteins such as bax, bim, bak, bad, puma, and noxa is reduced [9]. IL-4 and IL-6 derived from cancer cells can also function as anti-apoptotic factors. IL-4 production by primary epithelial cancer cells obtained from human colon, lung, and breast tissues elevates the expressions of anti-apoptotic proteins and increases the therapeutic resistance to anticancer agents, such as oxaliplatin, doxorubicin, and etoposide, in vitro and in vivo [10]. IL-6 secretion by ovarian, prostate, and lung cancer cells makes them resistant to cisplatin in vitro [11]. Moreover, therapy-induced apoptosis of cancer cells may promote tumor growth. Apoptotic extracellular vesicles (ApoEVs) released from glioblastoma (GBM) cells after chemotherapy or radiotherapy can cause the transformation of adjacent cells into a therapy-resistant population via alteration in the splicing process in mice [12]. Sphingosine-1 phosphate (S1P), which is secreted by cancer cells during apoptosis, was shown to stimulate S1P1 on primary human macrophages and to activate the angiogenic program [13]. Therefore, alternative strategies are needed to treat patients with apoptosis-resistant cancer.
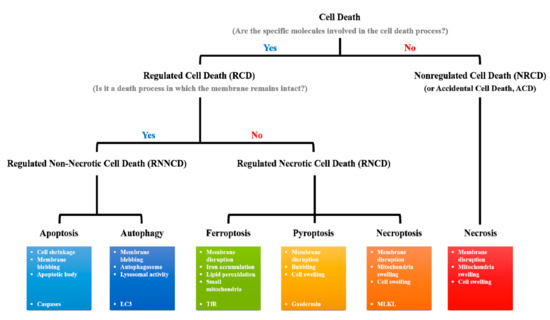
Figure 1.
Classification of cell death types. Cell death types can be classified according to whether they are regulated by a specific signal pathway. Except for necrosis, most types of cell death are included in the RCD category. The type of RCD can also be classified according to membrane disruption. Apoptotic- and autophagic-cell death does not induce additional inflammatory responses, because their cellular components are sequestered within the membrane structures, such as apoptotic bodies and autophagosomes, respectively, during cell death processes. However, since RNCDs, including ferroptosis, pyroptosis, and necroptosis, are death types accompanied by membrane disruption, RNCDs can induce inflammatory responses in the tissues.
Autophagy is an intracellular catabolic process that responds to various cellular stresses [14]. In cancer therapy, autophagy is involved in the survival and death of cancer cells, and the activation of a certain cell death type may be beneficial or detrimental to the cancer cells depending on the properties of the cancer microenvironment [15]. The induction of autophagy has been used as an alternative strategy to treat apoptosis-resistant cancer cells. TLRagonist-induced autophagy in tumors enhanced the efficiency of radiotherapy and improved anticancer immunity in vitro and in vivo [16,17]. Alkaloid compounds caused cell death in apoptosis-defective human cancer cell lines through the activation of autophagy [18]. Furthermore, the inhibition of autophagy may induce apoptosis in cancer cells. Chloroquine, an autophagy flux inhibitor, increased cyclophosphamide-induced apoptosis in mouse lymphoma cells lacking p53 activity and enhanced the radiosensitivity of human urinary bladder cancer cell lines, EJ and T24, via apoptosis [19,20]. Additionally, 3-methyladenine (3-MA), an autophagy inhibitor, promoted cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells [21]. However, in contrast to these results, the inhibition of autophagy may make cancer cells more susceptible to cancer therapies. 3-MA significantly reduced the survival of radiation-exposed MDA-MB-231 cells, a human breast cancer cell line, and vincristine-treated leukemia cells [22,23]. These data suggest that the activation of autophagic mechanisms can result in various consequences for cellular fate. In addition to regulated non-necrotic cell death including apoptosis and autophagy, the induction of regulated necrotic cell death (RNCD) has been evaluated in recent studies to improve cancer therapy. The types of RNCD and corresponding therapeutic strategies will be discussed in this review.
2. Types of Regulated Necrotic Cell Death
Cell death is one of the major homeostatic mechanisms, and the appropriate regulation of cell death in cancer therapy is paramount [24]. Since accidental cell death (ACD) such as necrosis can promote the development and progression of cancer, the induction of regulated cell death (RCD) may be more effective for cancer therapy [25,26]. Unlike ACD, RCD is a cell death type controlled by specific proteins [27]. For decades, researchers in the field of anticancer therapeutics mainly studied apoptotic and autophagic types of cell death. However, resistance to apoptosis has been detected in various types of cancers during radiotherapy and chemotherapy [28]. New alternative therapeutic strategies are required, because the basal level of autophagy is dependent on the cancer type and can result in completely different treatment results [29]. RNCD is controlled by specific proteins similar to apoptosis or autophagy; however, the morphological features of RNCD are different, because it causes plasma membrane disruption [30]. Another significant difference is that RNCD induces higher inflammatory responses compared to those induced by apoptosis or autophagy [31].
Recent studies demonstrated that inflammatory signals induced by RNCD delay tumor progression and promote anticancer immune responses in mice and human cancer cells [32,33,34]. These results indicate that the induction of RNCD, including necroptosis, pyroptosis, and ferroptosis, may be a useful strategy of cancer therapy. Necroptosis, pyroptosis, and ferroptosis are commonly regulated by specific proteins and are accompanied by disruption of the cellular membrane (Figure 2).
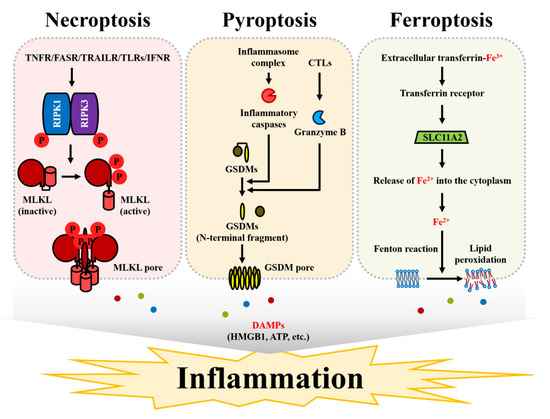
Figure 2.
Types of RNCD. Necroptosis can be induced by the signals of TNF receptors, FAS receptors, TRAIL receptors, toll-like receptors, and interferon receptors. In the absence of caspase-8 activity, the phosphorylation of RIPK1 and RIPK3 induces phosphorylation and the subsequent activation of inactive MLKL. Activated MLKL is translocated into the plasma membrane and forms the pores approximately 4 nm in diameter. Pyroptosis is caused by gasdermin-mediated pore formation. The activation of the inflammasome complex induces the enzymatic activity of caspase-1, and caspase-1 cleaves gasdermin D. The N-terminal region of cleaved gasdermin D is translocated into the plasma membrane and forms the pores approximately 10–15 nm in diameter. In addition to caspase-1, gasdermin proteins can be cleaved by caspase-4, caspase-5, and caspase-11. Moreover, gasdermin E can be cleaved by granzyme B, which is released by cytotoxic CD8 T cells and natural killer (NK) cells. Ferroptosis is caused by membrane disruption induced by an iron-mediated lipid peroxidation process known as the Fenton reaction. Transferrin receptor transfers Fe3+ bound to extracellular transferrin into the intracellular endosomal regions. Fe3+ is reduced to Fe2+ by STEAP3, a ferrireductase, and generated Fe2+ is released into the cytoplasm by SLC11A2 (also known as divalent metal transporter 1, DMT1). The release of Fe2+ into the cytosol causes lipid peroxidation in the plasma membrane. As a result of RNCD induction, DAMPs, such as HMGB1 and ATP, can be released into the extracellular regions to induce inflammatory responses in the tissues.
2.1. Necroptosis
Necroptosis is a type of programmed necrosis and its morphological features include cell death induced by cell expansion and plasma membrane rupture [35]. However, unlike necrosis, cell membrane permeability is tightly controlled during necroptotic cell death. Necroptosis can be induced by the TNF receptor superfamily, TLRs, interferon receptors, etc. [36]. In general, stimulatory factors, including ligands of these receptor or various stressors, can trigger extrinsic apoptosis through the activation of caspase-8 [37,38,39]. However, caspase-8 cleaves and thus inhibits the function of RIPK1 and RIPK3 proteins that are essential for the execution of necroptosis [40]. Therefore, the initiation of necroptosis can depend on the expression and activity of caspase-8 [41]. In most cancer cells, the expression or activity of caspase-8 can be suppressed or turned off [42]. Thus, cancer cells can be resistant to anticancer drugs that cause apoptosis [43]. When caspase-8 is absent or inactivated, RIPK1 and RIPK3 are phosphorylated and subsequently form a necrosome [44]. Activated RIPK3 phosphorylates the serine/threonine residues in MLKL to induce MLKL oligomerization [45]. The structural stability of the MLKL polymer depends on the disulfide bond between the molecules [46]. However, disulfide bonds are important for tetramerization of MLKL, but not for octamerization, which is important for membrane disruption [47]. Several mechanistic models of membrane collapse induced by MLKL have been proposed; however, specific consensus has not been reached [48]. After the phosphorylation of MLKL, the N-terminal domain is apparently translocated into the cell membrane [49]. MLKL pores have been confirmed to act as cation channels in liposomes [50]. In particular, the MLKL channel was shown to have higher permeability to Mg2+ than that to Ca2+ in the presence of Na+ and K+ [51]. Thus, insertion of an MLKL oligomer into the cell membrane can initiate necroptosis and subsequent release of damage-associated molecular patterns (DAMPs) containing intracellular components from the cells.
2.2. Pyroptosis
Both apoptosis and pyroptosis are mediated by caspases; however, unlike apoptosis, pyroptosis involves inflammatory caspase-related cell death [52]. Pyroptosis is distinguished from necroptosis, another RNCD, by involvement of caspases in cell death events. Unlike necroptis, pyroptosis involves plasma membrane disruption mediated by gasdermin (GSDM) proteins [53]. In general, pyroptosis includes inflammasome activation and maturation of IL-1β and IL-18 [54]. The assembly of the inflammasome complex is triggered by sensing cytoplasmic pathogen-associated molecular patterns (PAMPs) or DAMPs [55]. The NLR family proteins, such as NLRP1, NLRP3, and NLRC4, are responsible for the detection of these patterns [56]. Activated NLRs are oligomerized through the interaction between their NACHT domains, and then the CARD domain interacts with the CARD domain of pro-caspase-1 [57] to recruit pro-caspase-1 into the NLR oligomer [58]. Some NLRs have the PYD domain instead of the CARD domain [59]. These NLRs require ASC, an adaptor protein with the CARD domain, to recruit pro-caspase-1 [60]. In this case, the CARD domain of ASC interacts with the CARD domain in pro-caspase-1 [61]. Pro-caspase-1 in the inflammasome complex can be cleaved into two p20 and two p10 molecules, and the cleaved molecules bind to each other and act as a heterodimeric enzyme [62]. Activated caspase-1 cleaves IL-1β, IL-18, and GSDMD, an executor of pyroptotic cell death [63]. Furthermore, in humans, GSDMD can be cleaved by caspase-4, -5, and -11 [64]. The N-terminal domain of cleaved GSDMD is translocated into the plasma membrane and then oligomerized [65]. Finally, gasdermin pores mediated the K+ efflux and release of DAMPs [66]. In addition to GSDMD, GSDME can mediate pyroptosis without the formation of inflammasome complexes [67].
2.3. Ferroptosis
Ferroptosis is a type of RNCD caused by disruption of the plasma membrane [68]. However, unlike necroptosis and pyroptosis, ferroptotic cell death is not mediated by specific protein-related pores. Ferroptosis is caused by membrane disruption due to the accumulation of iron-dependent lipid peroxides [69]. Ferroptotic cell death depends on iron levels in the cells and can also be suppressed by iron chelators [70]. Extracellular Fe3+ atoms are bound to transferrin, a monomeric glycoprotein, and Fe3+ can be transferred into the endosome via the transferrin receptor (TfR) on the plasma membrane [71]. In the endosomes, Fe3+ is reduced to Fe2+ by endosomal ferrireductase [72]. Endosomal Fe2+ is released into the cytoplasm via a divalent metal transporter, SLC11A2 [73]. Cytosolic Fe2+ is oxidized to Fe3+ in the reaction with H2O2, resulting in the formation of •OH and HO- in a process known as the Fenton reaction [74]. These radicals generated by the chemical reaction disrupt the plasma membrane through the peroxidation of membrane lipids [75]. Ferroptosis is closely associated with imbalance in the iron metabolism; however, it can also be caused by NADPH-mediated lipid peroxidation [76]. Moreover, the inhibition of glutathione, a homeostatic molecule scavenging intracellular reactive oxygen species (ROS), can induce ferroptosis [77]. Overall, peroxidation of membrane lipids due to excessive ROS generation can cause ferroptosis.
3. Regulated Necrotic Cell Death and Tumor Therapy
Applications of the strategies for induction of apoptotic and autophagic cell deaths have been investigated in the treatment of various cancer types. However, since cancer can be resistant to apoptosis, new therapeutic approaches should be investigated. In particular, unlike apoptosis and autophagy, RNCD includes cell death types that induce inflammatory responses; hence, it is necessary to consider whether RNCDs are suitable for cancer treatment.
3.1. Necroptosis in Tumor Therapy
Necroptosis has been reported to induce the inhibition of tumor growth in various cancer types. Ionizing radiation caused necroptosis in human thyroid and adrenocortical cancers [78]. Moreover, radiation regulated the repopulation of human colorectal cancer cells via the RIP1/RIP3/MLKL/JNK/IL-8 pathway [79]. Chemoresistance of cancer cells is strongly associated with high levels of enzymatic activity of aldehyde dehydrogenase (ALDH) [80]. ALDH1A family inhibitors (ALDH1Ai), such as DEAB, 673A, and CM10, increased the sensitivity of chemotherapy-resistant human ovarian cancer cells, SKOv3, to cisplatin through the induction of calcium-dependent necroptosis [81]. In particular, a Smac mimetic necroptosis inducer increased the sensitivity of apoptosis-resistant cells, including RIPK3-expressing KKU213, RMCCA-1, and HuCCT-1 cell lines, to TNF in vitro [82]. Staurosporine, shikonin, proteasome inhibitors (MG132 and bortezomib), and Smac mimetics suppressed the viability of leukemia cells by inducing necroptosis in vitro and in vivo [83,84,85]. Anticancer agents, including 5-fluorouracil (5-FU) and resibufogenin, caused necroptosis-mediated inhibition of human colorectal cancer cells in vitro and in vivo [86,87]. These results imply that the induction of necroptosis in vitro and in vivo may be an effective strategy for cancer therapy, at least according to the animal studies. However, clinical data based on the expression of necroptosis-related proteins in various types of cancers are characterized by variable associations with treatment prognosis. The expression of RIPK3 is decreased in patients with melanoma, breast cancer, and colorectal cancer, and high expression of RIPK3 in the tumors of these patients is strongly associated with survival [88,89,90]. In contrast to the clinical expression of RIPK3, high expression of RIPK1 in patients with lung cancer or pancreatic ductal adenocarcinoma is highly associated with poor prognosis [91,92]. Furthermore, reduced expression of MLKL has been reported to be associated with tumor metastasis into the lymph nodes [93]. Additionally, low expression of MLKL is strongly associated with poor prognosis in ovarian cancer patients [94]. Therefore, the correlation between necroptosis induction and its therapeutic effects in various types of cancer patients requires additional investigation.
3.2. Pyroptosis in Tumor Therapy
The major modulators of the pyroptosis process include the gasdermin family proteins [95]. GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5), and DFNB59 constitute the gasdermin family and mediate pyroptosis in various types of cancer cells [96]. In particular, expression of GSDMA, GSDMB, GSDMC, or GSDME on the cell surface may lead to pore formation on the plasma membrane, e.g., in GSDMD-mediated pyroptosis [97]. The N-terminal domain of GSDMB was shown to form pores in an in vitro study; however, evidence is found unclear [98]. In general, GSDMA, GSDMC, GSDMD, and GSDME are known as tumor suppressors, and GSDMB was shown to function as an oncogene [99]. Since the differential expression of each gasdermin protein depends on cancer type, and remarkable data have been reported in the clinical studies, additional investigations are needed [100]. For example, enhanced expression of GSDMA and GSDMC was observed in human colorectal cancer progression [101]. Moreover, high expression of GSDMC in melanoma and lung adenocarcinoma was associated with a poor prognosis [102,103]. High levels of GSDMD are expressed in human NSCLC tissues, and knockdown of GSDMD in NSCLC cell lines (PC9, H1703, A549, SPC-A1, H1915, H1975, H1299 and H1650) attenuated their proliferation [104]. Elevated expression of GSDMD was observed in the tissues of patients with endometrioid carcinoma; however, other studies have shown that the tumor volume in groups treated with hydrogen-rich water was reduced by GSDMD-mediated pyroptosis compared to that in control mice [105]. High expression of GSDMB in cervical, uterine, esophageal, gastric, liver, and colon cancer tissues is significantly associated with a poor prognosis [98]. These results indicate that high expression of gasdermin proteins in the tumor tissues might not lead to favorable survival outcomes in cancer patients. Previous in vitro studies induced pyroptosis in cancer cells by treatment with various drugs. Metformin reduced cell viability by inducing pyroptosis caused by the cleavage of GSDMD in esophageal squamous cell carcinoma (ESCC) patients and human ESCC cell lines, including KYS510 and KYSE140 [106]. Nobiletin suppressed cell growth via GSDMD-mediated pyroptosis in ovarian cancer cells [107]. Navitoclax induced GSDME-related pyroptosis in human colorectal carcinoma HCT 116 cell [108]. In addition to the induction of pyroptosis by drug treatment, granzyme B secreted by cytotoxic T lymphocytes induced GSDME-mediated pyroptotic cell death without the formation of inflammasomes in mouse and human tumor cells [67]. Experimental data indicate that the induction of pyroptosis in the tumors may cause tumor suppression; however, additional clinical outcomes should be investigated in patients with various tumor types.
3.3. Ferroptosis in Tumor Therapy
Glutathione is an important molecule that regulates excessive ROS levels in the cells [109]. In particular, elevated activation of the KEAP1/NRF2/SLC7A11 axis of glutathione synthesis has been detected in various cancer cells and patients with breast cancer, bladder cancer, gastric carcinoma, glioma, and melanoma; this mechanism plays a major role in the resistance of cancer cells to cancer therapies [110,111]. Considering that ferroptosis is one of the types of RNCD induced by ROS-mediated lipid peroxidation process, i.e., the Fenton reaction, targeting this axis in cancer cells may be an alternative strategy for effective cancer treatment [112]. The induction of ferroptosis via inhibition of the glutathione synthesis pathway in various types of cancer cells has been investigated, and the results demonstrated that ferroptosis can effectively induce cell death in tumors resistant to radiotherapy and chemotherapy [34,113]. In previous studies, radiation alone induced lipid peroxidation and ferroptosis in a patient-derived xenograft (PDX) model [114]. In particular, iron chelation by desferrioxamine treatment of human glioma cells promoted the radioresistance of the cells in vitro [115]. These results indicate that ferroptosis may be an important strategy for the enhancement of radiosensitivity of tumors during radiotherapy. Ferroptosis inducers, such as erastin, sulfasalazine, and sorafenib, induced glutathione starvation and increased radiosensitivity in vitro and in vivo [116,117,118]. These ferroptosis inducers increase the sensitivity to anticancer drugs in various types of cancer during chemotherapy. For example, the induction of ferroptosis in cisplatin-resistant human cancer cells, such as head and neck cancer cells, non-small-cell lung cancer cells, and osteosarcoma cells, enhanced their sensitivity to cisplatin [119,120,121]. In clinical studies, high expression of SLC7A11 in patients with pulmonary cancer, pancreatic cancer, liver carcinoma, thyroid carcinoma, melanoma, or glioma was associated with a poor prognosis [122,123,124,125,126,127]. Therefore, the induction of ferroptosis may be an effective strategy for the treatment of therapy-resistant cancer types.
4. Conclusions
The induction of cell death in cancer cells is a major therapeutic strategy for tumor patients. Historically, the induction of apoptosis in various cancer cells has been tested in tumor therapies, and this strategy remains effective [128,129]. However, these approaches do not elicit a therapeutic effect in apoptosis-resistant tumors. To solve this problem, the induction of autophagic cell death has been attempted as a new strategy against apoptosis-resistant tumor cells [18,130]. The induction of autophagy in the cells can significantly reduce tumor volumes and may be a useful therapeutic strategy [131,132,133]. However, resistance to radiotherapy and chemotherapy can be induced by activation of autophagy, and autophagy has certain limitations in inducing cell death in various cancer types [134,135]. Therefore, the mechanisms of inflammatory cell death, such as RNCD, have been suggested for cancer treatment and were evaluated in various tumor studies.
RNCD has been studied in a variety of tumors. Necroptosis, pyroptosis, ferroptosis, parthanatos, and entotic cell death are the types of RNCD. Necroptosis, pyroptosis, and ferroptosis are discussed in this review. These types of cell death are caused by membrane disruption induced by specific signal cascades [136], which can elicit an inflammatory response within the tumor tissue [137]. The induction of RNCD in tumor tissues does not necessarily lead to positive results in cancer patients, since the inflammatory response in the tissues can have favorable or adverse effect depending on the tumor type [138]. However, significant antitumor effects of RNCD inducers were observed in anticancer drug-resistant cancer cells. FDA-approved homoharringtonine induced necroptosis in human colorectal and pancreatic cancers [139]. Simvastatin inhibited the proliferation and migration of human non-small-cell lung cancer through the induction of pyroptosis [140]. Moreover, simvastatin was administered to prevent the risk of recurrence in female patients after the breast cancer surgery [141]. Dihydrotanshinone, a component of Danshen (Salvia miltiorrhiza Bunge), was shown to induce ferroptosis in breast cancer cells and was reported to improve the survival of breast cancer patients [142]. Additionally, various RNCD inducers may inhibit tumor survival (Figure 3). Therefore, clinically meaningful results can be obtained in various additional studies.
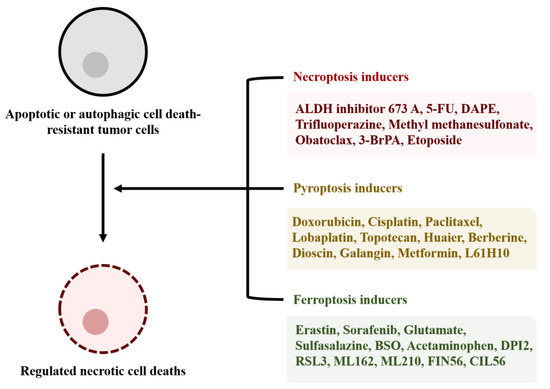
Figure 3.
Inducers of RNCD. RNCD inducers can be used to induce cell death of tumor cells that are resistant to apoptotic or autophagic cell death. RNCD inducers can cause membrane disruption of the tumor cells and subsequent inflammatory responses in the tumor tissues. The appropriate administration of the inducers according to tumor types may be effective tumor therapies. Aldehyde dehydrogenase (ALDH1A) inhibitor; 5-fluorouracil (5-FU); 1,2-diarachidonoyl-sn-glycero-3-phosphoethanolamine (DAPE); 3-bromopyruvate (3-BrPA); buthionine sulfoximine (BSO); dihydroxyphenyl-imino-2-imidazolidine (DPI2); (1S,3R)-methyl 2-(2-chloroacetyl)-2,3,4,9-tetrahydro-1-[4-(methoxycarbonyl)phenyl]-1H-pyrido[3¨C-b]indole-3-carboxylate (RSL3); α-[(2-chloroacetyl)(3-chloro-4-methoxyphenyl)amino]-N-(2-phenylethyl)-2-thiopheneacetamide (ML162); [4-[bis(4-chlorophenyl)methyl]-1-piperazinyl](5-methyl-4-nitro-3-isoxazolyl)methanone (ML210); N2,N7-dicyclohexyl-9-(hydroxyimino)-9H-fluorene-2,7-disulfonamide (FIN56); 2,7-bis(1-piperidinylsulfonyl)-9H-fluoren-9-one, oxime (CIL56).
To effectively induce RNCDs in cancer cells resistant to the apoptotic and autophagic pathways, the expression levels of mRNAs and proteins associated with RNCDs must be evaluated in patient tissues; then, the sensitivity of cancer cells to RNCDs must be enhanced by a combination of various therapeutic strategies; finally, the agents that directly activate RNCDs must be identified [143]. The results of the previous studies indicate that the expression levels of RNCD-related molecules in cancer patients differ depending on cancer types or individual genetic traits. The expression levels of MLKL were analyzed by immunohistochemistry in 613 patients with various cancers, including breast cancer, gastric cancer, cervical squamous cell carcinoma, ovarian cancer, colon cancer, and pancreatic adenocarcinoma; differences in the expression levels of MLKL were observed in patients with the same type of cancer, and low levels of MLKL expression were strongly associated with the advanced tumor stage and metastasis to the lymph nodes [93]. The expression level of GSDME was barely detectable or low in human breast cancer and melanoma cells, and low levels of GSDME in the cells were associated with resistance to anti-cancer therapies [144,145]. Moreover, in 89 gastric cancer patient tissues, gasdermin E gene was hypermethylated in approximately 50% of the samples, and 5-aza-2′deoxycytidine, a methyltrasferase inhibitor, suppressed the growth of primary gastric cancer cells [146]. Recently, cell–cell interactions have been reported to regulate ferroptosis. In human colorectal cancer cell lines, high density of the cells promoted the resistance to erastin, a ferroptosis inducer, via activation of the E-cadherin/Hippo pathway [147]. These results suggest that analysis of RNCD-related molecules in RNCD-resistant tumor tissues is important for effective anti-cancer therapy. The combination therapy for necroptosis induction has been extensively studied in acute myeloid leukemia (AML). The combination of emricasan, a stable and synergistic therapeutic effect in vitro and in vivo [148]. Cotreatment of ESCC with cisplatin and BI2536, a polo-like kinase 1 (PLK1) inhibitor, caused pyroptosis in vitro and in vivo [149]. Combined treatment of hypopharyngeal squamous carcinoma (HPSCC) cells with paclitaxel and RSL3 reduced the viability of the cells via ferroptosis [150]. Therefore, combination therapy adjusted to cancer properties may be an alternative therapeutic strategy for drug-resistant cancer types. Various drugs that directly induce RNCDs have been developed. HS-173, a potent PI3Kα inhibitor, induced necroptosis in human lung cancer cells in a RIP3-dependent manner [151]. A diazepin-quinazolin-amine derivative BIX-01294 enhanced GSDME-mediated pyroptosis in human gastric adenocarcinoma cell lines [152]. Talaroconvolutin isolated from the endophytic fungus Talaromyces wortmanni caused ferroptosis in human colorectal cancer in vitro and in vivo [153]. However, the number of clinical studies on the use of the induction of RNCDs in cancer patients is very limited, and additional investigation and clinical trials are required.
The induction of inflammatory responses in the tumor tissue may have beneficial or adverse effects on the host. Pro-inflammatory cytokines, including TNF-α, IL-1α, IL-1β, and IL-6, in the tumor microenvironment have been reported to promote tumor progression, metastasis, and invasion [154]. Additionally, the inflammatory responses caused by necroptosis, pyroptosis, and ferroptosis in the tumor cells can promote pro-tumor effects [101,155,156]. Therefore, there is no guarantee that the inflammatory response induced by RNCDs in the tumor microenvironment will be beneficial for the host. However, the antitumor immune response in the patient tumor tissues often depends on the immunogenicity of the tumor [157]. Induction of immunogenic cell death (ICD) alone is not sufficient to completely destroy the tumor microenvironment; however, improvement of immunogenicity in the tumor tissues is important for triggering dendritic cell-mediated anticancer immune responses [158]. Therefore, induction of ICD in the tumors may be an important strategy for anti-tumor therapy. Necroptosis, pyroptosis, and ferroptosis are the representative ICD mechanisms [137]. ICD in the tumor microenvironment plays an important role in the stimulation of the anti-tumor immune system. Immature dendritic cells that are chronically exposed to DAMPs released by ICD can mature and stimulate T cell responses that kill cancer cells via antigen presentation-dependent mechanisms [159]. Therefore, combination of RNCD inducers with appropriate immunotherapy may be an effective anticancer treatment strategy.
Beclin-1 is involved in the balance between apoptosis and autophagy [160]. The activity of caspase-8 is important for discrimination between apoptosis and necroptosis [161]. Caspase-3 activated in the process of apoptosis mediates pyroptosis [108]. Autophagy regulates ferroptosis through ferritin degradation [162]. These results indicate that the fate of cancer cells is not affected by only a single type of cell death. Therefore, tumor therapies can induce various types of cell death in tumor tissues, and cancer cells may activate resistance mechanisms to survive these challenges. Thus, effective suppression of cancer viability in tumor patients requires consideration of inherent characteristics of cancer to determine cell types that must be activated. In this review, the importance of RNCD for the induction of cell death of cancers resistant to apoptosis and autophagy inducers was discussed. Thus, although various trials investigating follow-up clinical outcomes of RNCD are needed, the use of RNCD inducers in patients with anticancer-drug resistant tumors may be a promising alternative therapeutic strategy.
Author Contributions
Conceptualization: Y.W. and Y.-J.J.; writing—original draft preparation: Y.W. and Y.-J.J.; investigation: Y.W. and H.-J.L.; funding: Y.M.J. and Y.-J.J.; supervision: Y.-J.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science, and Technology (2018R1D1A1B07049097), and by a Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (2020R1A6C101A195).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Copur, M.S. State of Cancer Research Around the Globe. Oncology (Williston Park) 2019, 33, 181–185. [Google Scholar] [PubMed]
- Parsa, N. Environmental factors inducing human cancers. Iran. J. Public Health 2012, 41, 1–9. [Google Scholar]
- Kerschbaum, E.; Nussler, V. Cancer Prevention with Nutrition and Lifestyle. Visc. Med. 2019, 35, 204–209. [Google Scholar] [CrossRef]
- Arruebo, M.; Vilaboa, N.; Saez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; Gonzalez-Fernandez, A. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef]
- Zimmerman, M.A.; Huang, Q.; Li, F.; Liu, X.; Li, C.Y. Cell death-stimulated cell proliferation: A tissue regeneration mechanism usurped by tumors during radiotherapy. Semin. Radiat. Oncol. 2013, 23, 288–295. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef]
- Todaro, M.; Lombardo, Y.; Francipane, M.G.; Alea, M.P.; Cammareri, P.; Iovino, F.; Di Stefano, A.B.; Di Bernardo, C.; Agrusa, A.; Condorelli, G.; et al. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 2008, 15, 762–772. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, S.; Halstensen, T.S. Increased interleukin-6 expression is associated with poor prognosis and acquired cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2016, 35, 3265–3274. [Google Scholar] [CrossRef][Green Version]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135.e10. [Google Scholar] [CrossRef]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brune, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Cho, J.H.; Lee, H.J.; Ko, H.J.; Yoon, B.I.; Choe, J.; Kim, K.C.; Hahn, T.W.; Han, J.A.; Choi, S.S.; Jung, Y.M.; et al. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget 2017, 8, 24932–24948. [Google Scholar] [CrossRef]
- Kang, S.-J.; Tak, J.-H.; Cho, J.-H.; Lee, H.-J.; Jung, Y.-J. Stimulation of the endosomal TLR pathway enhances autophagy-induced cell death in radiotherapy of breast cancer. Genes 2010, 32, 599–606. [Google Scholar] [CrossRef]
- Law, B.Y.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef]
- Maclean, K.H.; Dorsey, F.C.; Cleveland, J.L.; Kastan, M.B. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J. Clin. Investig. 2008, 118, 79–88. [Google Scholar] [CrossRef]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H.; et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell. Physiol. Biochem. 2018, 45, 54–66. [Google Scholar] [CrossRef]
- Liu, D.; Yang, Y.; Liu, Q.; Wang, J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 2011, 28, 105–111. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, X.; Zhou, X.; Wang, D.; Wang, L.; Li, C. Effect of autophagy inhibition on cell viability and cell cycle progression in MDAMB231 human breast cancer cells. Mol. Med. Rep. 2014, 10, 625–630. [Google Scholar] [CrossRef]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011, 25, 23–31. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Oncogene 2019, 38, 1920–1935. [Google Scholar] [CrossRef]
- Fulda, S. Regulation of cell death in cancer-possible implications for immunotherapy. Front. Oncol. 2013, 3, 29. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Lu, J.V.; Walsh, C.M. Programmed necrosis and autophagy in immune function. Immunol. Rev. 2012, 249, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Ros, U.; Pena-Blanco, A.; Hanggi, K.; Kunzendorf, U.; Krautwald, S.; Wong, W.W.; Garcia-Saez, A.J. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017, 19, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Gamero, A.M. Interferons as inducers of apoptosis in malignant cells. J. Interferon Cytokine Res. 2013, 33, 162–170. [Google Scholar] [CrossRef]
- Ndebele, K.; Gona, P.; Jin, T.G.; Benhaga, N.; Chalah, A.; Degli-Esposti, M.; Khosravi-Far, R. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induced mitochondrial pathway to apoptosis and caspase activation is potentiated by phospholipid scramblase-3. Apoptosis 2008, 13, 845–856. [Google Scholar] [CrossRef]
- Someda, M.; Kuroki, S.; Miyachi, H.; Tachibana, M.; Yonehara, S. Caspase-8, receptor-interacting protein kinase 1 (RIPK1), and RIPK3 regulate retinoic acid-induced cell differentiation and necroptosis. Cell Death Differ. 2020, 27, 1539–1553. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Komuves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cina, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Fiandalo, M.V.; Kyprianou, N. Caspase control: Protagonists of cancer cell apoptosis. Exp. Oncol. 2012, 34, 165–175. [Google Scholar] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef]
- Huang, D.; Zheng, X.; Wang, Z.A.; Chen, X.; He, W.T.; Zhang, Y.; Xu, J.G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Su, L.; Quade, B.; Wang, H.; Sun, L.; Wang, X.; Rizo, J. A plug release mechanism for membrane permeation by MLKL. Structure 2014, 22, 1489–1500. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef]
- Lim, Y.; Kumar, S. A single cut to pyroptosis. Oncotarget 2015, 6, 36926–36927. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Brydges, S.D.; Broderick, L.; McGeough, M.D.; Pena, C.A.; Mueller, J.L.; Hoffman, H.M. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J. Clin. Investig. 2013, 123, 4695–4705. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Dorfleutner, A.; Chu, L.; Stehlik, C. Inhibiting the inflammasome: One domain at a time. Immunol. Rev. 2015, 265, 205–216. [Google Scholar] [CrossRef]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef]
- Hafner-Bratkovic, I.; Susjan, P.; Lainscek, D.; Tapia-Abellan, A.; Cerovic, K.; Kadunc, L.; Angosto-Bazarra, D.; Pelegrin, P.; Jerala, R. NLRP3 lacking the leucine-rich repeat domain can be fully activated via the canonical inflammasome pathway. Nat. Commun. 2018, 9, 5182. [Google Scholar] [CrossRef]
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 2009, 182, 3173–3182. [Google Scholar] [CrossRef]
- Lu, A.; Li, Y.; Schmidt, F.I.; Yin, Q.; Chen, S.; Fu, T.M.; Tong, A.B.; Ploegh, H.L.; Mao, Y.; Wu, H. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat. Struct. Mol. Biol. 2016, 23, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shi, J.; Shao, F. Inflammatory Caspases: Activation and Cleavage of Gasdermin-D In Vitro and During Pyroptosis. Methods Mol. Biol. 2018, 1714, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 2019, 51, 43–49.e4. [Google Scholar] [CrossRef] [PubMed]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Nunez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–4975. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Hirayama, T.; Oomoto, I.; Wang, D.O.; Nagasawa, H. Fe(II) Ion Release during Endocytotic Uptake of Iron Visualized by a Membrane-Anchoring Fe(II) Fluorescent Probe. ACS Chem. Biol. 2018, 13, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thevenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C. Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci. Rep. 2016, 6, 30033. [Google Scholar] [CrossRef]
- Nehs, M.A.; Lin, C.I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D., Jr.; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, M.; He, S.; Luo, Y.; Zhao, Y.; Cheng, J.; Gong, Y.; Xie, J.; Wang, Y.; Hu, B.; et al. Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 461. [Google Scholar] [CrossRef]
- Clark, D.W.; Palle, K. Aldehyde dehydrogenases in cancer stem cells: Potential as therapeutic targets. Ann. Transl. Med. 2016, 4, 518. [Google Scholar] [CrossRef]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e6. [Google Scholar] [CrossRef] [PubMed]
- Akara-Amornthum, P.; Lomphithak, T.; Choksi, S.; Tohtong, R.; Jitkaew, S. Key necroptotic proteins are required for Smac mimetic-mediated sensitization of cholangiocarcinoma cells to TNF-alpha and chemotherapeutic gemcitabine-induced necroptosis. PLoS ONE 2020, 15, e0227454. [Google Scholar] [CrossRef] [PubMed]
- Dunai, Z.A.; Imre, G.; Barna, G.; Korcsmaros, T.; Petak, I.; Bauer, P.I.; Mihalik, R. Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PLoS ONE 2012, 7, e41945. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. Regulation of RIPK3- and RHIM-dependent Necroptosis by the Proteasome. J. Biol. Chem. 2016, 291, 5948–5959. [Google Scholar] [CrossRef] [PubMed]
- Oliver Metzig, M.; Fuchs, D.; Tagscherer, K.E.; Grone, H.J.; Schirmacher, P.; Roth, W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene 2016, 35, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Ma, Y.; Wang, H.; Dai, Y.; Chen, C.; Liu, Y.; Jing, L.; Sun, X. Resibufogenin suppresses colorectal cancer growth and metastasis through RIP3-mediated necroptosis. J. Transl. Med. 2018, 16, 201. [Google Scholar] [CrossRef]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar] [CrossRef]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Orlowski, G.M.; Chan, F.K. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015, 6, e1636. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, W.; Xu, X.; Li, B.; He, W.; Padilla, M.T.; Jang, J.H.; Nyunoya, T.; Amin, S.; Wang, X.; et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis 2013, 34, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Marinis, J.M.; Lich, J.D.; Berger, S.B.; Chirala, A.; Cox, J.A.; Eidam, P.M.; Finger, J.N.; Gough, P.J.; Jeong, J.U.; et al. Identification of a RIP1 Kinase Inhibitor Clinical Candidate (GSK3145095) for the Treatment of Pancreatic Cancer. ACS Med. Chem. Lett. 2019, 10, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Shi, D.; Lv, X.; Chen, S.; Huang, Q.; Xie, M.; Shao, Z. Prognostic and clinicopathological significance of MLKL expression in cancer patients: A meta-analysis. BMC Cancer 2018, 18, 736. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Peng, K.; Liu, Y.; Xiong, J.; Zhu, F.F. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco. Targets Ther. 2013, 6, 1539–1543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ramos-Junior, E.S.; Morandini, A.C. Gasdermin: A new player to the inflammasome game. Biomed. J. 2017, 40, 313–316. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell. Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef]
- Li, L.; Li, Y.; Bai, Y. Role of GSDMB in Pyroptosis and Cancer. Cancer Manag. Res. 2020, 12, 3033–3043. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Tamura, M.; Tanaka, S.; Fujii, T.; Aoki, A.; Komiyama, H.; Ezawa, K.; Sumiyama, K.; Sagai, T.; Shiroishi, T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 2007, 89, 618–629. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-”host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Watabe, K.; Ito, A.; Asada, H.; Endo, Y.; Kobayashi, T.; Nakamoto, K.; Itami, S.; Takao, S.; Shinomura, Y.; Aikou, T.; et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn. J. Cancer Res. 2001, 92, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xu, Z.; Chen, X.; Wang, X.; Zeng, S.; Qian, L.; Yang, X.; Ou, C.; Lin, W.; Gong, Z.; et al. Overexpression of GSDMC is a prognostic factor for predicting a poor outcome in lung adenocarcinoma. Mol. Med. Rep. 2020, 21, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qiu, X.; Xi, G.; Liu, H.; Zhang, F.; Lv, T.; Song, Y. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 2018, 40, 1971–1984. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, P.Y.; Bao, W.; Chen, S.J.; Wu, F.S.; Zhu, P.Y. Hydrogen inhibits endometrial cancer growth via a ROS/NLRP3/caspase-1/GSDMD-mediated pyroptotic pathway. BMC Cancer 2020, 20, 28. [Google Scholar] [CrossRef]
- Wang, L.; Li, K.; Lin, X.; Yao, Z.; Wang, S.; Xiong, X.; Ning, Z.; Wang, J.; Xu, X.; Jiang, Y.; et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 2019, 450, 22–31. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, J.; Mao, L.; Guo, Y.; Hao, Y.; Deng, Y.; Han, X.; Li, Q.; Liao, W.; Yuan, M. Nobiletin Triggers Reactive Oxygen Species-Mediated Pyroptosis through Regulating Autophagy in Ovarian Cancer Cells. J. Agric. Food Chem. 2020, 68, 1326–1336. [Google Scholar] [CrossRef]
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 281. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(-) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox. Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. mTOR-Mediated Antioxidant Activation in Solid Tumor Radioresistance. J. Oncol. 2019, 2019, 5956867. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Buha, A.; Telkoparan-Akillilar, P.; Cevik, D.; Kouretas, D.; Veskoukis, A.; Skaperda, Z.; Tsatsakis, A.; Wallace, D.; Suzen, S.; et al. Potential Applications of NRF2 Modulators in Cancer Therapy. Antioxidants 2020, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Z.; Miao, Y.; Liu, Z.; Chen, F.; Hu, X.; Feng, J.; Chen, H.; Liu, P.; Zhang, H. Radiosensitizing effect of deferoxamine on human glioma cells. Int. J. Radiat. Res. 2019, 17, 325–333. [Google Scholar] [CrossRef]
- Shibata, Y.; Yasui, H.; Higashikawa, K.; Miyamoto, N.; Kuge, Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 2019, 14, e0225931. [Google Scholar] [CrossRef]
- Pan, X.; Lin, Z.; Jiang, D.; Yu, Y.; Yang, D.; Zhou, H.; Zhan, D.; Liu, S.; Peng, G.; Chen, Z.; et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol. Lett. 2019, 17, 3001–3008. [Google Scholar] [CrossRef]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Li, Y.; Yan, H.; Xu, X.; Liu, H.; Wu, C.; Zhao, L. Erastin/sorafenib induces cisplatin-resistant non-small cell lung cancer cell ferroptosis through inhibition of the Nrf2/xCT pathway. Oncol. Lett. 2020, 19, 323–333. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, K.; Huang, X.; Zhao, C.; Mei, Y.; Li, X.; Jiao, L.; Yang, H. lncRNA SLC7A11-AS1 Promotes Chemoresistance by Blocking SCF(beta-TRCP)-Mediated Degradation of NRF2 in Pancreatic Cancer. Mol. Ther. Nucleic Acids 2020, 19, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, Y.; Ling, J.; Zhuo, W.; Yu, Z.; Luo, Y.; Zhu, Y. Overexpression of SLC7A11: A novel oncogene and an indicator of unfavorable prognosis for liver carcinoma. Future Oncol. 2018, 14, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Qian, C.; Cao, H.; Wang, Z.; Luo, T.; Liang, C. Upregulation of the solute carrier family 7 genes is indicative of poor prognosis in papillary thyroid carcinoma. World J. Surg. Oncol. 2018, 16, 235. [Google Scholar] [CrossRef]
- Galvan, I.; Inacio, A.; Danino, M.; Corbi-Llopis, R.; Monserrat, M.T.; Bernabeu-Wittel, J. High SLC7A11 expression in normal skin of melanoma patients. Cancer Epidemiol. 2019, 62, 101582. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Buckingham, S.C.; Campbell, S.L.; Robel, S.; Holt, K.T.; Ogunrinu-Babarinde, T.; Warren, P.P.; White, D.M.; Reid, M.A.; Eschbacher, J.M.; et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl. Med. 2015, 7, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhang, M.; Wu, J.; Chen, F.; Wang, Y.; Ma, Y.; Dai, Z. Glioma progression is suppressed by Naringenin and APO2L combination therapy via the activation of apoptosis in vitro and in vivo. Investig. New Drugs 2020. [Google Scholar] [CrossRef]
- Guo, R.; Li, Y.; Xue, Y.; Chen, Y.; Li, J.; Deng, X.; Su, J.; Liu, Y.; Sun, L. SIRT3 increases cisplatin sensitivity of small-cell lung cancer through apoptosis. Gene 2020, 745, 144629. [Google Scholar] [CrossRef]
- Wong, V.K.; Li, T.; Law, B.Y.; Ma, E.D.; Yip, N.C.; Michelangeli, F.; Law, C.K.; Zhang, M.M.; Lam, K.Y.; Chan, P.L.; et al. Saikosaponin-d, a novel SERCA inhibitor, induces autophagic cell death in apoptosis-defective cells. Cell Death Dis. 2013, 4, e720. [Google Scholar] [CrossRef]
- Muhammad, N.; Steele, R.; Isbell, T.S.; Philips, N.; Ray, R.B. Bitter melon extract inhibits breast cancer growth in preclinical model by inducing autophagic cell death. Oncotarget 2017, 8, 66226–66236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kim, H.S.; Jin, T.; Woo, J.; Piao, Y.J.; Moon, W.K. Near-infrared photothermal therapy using anti-EGFR-gold nanorod conjugates for triple negative breast cancer. Oncotarget 2017, 8, 86566–86575. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O’Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Regulated necrotic cell death: The passive aggressive side of Bax and Bak. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Philipp, S.; Sosna, J.; Plenge, J.; Kalthoff, H.; Adam, D. Homoharringtonine, a clinically approved anti-leukemia drug, sensitizes tumor cells for TRAIL-induced necroptosis. Cell. Commun. Signal. 2015, 13, 25. [Google Scholar] [CrossRef]
- Wang, F.; Liu, W.; Ning, J.; Wang, J.; Lang, Y.; Jin, X.; Zhu, K.; Wang, X.; Li, X.; Yang, F.; et al. Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis. Int. J. Biol. Sci. 2018, 14, 406–417. [Google Scholar] [CrossRef]
- Higgins, M.J.; Prowell, T.M.; Blackford, A.L.; Byrne, C.; Khouri, N.F.; Slater, S.A.; Jeter, S.C.; Armstrong, D.K.; Davidson, N.E.; Emens, L.A.; et al. A short-term biomarker modulation study of simvastatin in women at increased risk of a new breast cancer. Breast Cancer Res. Treat. 2012, 131, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Shen, Y.C.; Wu, C.Y.; Tsai, Y.Y.; Yang, Y.H.; Lin, Y.Y.; Kuan, F.C.; Lu, C.N.; Chang, G.H.; Tsai, M.S.; et al. Danshen Improves Survival of Patients with Breast Cancer and Dihydroisotanshinone I Induces Ferroptosis and Apoptosis of Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1226. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell. Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Gao, W.Q.; Shi, X.Y.; Ding, J.J.; Liu, W.; He, H.B.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Lage, H.; Helmbach, H.; Grottke, C.; Dietel, M.; Schadendorf, D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001, 494, 54–59. [Google Scholar] [CrossRef]
- Akino, K.; Toyota, M.; Suzuki, H.; Imai, T.; Maruyama, R.; Kusano, M.; Nishikawa, N.; Watanabe, Y.; Sasaki, Y.; Abe, T.; et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007, 98, 88–95. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, F.; Li, Y.; Qian, W.; Ding, W.; Ye, X. Bypassing drug resistance by triggering necroptosis: Recent advances in mechanisms and its therapeutic exploitation in leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 310. [Google Scholar] [CrossRef]
- Pezuk, J.A. Pyroptosis in combinatorial treatment to improve cancer patients’ outcome, is that what we want? Ebiomedicine 2019, 41, 17–18. [Google Scholar] [CrossRef]
- Wu, Y.N.; Yu, C.C.; Luo, M.; Cen, C.; Qiu, J.L.; Zhang, S.Z.; Hu, K.M. Ferroptosis in Cancer Treatment: Another Way to Rome. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, K.H.; Kim, S.J.; Yoon, Y.C.; Yan, H.H.; Fang, Z.; Lee, J.E.; Lim, J.H.; Mah, S.; Hong, S.; et al. HS-173 as a novel inducer of RIP3-dependent necroptosis in lung cancer. Cancer. Lett. 2019, 444, 94–104. [Google Scholar] [CrossRef]
- Deng, B.B.; Jiao, B.P.; Liu, Y.J.; Li, Y.R.; Wang, G.J. BIX-01294 enhanced chemotherapy effect in gastric cancer by inducing GSDME-mediated pyroptosis. Cell. Biol. Int. 2020, 44, 1890–1899. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, S.; Li, C.; Ai, Z.; Shen, W.; Ren, W.; Yang, X. Discovery of a novel ferroptosis inducer-talaroconvolutin A-killing colorectal cancer cells in vitro and in vivo. Cell Death Dis. 2020, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Inflammation: A driving force speeds cancer metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Qiao, M.; Zhao, C.; Deng, J.; Li, X.; Zhou, C. Targeting ferroptosis for cancer therapy: Exploring novel strategies from its mechanisms and role in cancers. Transl. Lung Cancer Res. 2020, 9, 1569–1584. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef]
- Montico, B.; Nigro, A.; Casolaro, V.; Dal Col, J. Immunogenic Apoptosis as a Novel Tool for Anticancer Vaccine Development. Int. J. Mol. Sci. 2018, 19, 594. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).