Role of TGF-β in Skin Chronic Wounds: A Keratinocyte Perspective


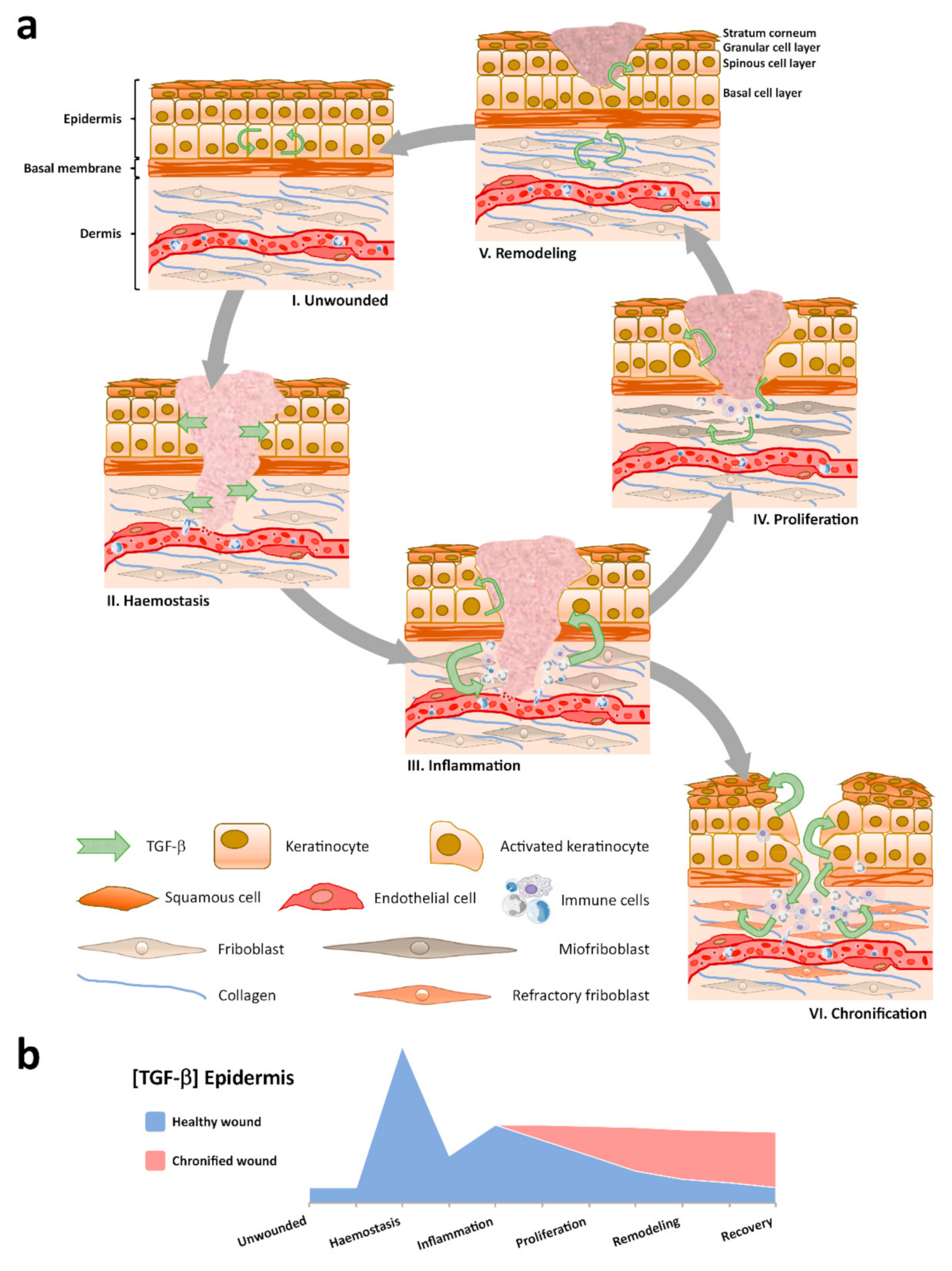{kind=link}
Abstract
:1. Introduction
2. TGF-β Signaling: A Context Dependent Mechanism
3. TGF-β Expression in Healthy and Healing Epidermis
4. Paradoxical Role of TGF-β in Inflammation
5. Animal Models: Relevant but Potentially Misleading Evidence
6. TGF-β in Human Chronic Wounds
7. Characteristic TGF-β Responses of Skin Cells
8. Reaction to Exacerbated TGF-β Levels in Wound Healing
9. Epithelial to Mesenchymal Transition During Wound Healing and the Involvement of TGF-β
10. Future Directions
11. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the Keratinocyte Activation Cycle. J. Investig. Dermatol. 2001, 116, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, G.; Bernabé-García, Á.; Moraleda, J.M.; Nicolás, F.J. Amniotic membrane application for the healing of chronic wounds and ulcers. Placenta 2017, 59, 146–153. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, S.; Ferguson, M.W. Transforming growth factor βs and wound healing. Int. J. Biochem. Cell Biol. 1997, 29, 63–78. [Google Scholar] [CrossRef]
- Bielefeld, K.A.; Amini-Nik, S.; Alman, B.A. Cutaneous wound healing: recruiting developmental pathways for regeneration. Cell Mol. Life Sci. 2013, 70, 2059–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-G.; Massagué, J.; Hata, A.; Lo, R.S.; Wotton, D.; Shi, Y.; Pavletich, N. Determinants of specificity in TGF-β signal transduction. Genes Dev. 1998, 12, 2144–2152. [Google Scholar] [CrossRef] [Green Version]
- Kiritsi, D.; Nyström, A. The role of TGFβ in wound healing pathologies. Mech. Ageing Dev. 2018, 172, 51–58. [Google Scholar] [CrossRef]
- Hinck, A.P.; Archer, S.J.; Qian, S.W.; Roberts, A.B.; Sporn, M.B.; Weatherbee, J.A.; Tsang, M.L.-S.; Lucas, R.; Zhang, B.-L.; Wenker, J.; et al. Transforming Growth Factor β1: Three-Dimensional Structure in Solution and Comparison with the X-ray Structure of Transforming Growth Factor β2. Biochemistry 1996, 35, 8517–8534. [Google Scholar] [CrossRef]
- Mittl, P.R.E.; Priestle, J.P.; Cox, D.A.; McMaster, G.; Cerletti, N.; Grütter, M.G. The crystal structure of TGF-β3 and comparison to TGF-β2: Implications for receptor binding. Protein Sci. 1996, 5, 1261–1271. [Google Scholar] [CrossRef]
- Cheifetz, S.; Hernandez, H.; Laiho, M.; Dijke, P.T.; Iwata, K.K.; Massagué, J. Distinct transforming growth factor-β (TGF-β) receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J. Biol. Chem. 1990, 265, 20533–20538. [Google Scholar]
- Baardsnes, J.; Hinck, C.S.; Hinck, A.P.; O’Connor-McCourt, M.D. TβR-II Discriminates the High- and Low-Affinity TGF-β Isoforms via Two Hydrogen-Bonded Ion Pairs. Biochemistry 2009, 48, 2146–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheifetz, S.; Bassols, A.; Stanley, K.; Ohta, M.; Greenberger, J.; Massagué, J. Heterodimeric transforming growth factor beta. Biological properties and interaction with three types of cell surface receptors. J. Biol. Chem. 1988, 263, 10783–10789. [Google Scholar] [PubMed]
- Ogawa, Y.; Schmidt, D.K.; Dasch, J.R.; Chang, R.J.; Glaser, C.B. Purification and characterization of transforming growth factor-β2.3 and -β1.2 heterodimers from bovine bone. J. Biol. Chem. 1992, 267, 2325–2328. [Google Scholar] [PubMed]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFβ activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Soo, C.; Hu, F.-Y.; Zhang, X.; Wang, Y.; Beanes, S.R.; Lorenz, H.P.; Hedrick, M.H.; MacKool, R.J.; Plaas, A.; Kim, S.-J.; et al. Differential Expression of Fibromodulin, a Transforming Growth Factor-β Modulator, in Fetal Skin Development and Scarless Repair. Am. J. Pathol. 2000, 157, 423–433. [Google Scholar] [CrossRef]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix. Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [Green Version]
- Bruce, D.L.; Sapkota, G.P. Phosphatases in SMAD regulation. FEBS Lett. 2012, 586, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- García-Vizcaíno, E.M.; Liarte, S.; Alonso-Romero, J.L.; Nicolás, F.J. Sirt1 interaction with active Smad2 modulates transforming growth factor-β regulated transcription. Cell Commun. Signal. 2017, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.-H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landstrom, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Attisano, L. The TGFβ superfamily signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 47–63. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal integration in TGF-β, WNT, and Hippo pathways. F1000Prime Rep. 2013, 5. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Xi, Q. Crosstalk between TGF-β signaling and epigenome. Acta Biochim. Biophys. Sin. 2018, 50, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef] [Green Version]
- Kane, C.J.M.; Hebda, P.A.; Mansbridge, J.N.; Hanawalt, P.C. Direct evidence for spatial and temporal regulation of transforming growth factor β1 expression during cutaneous wound healing. J. Cell. Physiol. 1991, 148, 157–173. [Google Scholar] [CrossRef]
- Schmid, P.; Cox, D.; Bilbe, G.; McMaster, G.; Morrison, C.; Stähelin, H.; Lüscher, N.; Seiler, W. TGF-βS and TGF-β type II receptor in human epidermis: Differential expression in acute and chronic skin wounds. J. Pathol. 1993, 171, 191–197. [Google Scholar] [CrossRef]
- Jiang, C.K.; Tomić-Canić, M.; Lucas, D.J.; Simon, M.; Blumenberg, M. TGF beta promotes the basal phenotype of epidermal keratinocytes: transcriptional induction of K#5 and K#14 keratin genes. Growth Factors 1995, 12, 87–97. [Google Scholar] [PubMed]
- Ramírez, H.; Patel, S.B.; Pastar, I. The Role of TGFβ Signaling in Wound Epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, R.W.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Madlener, M.; Werner, S. Transforming Growth Factors 1, 2, and 3 and Their Receptors Are Differentially Regulated during Normal and Impaired Wound Healing. J. Biol. Chem. 1996, 271, 10188–10193. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Qiu, C.X.; Ludlow, A.; Ferguson, M.W.; Brunner, G. Active transforming growth factor-beta in wound repair: determination using a new assay. Am. J. Pathol. 1999, 154, 105–111. [Google Scholar] [CrossRef]
- Wang, X.-J.; Han, G.; Owens, P.; Siddiqui, Y.; Li, A.G. Role of TGFβ-mediated inflammation in cutaneous wound healing. J. Investig. Dermatol. Symp. Proc. 2006, 11, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Perspective Article: Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Valluru, M.; Staton, C.A.; Reed, M.W.R.; Brown, N.J. Transforming Growth Factor-β and Endoglin Signaling Orchestrate Wound Healing. Front. Physiol. 2011, 2, 89. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Mauviel, A. Transforming growth factor-beta signaling in skin: stromal to epithelial cross-talk. J. Investig. Dermatol. 2009, 129, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Le Poole, B. Keratinocytes suppress transforming growth factor-β1 expression by fibroblasts in cultured skin substitutes. Br. J. Dermatol. 1999, 140, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Li, A.G.; Lu, S.-L.; Han, G.; Hoot, K.E.; Wang, X.-J. Role of TGFβ in skin inflammation and carcinogenesis. Mol. Carcinog. 2006, 45, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, L.; Levine, A.D. TGF-β Inhibits IL-2 Production and Promotes Cell Cycle Arrest in TCR-Activated Effector/Memory T Cells in the Presence of Sustained TCR Signal Transduction. J. Immunol. 2008, 180, 1490–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutcher, I.; Donkor, M.K.; Ma, Q.; Rudensky, A.Y.; Flavell, R.A.; Li, M.O. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity 2011, 34, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Li, F.; Singh, T.P.; Wolf, P.; Wang, X.-J. The Pro-inflammatory Role of TGFβ1: A Paradox? Int. J. Biol. Sci. 2012, 8, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Alexander, V.; Vijayachandra, K.; Bhogte, E.; Diamond, I.; Glick, A. Conditional epidermal expression of TGFβ1 blocks neonatal lethality but causes a reversible hyperplasia and alopecia. Proc. Natl. Acad. Sci. USA 2001, 98, 9139–9144. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.; Ghahary, A.; Demare, J.; Yang, L.; Iwashina, T.; Scott, P.G.; Tredget, E.E. Development, characterization, and wound healing of the keratin 14 promoted transforming growth factor-beta1 transgenic mouse. Wound Repair Regen. 2002, 10, 177–187. [Google Scholar] [CrossRef]
- Li, A.G.; Wang, D.; Feng, X.-H.; Wang, X.-J. Latent TGFβ1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J. 2004, 23, 1770–1781. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, G.S.; Yang, X.; Glick, A.B.; Weinstein, M.; Letterio, J.J.; Mizel, D.E.; Anzano, M.; Greenwell-Wild, T.; Wahl, S.M.; Deng, C.; et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nature 1999, 1, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Letterio, J.J.; Lechleider, R.J.; Chen, L.; Hayman, R.; Gu, H.; Roberts, A.B.; Deng, C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999, 18, 1280–1291. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Williams, C.A.; Salter, K.; Garl, P.J.; Li, A.G.; Wang, X.J. A role for TGFβ signaling in the pathogenesis of psoriasis. J. Invest. Dermatol. 2010, 130, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sferra, R.; Fargnoli, M.C.; Corbelli, E.; Pellegrini, C.; Peris, K.; Gaudio, E.; Vetuschi, A. Immunopathogenesis of psoriasis: a possible role of TGFβ/Smads pathway. Ital. J. Anat. Embryol. 2014, 119, 277–285. [Google Scholar] [PubMed]
- Zhang, Y.; Meng, X.-M.; Huang, X.-R.; Wang, X.-J.; Yang, L.; Lan, H.Y. Transforming growth factor-β1 mediates psoriasis-like lesions via a Smad3-dependent mechanism in mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 921–932. [Google Scholar] [CrossRef]
- Pastar, I.; Stojadinovic, O.; Yin, N.C.; Ramírez, H.; Nusbaum, A.G.; Sawaya, A.; Patel, S.B.; Khalid, L.; Isseroff, R.R.; Tomic-Canic, M. Epithelialization in Wound Healing: A Comprehensive Review. Adv. Wound Care 2014, 3, 445–464. [Google Scholar] [CrossRef] [Green Version]
- Finnson, K.W.; McLean, S.; Di Guglielmo, G.M.; Philip, A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv. Wound Care 2013, 2, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Mustoe, T.; Pierce, G.; Thomason, A.; Gramates, P.; Sporn, M.; Deuel, T. Accelerated healing of incisional wounds in rats induced by transforming growth factor-beta. Science 1987, 237, 1333–1336. [Google Scholar] [CrossRef]
- Pierce, G.F.; Mustoe, T.A.; Lingelbach, J.; Masakowski, V.R.; Gramates, P.; Deuel, T.F. Transforming growth factor beta reverses the glucocorticoid-induced wound-healing deficit in rats: possible regulation in macrophages by platelet-derived growth factor. Proc. Natl. Acad. Sci. USA 1989, 86, 2229–2233. [Google Scholar] [CrossRef] [Green Version]
- Salomon, G.D.; Kasid, A.; Bernstein, E.; Buresh, C.; Director, E.; A Norton, J. Gene expression in normal and doxorubicin-impaired wounds: importance of transforming growth factor-beta. Surgery 1990, 108, 318–322. [Google Scholar]
- Beck, L.S.; DeGuzman, L.; Lee, W.P.; Xu, Y.; A McFatridge, L.; Amento, E.P. TGF-beta 1 accelerates wound healing: reversal of steroid-impaired healing in rats and rabbits. Growth Factors 1991, 5, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Beck, L.S.; DeGuzman, L.; Lee, W.P.; Xu, Y.; Siegel, M.W.; Amento, E.P. One systemic administration of transforming growth factor-beta 1 reverses age- or glucocorticoid-impaired wound healing. J. Clin. Investig. 1993, 92, 2841–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowe, M.J.; Doetschman, T.; Greenhalgh, D.G. Delayed Wound Healing in Immunodeficient TGF-β1 Knockout Mice. J. Investig. Dermatol. 2000, 115, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.-Y.; Chesnoy, S.; Huang, L. Electroporatic Delivery of TGF-β1 Gene Works Synergistically with Electric Therapy to Enhance Diabetic Wound Healing in db/db Mice. J. Investig. Dermatol. 2004, 123, 791–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tredget, E.B.; Demare, J.; Chandran, G.; Tredget, E.E.; Yang, L.; Ghahary, A. Transforming growth factor-β and its effect on reepithelialization of partial-thickness ear wounds in transgenic mice. Wound Repair Regen. 2005, 13, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Saulis, A.S.; Liu, W.R.; Roy, N.K.; Chao, J.D.; Ledbetter, S.; Mustoe, T.A. The Temporal Effects of Anti-TGF-β1, 2, and 3 Monoclonal Antibody on Wound Healing and Hypertrophic Scar Formation. J. Am. Coll. Surg. 2005, 201, 391–397. [Google Scholar] [CrossRef] [PubMed]
- El Gazaerly, H.; Elbardisey, D.M.; Eltokhy, H.M. Effect of Transforming Growth Factor Beta 1 on Wound Healing in Induced Diabetic Rats. Int. J. Heal. Sci. 2013, 7, 160–172. [Google Scholar] [CrossRef]
- Wu, L.; Xia, Y.P.; Roth, S.I.; Gruskin, E.; Mustoe, T.A. Transforming growth factor-beta1 fails to stimulate wound healing and impairs its signal transduction in an aged ischemic ulcer model: importance of oxygen and age. Am. J. Pathol. 1999, 154, 301–309. [Google Scholar] [CrossRef]
- Koch, R.M.; Roche, N.S.; Parks, W.T.; Ashcroft, G.S.; Letterio, J.J.; Roberts, A.B. Incisional wound healing in transforming growth factor-β1 null mice. Wound Repair Regen. 2000, 8, 179–191. [Google Scholar] [CrossRef]
- Grose, R.; Werner, S. Wound-Healing Studies in Transgenic and Knockout Mice. Mol. Biotechnol. 2004, 28, 147–166. [Google Scholar] [CrossRef]
- Robson, M.C.; Phillip, L.G.; Cooper, D.M.; Lyle, W.G.; Robson, L.E.; Odom, L.; Hill, D.P.; Hanham, A.F.; Ksander, G.A. Safety and effect of transforming growth factor-β2 for treatment of venous stasis ulcers. Wound Repair Regen. 1995, 3, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.P.; Griffiths, G.D.; Schor, A.M.; Leese, G.P.; Schor, S.L. Growth factors in the treatment of diabetic foot ulcers. BJS 2003, 90, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.W.; Arany, P.R.; Philip, A. Transforming Growth Factor Beta Signaling in Cutaneous Wound Healing: Lessons Learned from Animal Studies. Adv. Wound Care 2013, 2, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittie, L.; Sachs, D.L.; Orringer, J.S.; Voorhees, J.J.; Fisher, G.J. Eccrine sweat glands are major contributors to reepithelialization of human wounds. Am. J. Pathol. 2013, 182, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, M.-L.; Escario, E.; Poblet, E.; Sánchez, D.; Buchón, F.-F.; Izeta, A.; Jimenez, F. Hair follicle–containing punch grafts accelerate chronic ulcer healing: A randomized controlled trial. J. Am. Acad. Dermatol. 2016, 75, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Poblet, E.; Jimenez, F.; Escario-Travesedo, E.; Hardman, J.A.; Hernandez-Hernandez, I.; Agudo-Mena, J.L.; Cabrera-Galvan, J.J.; Nicu, C.; Paus, R. Eccrine sweat glands associate with the human hair follicle within a defined compartment of dermal white adipose tissue. Br. J. Dermatol. 2018, 178, 1163–1172. [Google Scholar] [CrossRef]
- Hibino, T.; Nishiyama, T. Role of TGF-β2 in the human hair cycle. J. Dermatol. Sci. 2004, 35, 9–18. [Google Scholar] [CrossRef]
- Jamora, C.; Lee, P.; Kocieniewski, P.; Azhar, M.; Hosokawa, R.; Chai, Y.; Fuchs, E. A signaling pathway involving TGF-β2 and snail in hair follicle morphogenesis. PLoS Biol. 2005, 3, e11. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Aoi, N.; Yamauchi, Y.; Sato, T.; Suga, H.; Eto, H.; Kato, H.; Tabata, Y.; Yoshimura, K. TGF-β2 is specifically expressed in human dermal papilla cells and modulates hair folliculogenesis. J. Cell. Mol. Med. 2009, 13, 4643–4656. [Google Scholar] [CrossRef] [Green Version]
- Oshimori, N.; Fuchs, E. Paracrine TGF-β signaling counterbalances BMP-mediated repression in hair follicle stem cell activation. Cell Stem Cell 2012, 10, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Cowin, A.J.; Hatzirodos, N.; Holding, C.A.; Dunaiski, V.; Rayner, T.E.; Harries, R.H.; Fitridge, R.; Cooter, R.D.; Schultz, G.S.; Belford, D.A. Effect of Healing on the Expression of Transforming Growth Factor βs and their Receptors in Chronic Venous Leg Ulcers. J. Investig. Dermatol. 2001, 117, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Jude, E.B.; Blakytny, R.; Bulmer, J.; Boulton, A.J.M.; Ferguson, M.W.J. Transforming growth factor-beta 1, 2, 3 and receptor type I and II in diabetic foot ulcers. Diabet. Med. 2002, 19, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Faler, B.J.; A Macsata, R.; Plummer, D.; Mishra, L.; Sidawy, A.N. Transforming growth factor-beta and wound healing. Perspect. Vasc. Surg. Endovasc. Ther. 2006, 18, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Rosenbloom, J.; A Jimenez, S. Transforming growth factor β (TGFβ) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Cordeiro, M.F.; Bhattacharya, S.S.; Schultz, G.S.; Khaw, P.T. TGF-β1, -β2, and -β3 in vitro: biphasic effects on Tenon’s fibroblast contraction, proliferation, and migration. Invest. Ophthalmol. Vis. Sci. 2000, 41, 756–763. [Google Scholar]
- Desmoulière, A. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ferrer, M.; Afshar-Sherif, A.-R.; Uwamariya, C.; De Crombrugghe, B.; Davidson, J.M.; Bhowmick, N.A. Dermal Transforming Growth Factor-β Responsiveness Mediates Wound Contraction and Epithelial Closure. Am. J. Pathol. 2010, 176, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 108. [Google Scholar]
- Murata, H.; Zhou, L.; Ochoa, S.; Hasan, A.; Badiavas, E.; Falanga, V. TGF-β3 Stimulates and Regulates Collagen Synthesis Through TGF-β1-Dependent and Independent Mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, R.L. The evidence for the role of transforming growth factor-beta in the formation of abnormal scarring. Int. Wound J. 2011, 8, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-C.; Kim, H.T.; Park, S.H.; Cha, J.-S.; Yufit, T.; Kim, S.-J.; Falanga, V. Fibroblasts from chronic wounds show altered TGF-β-signaling and decreased TGF-β Type II Receptor expression. J. Cell. Physiol. 2003, 195, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.W.; Kim, I.S. TGF-beta1 enhances betaig-h3-mediated keratinocyte cell migration through the α3β1 integrin and PI3K. J. Cell Biochem. 2004, 92, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Landström, M.; Moustakas, A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J. Cell. Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef]
- Cho, H.-R.; Hong, S.-B.; Kim, Y.I.; Lee, J.-W.; Kim, N.-I. Differential Expression of TGF-β Isoforms During Differentiation of HaCaT Human Keratinocyte Cells: Implication for the Separate Role in Epidermal Differentiation. J. Korean Med Sci. 2004, 19, 853–858. [Google Scholar] [CrossRef]
- Duan, D.; Derynck, R. Transforming growth factor-β (TGF-β)-induced up-regulation of TGF-β receptors at the cell surface amplifies the TGF-β response. J. Biol. Chem. 2019, 294, 8490–8504. [Google Scholar] [CrossRef]
- Park, S.; Gonzalez, D.G.; Guirao, B.; Boucher, J.D.; Cockburn, K.; Marsh, E.D.; Mesa, K.R.; Brown, S.; Rompolas, P.; Haberman, A.M.; et al. Tissue-scale coordination of cellular behaviour promotes epidermal wound repair in live mice. Nature 2017, 19, 155–163. [Google Scholar]
- Aragona, M.; Dekoninck, S.; Rulands, S.; Lenglez, S.; Mascré, G.; Simons, B.D.; Blanpain, C. Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat. Commun. 2017, 8, 14684. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Gao, K.; Häkkinen, L.; Larjava, H.S. Mice lacking β6 integrin in skin show accelerated wound repair in dexamethasone impaired wound healing model. Wound Repair Regen. 2009, 17, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Sellheyer, K.; Bickenbach, J.R.; Rothnagel, J.A.; Bundman, D.; Longley, M.A.; Krieg, T.; Roche, N.S.; Roberts, A.B.; Roop, D.R. Inhibition of skin development by overexpression of transforming growth factor beta 1 in the epidermis of transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 5237–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chan, T.; Demare, J.; Iwashina, T.; Ghahary, A.; Scott, P.G.; Tredget, E.E. Healing of Burn Wounds in Transgenic Mice Overexpressing Transforming Growth Factor-β1 in the Epidermis. Am. J. Pathol. 2001, 159, 2147–2157. [Google Scholar] [CrossRef]
- Brown, R.L.; Ormsby, I.; Doetschman, T.C.; Greenhalgh, D.G. Wound healing in the transforming growth factor-beta1-deficient mouse. Wound Repair Regen. 1995, 3, 25–36. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Dijke, P.T. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Futur. Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [Green Version]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Naber, H.P.; Drabsch, Y.; Snaar-Jagalska, B.E.; Dijke, P.T.; Van Laar, T. Snail and Slug, key regulators of TGF-β-induced EMT, are sufficient for the induction of single-cell invasion. Biochem. Biophys. Res. Commun. 2013, 435, 58–63. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haensel, D.; Dai, X. Epithelial-to-mesenchymal transition in cutaneous wound healing: Where we are and where we are heading. Dev. Dyn. 2018, 247, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselle, P.; Braye, F.; Dayan, G. Re-epithelialization of adult skin wounds: Cellular mechanisms and therapeutic strategies. Adv. Drug Deliv. Rev. 2018, 146, 344–365. [Google Scholar] [CrossRef]
- Weber, C.E.; Li, N.Y.; Wai, P.Y.; Kuo, P.C. Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound healing and tissue remodeling after injury. J. Burn. Care Res. 2012, 33, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Haensel, D.; Gutierrez, G.; Du, H.; Dai, X.; Nie, Q. Intermediate cell states in epithelial-to-mesenchymal transition. Phys. Biol. 2019, 16, 021001. [Google Scholar] [CrossRef]
- Sader, F.; Denis, J.-F.; Laref, H.; Roy, S. Epithelial to mesenchymal transition is mediated by both TGF-β canonical and non-canonical signaling during axolotl limb regeneration. Sci. Rep. 2019, 9, 1144. [Google Scholar] [CrossRef]
- Lévesque, M.; Gatien, S.; Finnson, K.; Desmeules, S.; Villiard, É.; Pilote, M.; Philip, A.; Roy, S. Transforming Growth Factor: β Signaling Is Essential for Limb Regeneration in Axolotls. PLoS ONE 2007, 2, e1227. [Google Scholar]
- Ho, D.M.; Whitman, M. TGF-beta signaling is required for multiple processes during Xenopus tail regeneration. Dev. Biol. 2008, 315, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chablais, F.; Jazwinska, A. The regenerative capacity of the zebrafish heart is dependent on TGFβ signaling. Development 2012, 139, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denis, J.-F.; Levesque, M.; Tran, S.D.; Camarda, A.-J.; Roy, S. Axolotl as a Model to Study Scarless Wound Healing in Vertebrates: Role of the Transforming Growth Factor Beta Signaling Pathway. Adv. Wound Care 2013, 2, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, M.; Villiard, É.; Roy, S. Skin wound healing in axolotls: A scarless process. J. Exp. Zool. Part B Mol. Dev. Evol. 2010, 314, 684–697. [Google Scholar] [CrossRef]
- Godwin, J.W.; Rosenthal, N. Scar-free wound healing and regeneration in amphibians: Immunological influences on regenerative success. Differentiation 2014, 87, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Larson, B.J.; Longaker, M.T.; Lorenz, H.P. Scarless fetal wound healing: a basic science review. Plast. Reconstr. Surg. 2010, 126, 1172–1180. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, M.W.J.; O’Kane, S. Scar-free healing: from embryonic mechanisms to adult therapeutic intervention. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Schrementi, M.E.; Ferreira, A.M.; Zender, C.; DiPietro, L.A. Site-specific production of TGF-? in oral mucosal and cutaneous wounds. Wound Repair Regen. 2008, 16, 80–86. [Google Scholar] [CrossRef]
- Zhao, J.L.J.; Liu, J.Z.J. The Expression Level of TGF-β1, TGF-β3 and VEGF in Transplanted Oral Mucosal and Cutaneous Wounds. Clin. Microbiol. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.-L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [Green Version]
- Bucur, M.; Dinca, O.; Vladan, C.; Popp, C.; Nichita, L.; Cioplea, M.; Stînga, P.; Mustatea, P.; Zurac, S.; Ionescu, E. Variation in Expression of Inflammation-Related Signaling Molecules with Profibrotic and Antifibrotic Effects in Cutaneous and Oral Mucosa Scars. J. Immunol. Res. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- So, K.; McGrouther, D.A.; Bush, J.A.; Durani, P.; Taylor, L.; Skotny, G.; Mason, T.; Metcalfe, A.; Oʼkane, S.; Ferguson, M.W.J. Avotermin for Scar Improvement following Scar Revision Surgery: A Randomized, Double-Blind, Within-Patient, Placebo-Controlled, Phase II Clinical Trial. Plast. Reconstr. Surg. 2011, 128, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.; Lorenz, H.; Meuli, M.; Lin, R.Y.; Adzick, N. A model of scarless human fetal wound repair is deficient in transforming growth factor beta. J. Pediatr. Surg. 1995, 30, 198–203. [Google Scholar] [CrossRef]
- Caley, M.P.; Martins, V.L.; O’Toole, E.A. Metalloproteinases and Wound Healing. Adv. Wound Care 2015, 4, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-Hoff, M.H.; Ehrhart, E.J.; Kalia, M.; Jirtle, R.; Flanders, K.; Tsang, M.L. Immunohistochemical detection of active transforming growth factor-beta in situ using engineered tissue. Am. J. Pathol. 1995, 147, 1228–1237. [Google Scholar]
- Ewan, K.B.; Shyamala, G.; Ravani, S.A.; Tang, Y.; Akhurst, R.; Wakefield, L.; Barcellos-Hoff, M.H. Latent transforming growth factor-beta activation in mammary gland: regulation by ovarian hormones affects ductal and alveolar proliferation. Am. J. Pathol. 2002, 160, 2081–2093. [Google Scholar] [CrossRef]
- Yang, S.L.; Zhu, L.Y.; Han, R.; Sun, L.L.; Dou, J.T. Effect of Negative Pressure Wound Therapy on Cellular Fibronectin and Transforming Growth Factor-β1 Expression in Diabetic Foot Wounds. Foot Ankle Int. 2017, 38, 893–900. [Google Scholar] [CrossRef]
- Castellanos, G.; Bernabé-García, Á.; Insausti, C.G.; Piñero, A.; Moraleda, J.M.; Nicolás, F.J. The Use of Amniotic Membrane in the Management of Complex Chronic Wounds. Wound Healing 2016, 51868. [Google Scholar]
- Harris, I.R.; Yee, K.C.; Walters, C.E.; Cunliffe, W.J.; Kearney, J.N.; Wood, E.J.; Ingham, E. Cytokine and protease levels in healing and non-healing chronic venous leg ulcers. Exp. Dermatol. 1995, 4, 342–349. [Google Scholar] [CrossRef]
- Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Human Skin Keratinocytes on Sustained TGF-β Stimulation Reveal Partial EMT Features and Weaken Growth Arrest Responses. Cells 2020, 9, 255. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Role of TGF-β in Skin Chronic Wounds: A Keratinocyte Perspective. Cells 2020, 9, 306. https://doi.org/10.3390/cells9020306
Liarte S, Bernabé-García Á, Nicolás FJ. Role of TGF-β in Skin Chronic Wounds: A Keratinocyte Perspective. Cells. 2020; 9(2):306. https://doi.org/10.3390/cells9020306
Chicago/Turabian StyleLiarte, Sergio, Ángel Bernabé-García, and Francisco J. Nicolás. 2020. "Role of TGF-β in Skin Chronic Wounds: A Keratinocyte Perspective" Cells 9, no. 2: 306. https://doi.org/10.3390/cells9020306
APA StyleLiarte, S., Bernabé-García, Á., & Nicolás, F. J. (2020). Role of TGF-β in Skin Chronic Wounds: A Keratinocyte Perspective. Cells, 9(2), 306. https://doi.org/10.3390/cells9020306