A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of Cell Lines
2.3. CRISPR Cell Line Generation
2.4. Cell Stimulation Conditions
2.5. Western Blot Analysis
2.6. Crystal Violet Assay
2.7. Propidium Iodide Staining
2.8. Immunofluorescence Microscopy
2.9. Coimmunoprecipitation of Caspase-8-Bound Complexes
2.10. TNF-Fc Ligand Affinity Precipitation
2.11. Affymetrix GeneChip Oligoarray Analysis
2.12. Statistics
3. Results
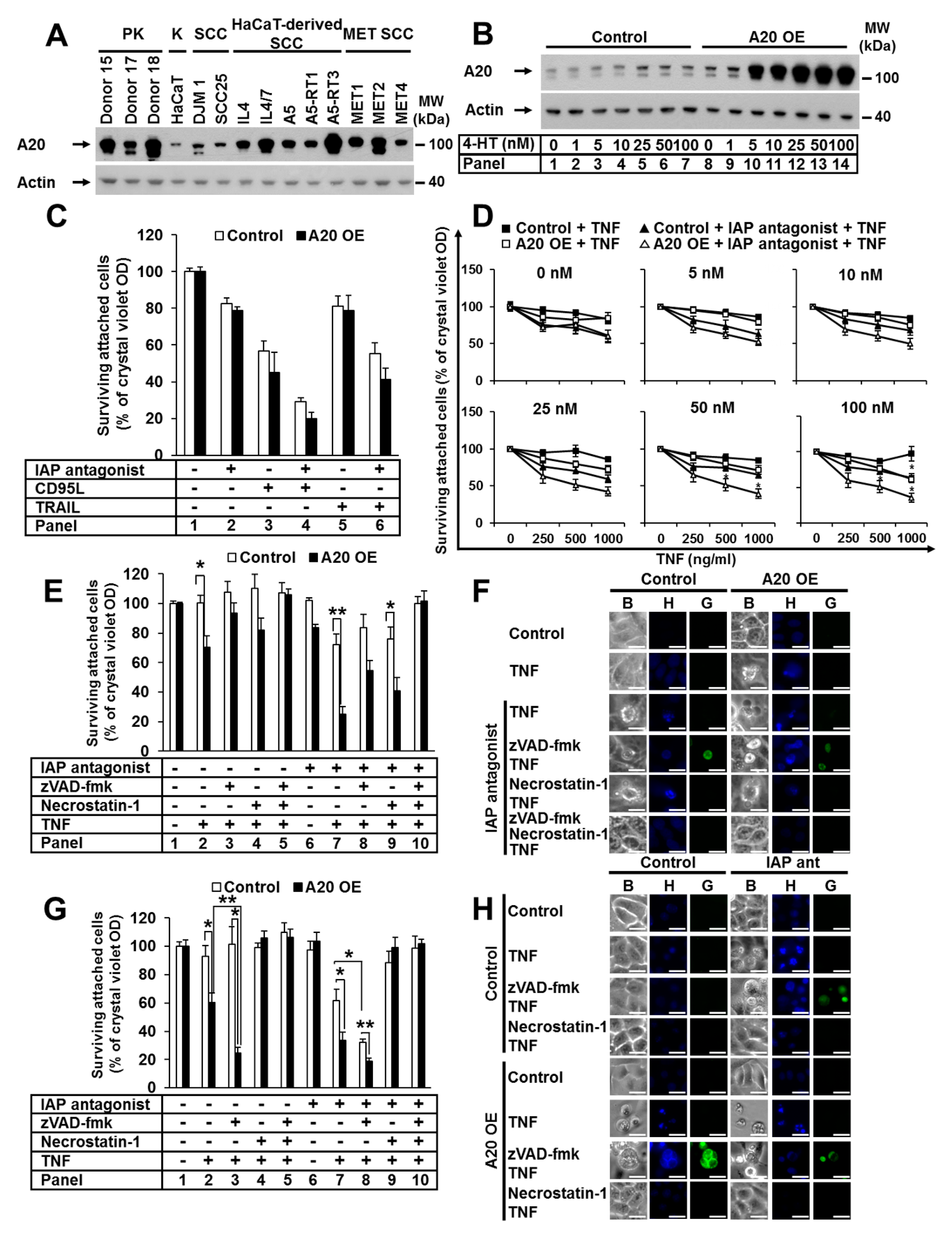
3.1. Elevated A20 Expression Sensitized Human and Murine Keratinocytes to TNF-Induced Cell Death
3.2. A20 Deficiency in Human Cells Did Not Change Their Sensitivity to TNF-Induced Cell Death
3.3. An Elevated Level of A20 Enhanced Ripoptosome Formation But Impairred TNF Complex I Formation
3.4. A20-Mediated TNF-Induced Cell Death in Keratinocytes Is Dependent on Canonical NF-κB Signaling and cIAP1/2
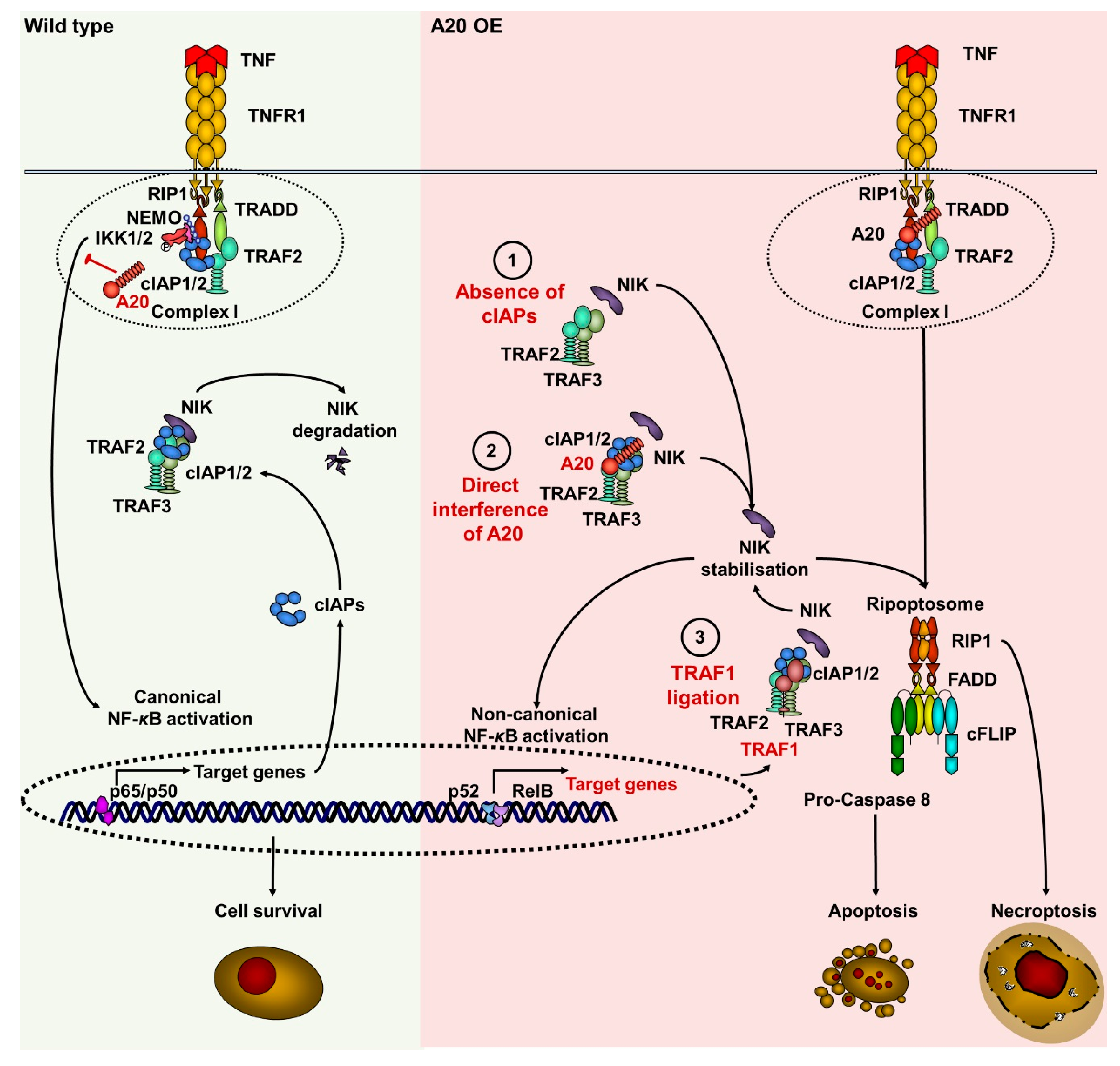
3.5. The Noncanonical NF-κB Pathway Was Activated in Keratinocytes with Elevated A20 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Diessenbacher, P.; Hupe, M.; Sprick, M.R.; Kerstan, A.; Geserick, P.; Haas, T.L.; Wachter, T.; Neumann, M.; Walczak, H.; Silke, J.; et al. NF-κB inhibition reveals differential mechanisms of TNF versus TRAIL-induced apoptosis upstream or at the level of caspase-8 activation independent of cIAP2. J. Investig. Dermatol. 2008, 128, 1134–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Ripk1 and haematopoiesis: A case for LUBAC and Ripk3. Cell Death Differ. 2018, 25, 1361–1363. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef] [Green Version]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.W., Jr.; Boguski, M.S.; Dixit, V.M. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J. Biol. Chem. 1990, 265, 14705–14708. [Google Scholar] [PubMed]
- Opipari, A.W., Jr.; Hu, H.M.; Yabkowitz, R.; Dixit, V.M. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J. Biol. Chem. 1992, 267, 12424–12427. [Google Scholar] [PubMed]
- Durkop, H.; Hirsch, B.; Hahn, C.; Foss, H.D.; Stein, H. Differential expression and function of A20 and TRAF1 in Hodgkin lymphoma and anaplastic large cell lymphoma and their induction by CD30 stimulation. J. Pathol. 2003, 200, 229–239. [Google Scholar] [CrossRef]
- Song, H.Y.; Rothe, M.; Goeddel, D.V. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc. Natl. Acad. Sci. USA 1996, 93, 6721–6725. [Google Scholar] [CrossRef] [Green Version]
- Heyninck, K.; De Valck, D.; Vanden Berghe, W.; Van Criekinge, W.; Contreras, R.; Fiers, W.; Haegeman, G.; Beyaert, R. The zinc finger protein A20 inhibits TNF-induced NF-κB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-κB-inhibiting protein ABIN. J. Cell Biol. 1999, 145, 1471–1482. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Kool, M.; van Loo, G.; Waelput, W.; De Prijck, S.; Muskens, F.; Sze, M.; van Praet, J.; Branco-Madeira, F.; Janssens, S.; Reizis, B.; et al. The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 2011, 35, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Tavares, R.M.; Turer, E.E.; Liu, C.L.; Advincula, R.; Scapini, P.; Rhee, L.; Barrera, J.; Lowell, C.A.; Utz, P.J.; Malynn, B.A.; et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 2010, 33, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Malynn, B.A. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 2012, 12, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, F.; Vacher, S.; Andrieu, C.; Espie, M.; Marty, M.; Lidereau, R.; Bieche, I. NF-κB genes have a major role in inflammatory breast cancer. BMC Cancer 2008, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Dong, H.; Liu, X.; Wang, C.; Liu, N.; Zhang, J.; Li, B.; Cao, W.; Ding, T.; Yang, Z.; et al. A20 is overexpressed in glioma cells and may serve as a potential therapeutic target. Expert Opin. Ther. Targets 2009, 13, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Codd, J.D.; Salisbury, J.R.; Packham, G.; Nicholson, L.J. A20 RNA expression is associated with undifferentiated nasopharyngeal carcinoma and poorly differentiated head and neck squamous cell carcinoma. J. Pathol. 1999, 187, 549–555. [Google Scholar] [CrossRef]
- Sohn, K.C.; Li, Z.J.; Choi, D.K.; Zhang, T.; Lim, J.W.; Chang, I.K.; Hur, G.M.; Im, M.; Lee, Y.; Seo, Y.J.; et al. Imiquimod induces apoptosis of squamous cell carcinoma (SCC) cells via regulation of A20. PLoS ONE 2014, 9, e95337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silke, J.; Kratina, T.; Chu, D.; Ekert, P.G.; Day, C.L.; Pakusch, M.; Huang, D.C.; Vaux, D.L. Determination of cell survival by RING-mediated regulation of inhibitor of apoptosis (IAP) protein abundance. Proc. Natl. Acad. Sci. USA 2005, 102, 16182–16187. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.; Gentle, I.E.; et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (TNF) to efficiently activate NF-κB and to prevent tnf-induced apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef] [Green Version]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [Green Version]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Kools, H.; de Groot, P.J.; Gavai, A.K.; Basnet, R.K.; Cheng, F.; Wu, J.; Wang, X.; Lommen, A.; Hooiveld, G.J.; et al. MADMAX—Management and analysis database for multiple ~omics experiments. J. Integr. Bioinform. 2011, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.L.; Blake, J.A.; Kadin, J.A.; Richardson, J.E.; Bult, C.J.; the Mouse Genome Database Group. Mouse Genome Database (MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res. 2018, 46, D836–D842. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef]
- Verstrepen, L.; Verhelst, K.; van Loo, G.; Carpentier, I.; Ley, S.C.; Beyaert, R. Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem. Pharmacol. 2010, 80, 2009–2020. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Declercq, W.; Van Herreweghe, F.; Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 2010, 3, re4. [Google Scholar] [CrossRef]
- Priem, D.; Devos, M.; Druwe, S.; Martens, A.; Slowicka, K.; Ting, A.T.; Pasparakis, M.; Declerq, W.; Vandenabeele, P.; van Loo, G.; et al. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis. 2019, 10, 692. [Google Scholar] [CrossRef]
- Verhelst, K.; Carpentier, I.; Kreike, M.; Meloni, L.; Verstrepen, L.; Kensche, T.; Dikic, I.; Beyaert, R. A20 inhibits LUBAC-mediated NF-κB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J. 2012, 31, 3845–3855. [Google Scholar] [CrossRef]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006, 6, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-κB signals induce the expression of c-FLIP. Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittkopf, N.; Gunther, C.; Martini, E.; He, G.; Amann, K.; He, Y.W.; Schuchmann, M.; Neurath, M.F.; Becker, C. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology 2013, 145, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Powolny-Budnicka, I.; Riemann, M.; Schmid, R.M.; Paxian, S.; Pfeffer, K.; Korner, H.; Weih, F. Rel/NF-κB family member RelA regulates NK1.1− to NK1.1+ transition as well as IL-15-induced expansion of NKT cells. Eur. J. Immunol. 2008, 38, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, S.; Kalita, M.; Fang, L.; Patel, K.V.; Tian, B.; Zhao, Y.; Edeh, C.B.; Brasier, A.R. Inducible tumor necrosis factor (TNF) receptor-associated factor-1 expression couples the canonical to the non-canonical NF-κB pathway in TNF stimulation. J. Biol. Chem. 2013, 288, 14612–14623. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Challa-Malladi, M.; Bratton, S.B.; Wright, C.W. Nuclear factor-κB-inducing kinase (NIK) contains an amino-terminal inhibitor of apoptosis (IAP)-binding motif (IBM) that potentiates NIK degradation by cellular IAP1 (c-IAP1). J. Biol. Chem. 2014, 289, 30680–30689. [Google Scholar] [CrossRef] [Green Version]
- Razani, B.; Reichardt, A.D.; Cheng, G. Non-canonical NF-κB signaling activation and regulation: Principles and perspectives. Immunol. Rev. 2011, 244, 44–54. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Boutaffala, L.; Bertrand, M.J.; Remouchamps, C.; Seleznik, G.; Reisinger, F.; Janas, M.; Benezech, C.; Fernandes, M.T.; Marchetti, S.; Mair, F.; et al. NIK promotes tissue destruction independently of the alternative NF-κB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ. 2015, 22, 2020–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feoktistova, M.; Leverkus, M. Programmed necrosis and necroptosis signalling. FEBS J. 2015, 282, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Panayotova-Dimitrova, D.; Leverkus, M. Pick your poison: The Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 2012, 11, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef] [Green Version]
- McQuade, T.; Cho, Y.; Chan, F.K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 2013, 456, 409–415. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Oyama, M.; Kozuka-Hata, H.; Inoue, J. Involvement of A20 in the molecular switch that activates the non-canonical NF-κB pathway. Sci. Rep. 2013, 3, 2568. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feoktistova, M.; Makarov, R.; Brenji, S.; Schneider, A.T.; Hooiveld, G.J.; Luedde, T.; Leverkus, M.; Yazdi, A.S.; Panayotova-Dimitrova, D. A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells 2020, 9, 351. https://doi.org/10.3390/cells9020351
Feoktistova M, Makarov R, Brenji S, Schneider AT, Hooiveld GJ, Luedde T, Leverkus M, Yazdi AS, Panayotova-Dimitrova D. A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells. 2020; 9(2):351. https://doi.org/10.3390/cells9020351
Chicago/Turabian StyleFeoktistova, Maria, Roman Makarov, Sihem Brenji, Anne T. Schneider, Guido J. Hooiveld, Tom Luedde, Martin Leverkus, Amir S. Yazdi, and Diana Panayotova-Dimitrova. 2020. "A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes" Cells 9, no. 2: 351. https://doi.org/10.3390/cells9020351
APA StyleFeoktistova, M., Makarov, R., Brenji, S., Schneider, A. T., Hooiveld, G. J., Luedde, T., Leverkus, M., Yazdi, A. S., & Panayotova-Dimitrova, D. (2020). A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells, 9(2), 351. https://doi.org/10.3390/cells9020351