TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression

 ,
,
Abstract
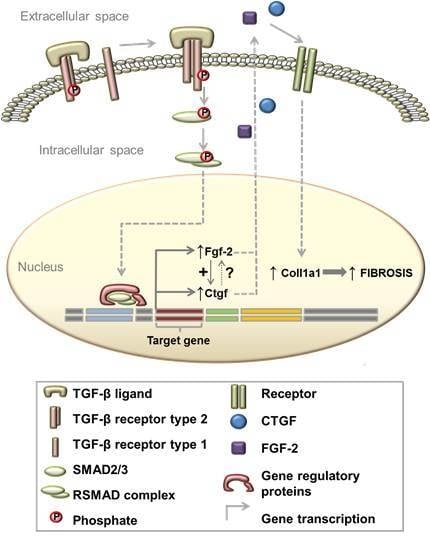
1. Introduction
2. Materials and Methods
2.1. C2C12 Cell Culture
2.2. Isolation of the Extensor Digitorum Longus (EDL) Muscle and Primary Myoblast Culture
2.3. Tgfbr1 and Acvr1b siRNA Assay
2.4. Ctgf and Fgf-2 siRNA Assay
2.5. RNA Isolation and Reverse Transcription
2.6. Quantitative Real Time PCR
2.7. Western Blotting
2.8. Immunofluorescence
2.9. Statistical Analysis
3. Results
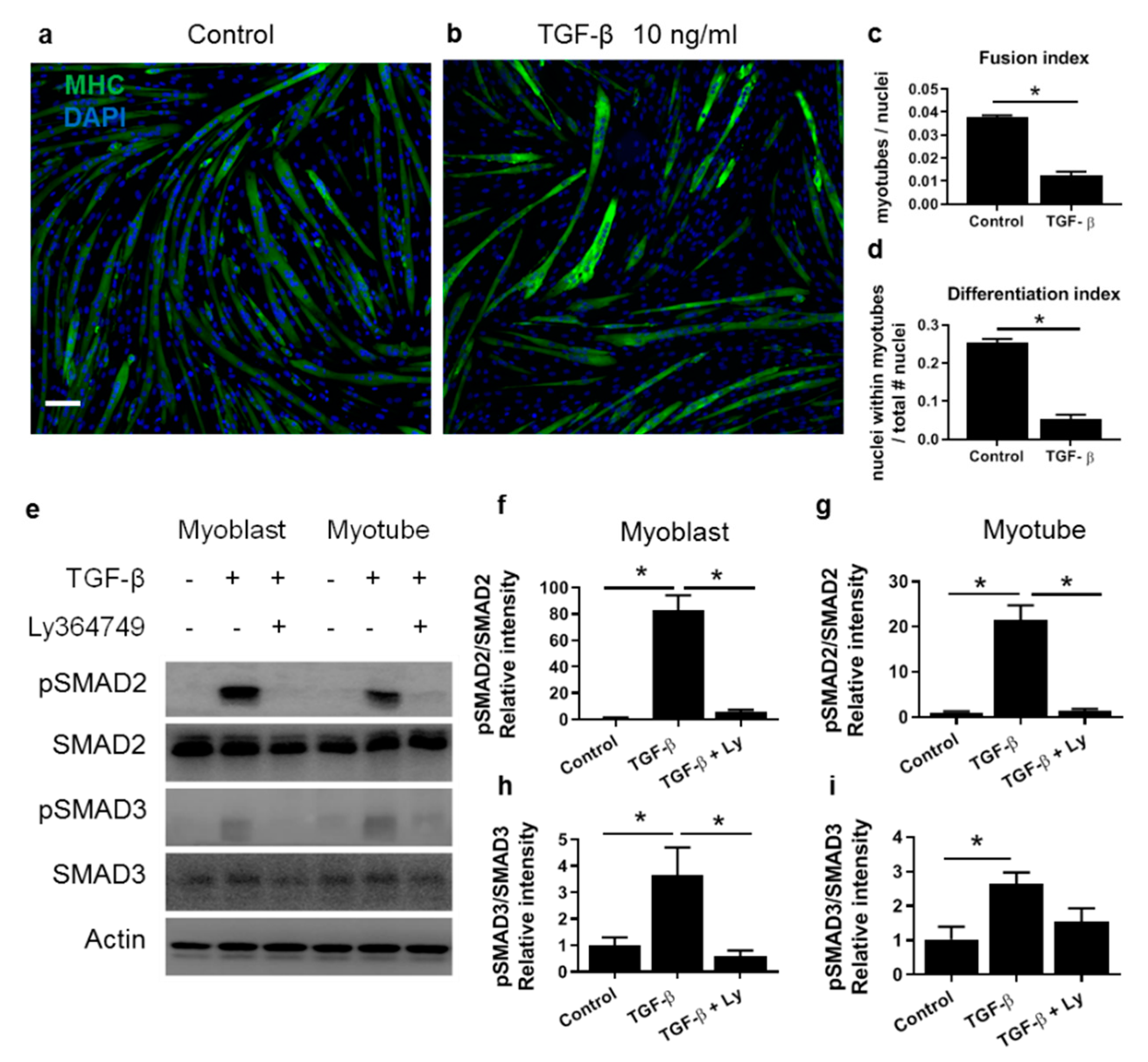
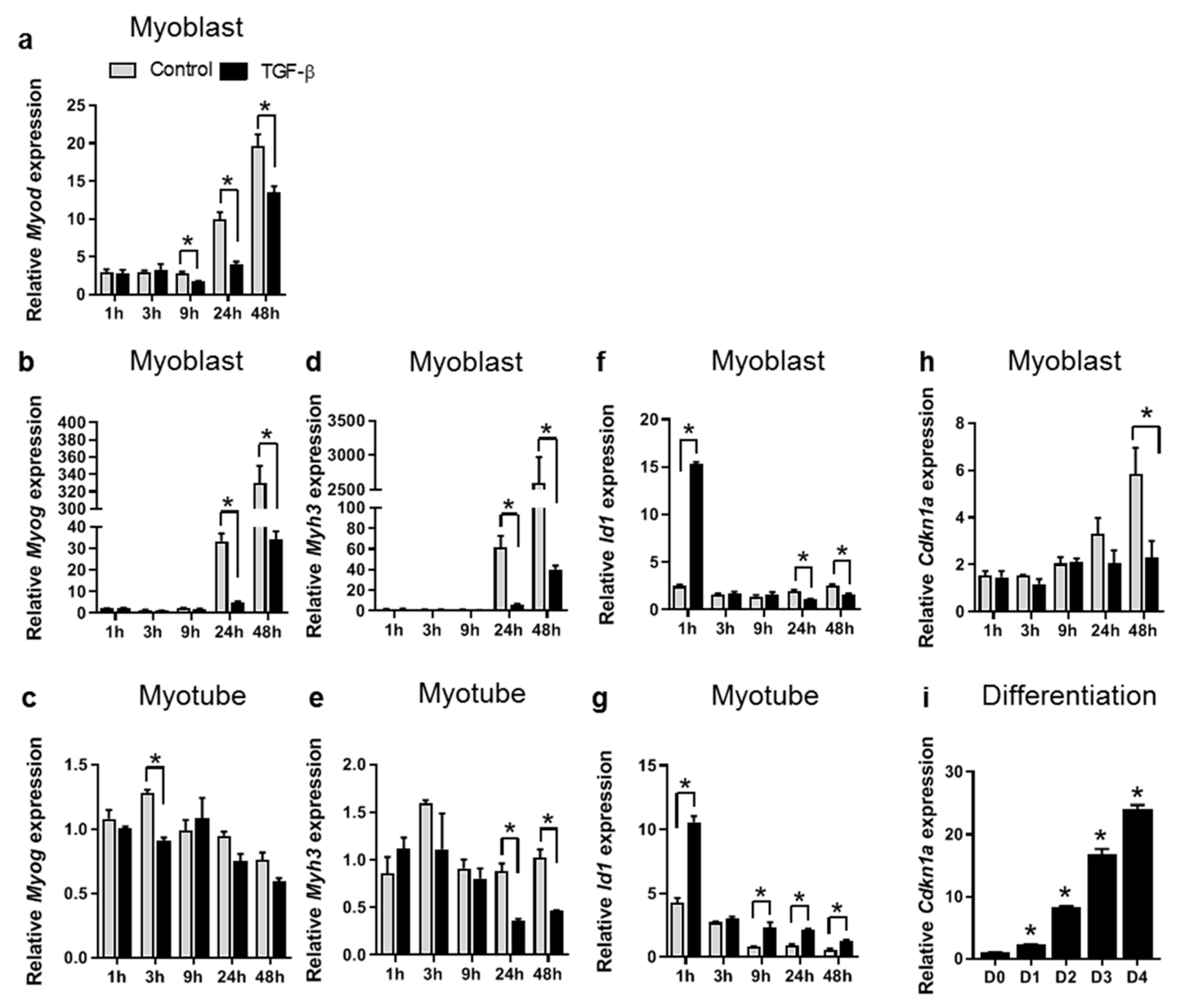
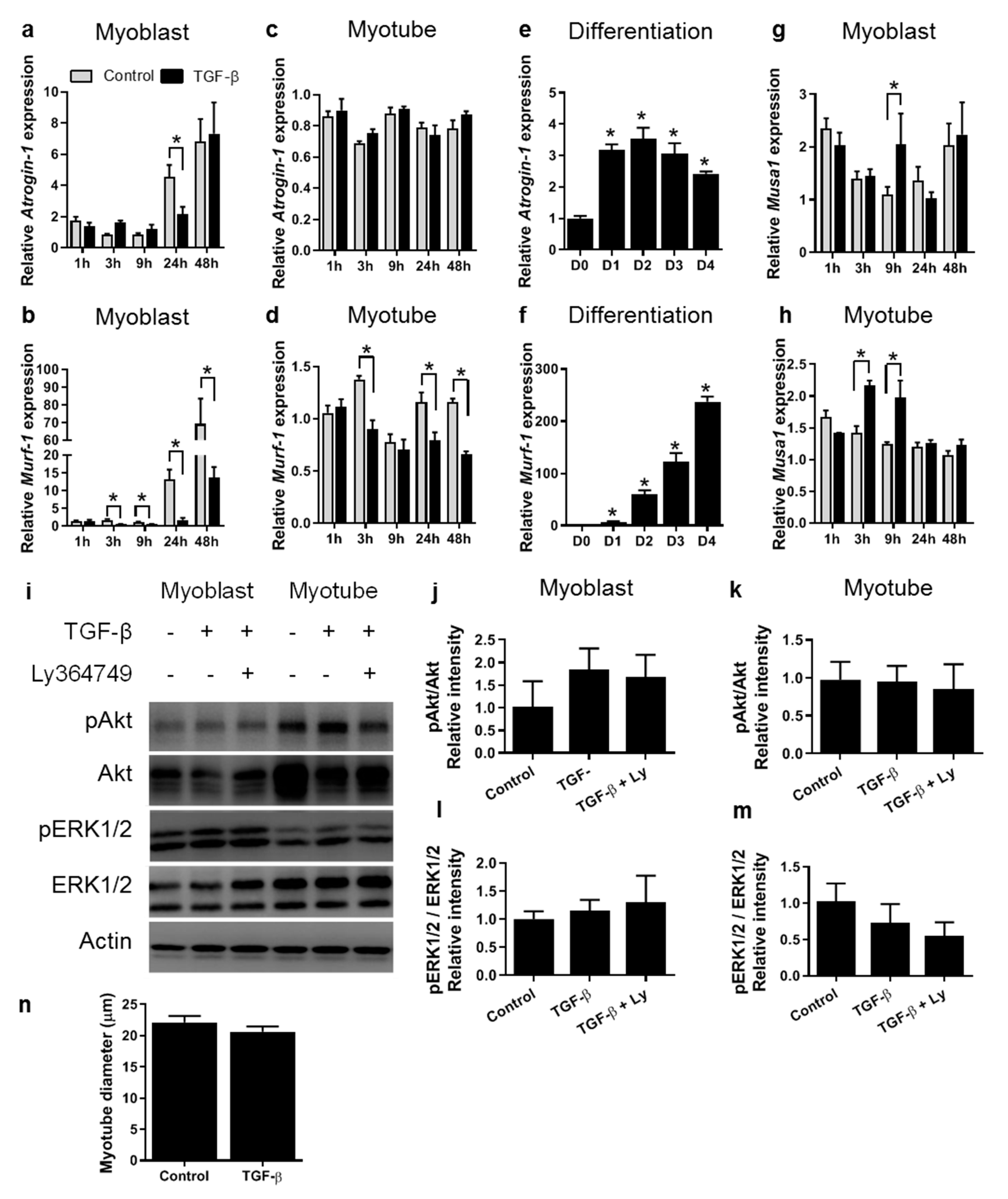
3.1. TGF-β Inhibits Expression of Myogenic Genes in both C2C12 Myoblasts and Myotubes
3.2. TGF-β Does Not Affect Myotube Size In Vitro
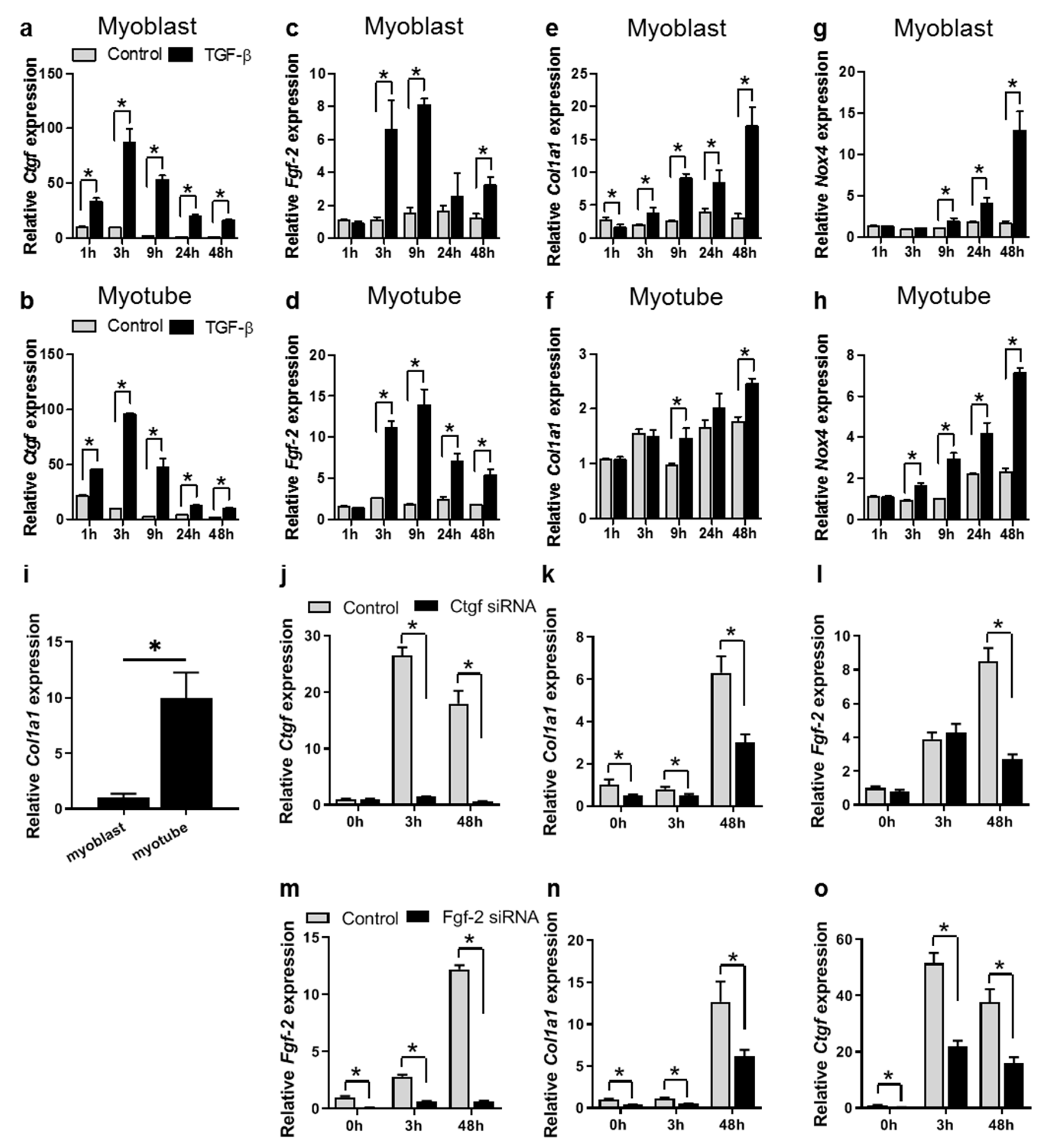
3.3. TGF-β Affects Fibrotic Gene Expression in a Time-Dependent Manner in Both Myoblasts and Myotubes
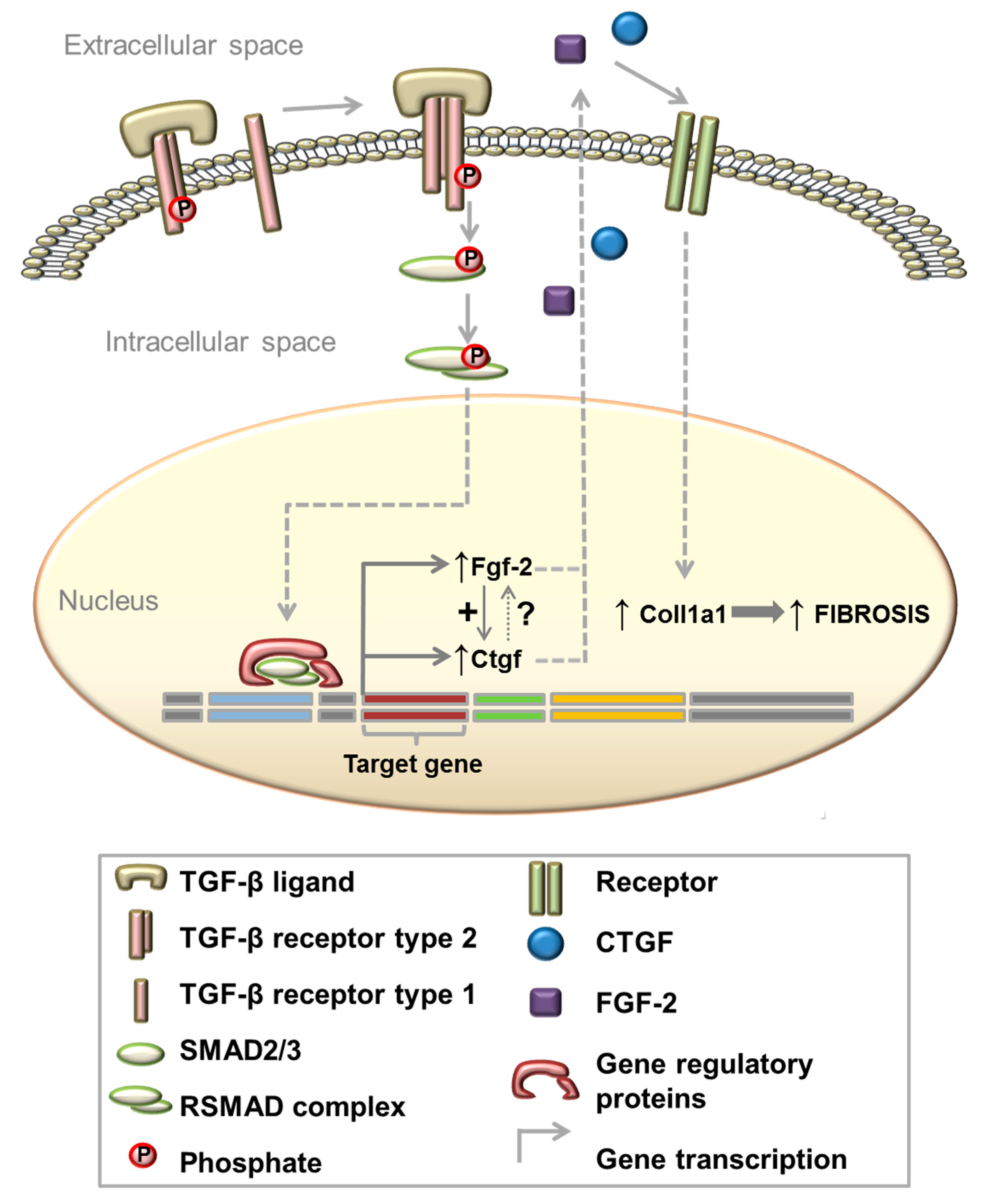
3.4. TGF-β Induces Col1a1 Expression via Ctgf and Fgf-2 in Myoblasts
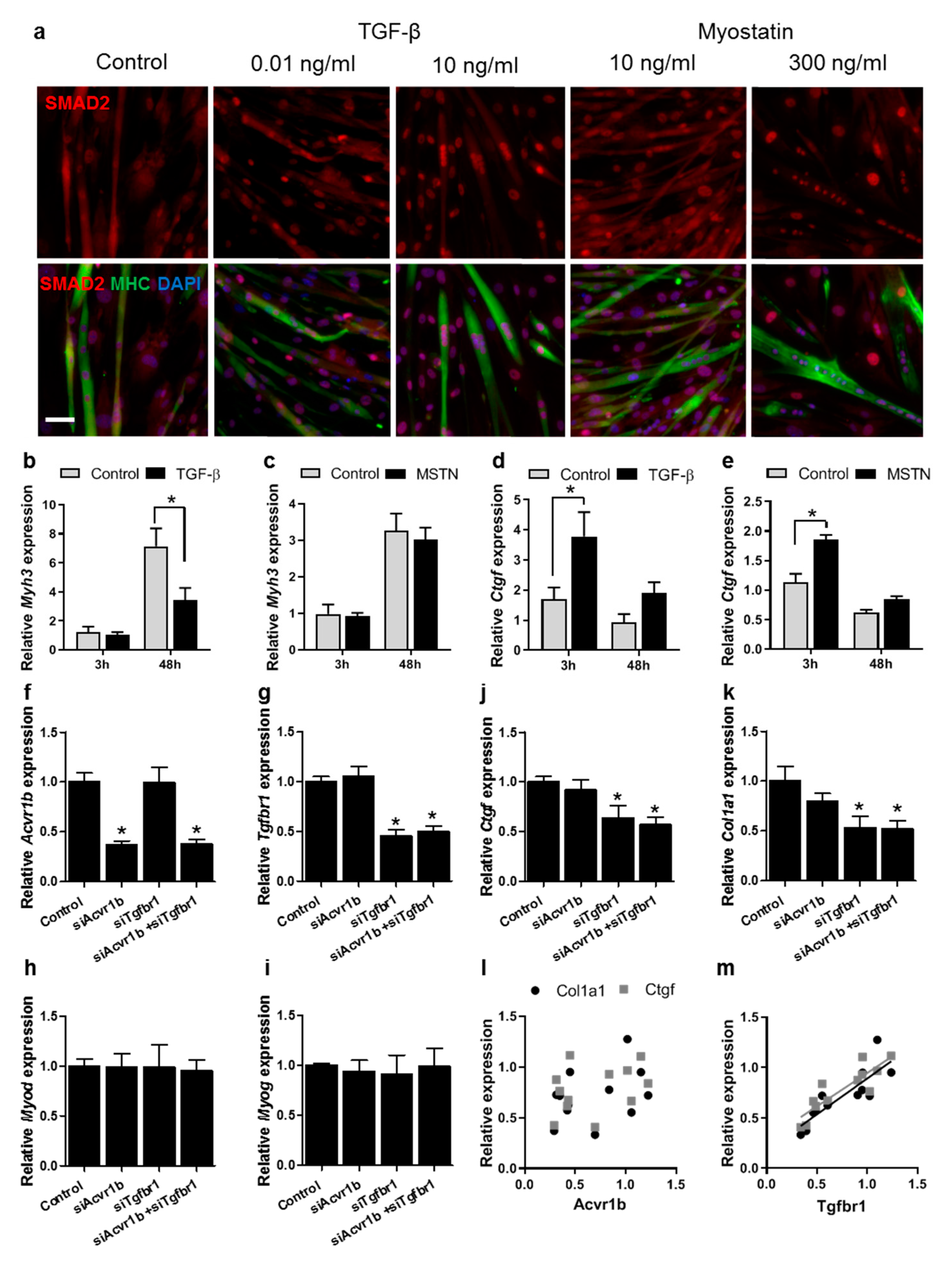
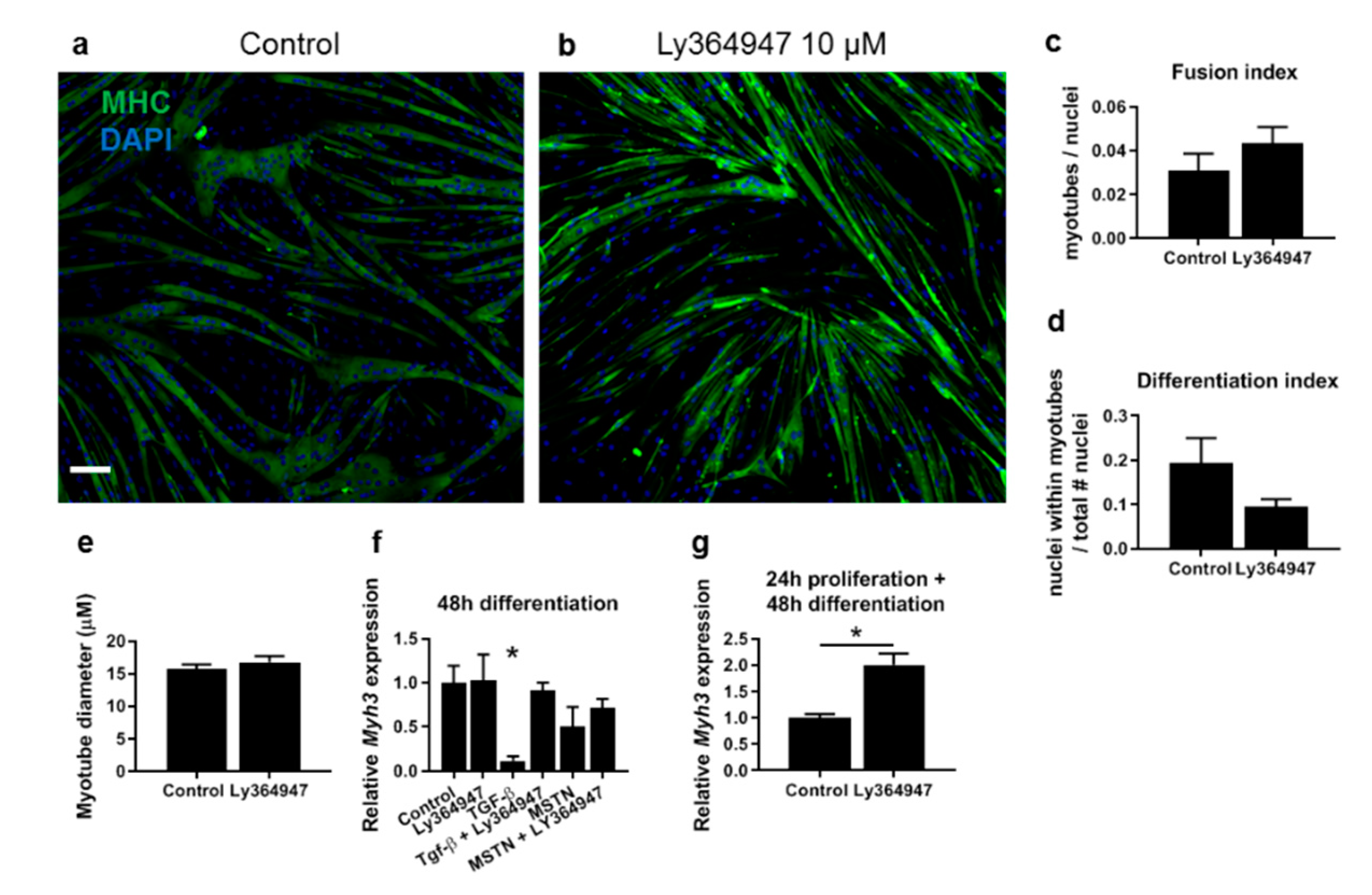
3.5. TGF-β Has a Larger Effect on Muscle Differentiation and Fibrosis than Myostatin
3.6. Tgfbr1 Levels Correlate with Ctgf and Col1a1 Expression Levels
4. Discussion
4.1. TGF-β Affects Myogenic Gene Expression in Both Myoblasts and Myotubes
4.2. TGF-β Does Not Affect Myotube Size In Vitro
4.3. TGF-β Contributes to Fibrosis by Stimulation of Fibrotic Gene Expression in Myoblasts and Myotubes
4.4. TGF-β Induces Col1a1 Expression via Autocrine Ctgf and Fgf-2 Signalling
4.5. TGF-β Has a More Pronounced Effect than Myostatin on Myoblast Differentiation and Fibrotic Gene Expression
4.6. Implications in Therapeutic Treatments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ryall, J.G.; Schertzer, J.D.; Lynch, G.S. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 2008, 9, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.; Simoes, E.; de Castro, G.; Morais, M.; de Matos-Neto, E.M.; Alves, M.J.; Pinto, N.I.; Figueredo, R.G.; Zorn, T.M.T.; Felipe-Silva, A.S.; et al. Tumour-derived transforming growth factor-beta signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1045–1059. [Google Scholar] [CrossRef]
- Bernasconi, P.; Torchiana, E.; Confalonieri, P.; Brugnoni, R.; Barresi, R.; Mora, M.; Cornelio, F.; Morandi, L.; Mantegazza, R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Investig. 1995, 96, 1137–1144. [Google Scholar] [CrossRef]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of tgfbeta in duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Robertson, T.A.; Maley, M.A.; Grounds, M.D.; Papadimitriou, J.M. The role of macrophages in skeletal muscle regeneration with particular reference to chemotaxis. Exp. Cell Res. 1993, 207, 321–331. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell. Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef]
- Lawrence, D.A.; Pircher, R.; Kryceve-Martinerie, C.; Jullien, P. Normal embryo fibroblasts release transforming growth factors in a latent form. J. Cell. Physiol. 1984, 121, 184–188. [Google Scholar] [CrossRef]
- Shur, I.; Lokiec, F.; Bleiberg, I.; Benayahu, D. Differential gene expression of cultured human osteoblasts. J. Cell. Biochem. 2001, 83, 547–553. [Google Scholar] [CrossRef]
- Zimowska, M.; Duchesnay, A.; Dragun, P.; Oberbek, A.; Moraczewski, J.; Martelly, I. Immunoneutralization of tgfbeta1 improves skeletal muscle regeneration: Effects on myoblast differentiation and glycosaminoglycan content. Int. J. Cell Biol. 2009, 2009, 659372. [Google Scholar] [CrossRef]
- Pasteuning-Vuhman, S.; Putker, K.; Tanganyika-de Winter, C.L.; Boertje-van der Meulen, J.W.; van Vliet, L.; Overzier, M.; Plomp, J.J.; Aartsma-Rus, A.; van Putten, M. Natural disease history of mouse models for limb girdle muscular dystrophy types 2d and 2f. PLoS ONE 2017, 12, e0182704. [Google Scholar] [CrossRef]
- Gonzalez, D.; Contreras, O.; Rebolledo, D.L.; Espinoza, J.P.; van Zundert, B.; Brandan, E. ALS skeletal muscle shows enhanced tgf-beta signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 2017, 12, e0177649. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef]
- Liu, D.; Black, B.L.; Derynck, R. Tgf-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by smad3. Genes Dev. 2001, 15, 2950–2966. [Google Scholar] [CrossRef]
- Olson, E.N.; Sternberg, E.; Hu, J.S.; Spizz, G.; Wilcox, C. Regulation of myogenic differentiation by type beta transforming growth factor. J. Cell Biol. 1986, 103, 1799–1805. [Google Scholar] [CrossRef]
- Massague, J.; Cheifetz, S.; Endo, T.; Nadal-Ginard, B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc. Natl. Acad. Sci. USA 1986, 83, 8206–8210. [Google Scholar] [CrossRef]
- Narola, J.; Pandey, S.N.; Glick, A.; Chen, Y.W. Conditional expression of tgf-beta1 in skeletal muscles causes endomysial fibrosis and myofibers atrophy. PLoS ONE 2013, 8, e79356. [Google Scholar] [CrossRef]
- Mendias, C.L.; Gumucio, J.P.; Davis, M.E.; Bromley, C.W.; Davis, C.S.; Brooks, S.V. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 2012, 45, 55–59. [Google Scholar] [CrossRef]
- Andreetta, F.; Bernasconi, P.; Baggi, F.; Ferro, P.; Oliva, L.; Arnoldi, E.; Cornelio, F.; Mantegazza, R.; Confalonieri, P. Immunomodulation of tgf-beta 1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: Implications for antifibrotic therapy. J. Neuroimmunol. 2006, 175, 77–86. [Google Scholar] [CrossRef]
- Li, Y.; Foster, W.; Deasy, B.M.; Chan, Y.; Prisk, V.; Tang, Y.; Cummins, J.; Huard, J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: A key event in muscle fibrogenesis. Am. J. Pathol. 2004, 164, 1007–1019. [Google Scholar] [CrossRef]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating myod expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef]
- Li, Z.B.; Kollias, H.D.; Wagner, K.R. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef]
- Rebbapragada, A.; Benchabane, H.; Wrana, J.L.; Celeste, A.J.; Attisano, L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol. Cell. Biol. 2003, 23, 7230–7242. [Google Scholar] [CrossRef]
- ten Dijke, P.; Yamashita, H.; Ichijo, H.; Franzen, P.; Laiho, M.; Miyazono, K.; Heldin, C.H. Characterization of type I receptors for transforming growth factor-beta and activin. Science 1994, 264, 101–104. [Google Scholar] [CrossRef]
- Kemaladewi, D.U.; de Gorter, D.J.; Aartsma-Rus, A.; van Ommen, G.J.; ten Dijke, P.; t Hoen, P.A.; Hoogaars, W.M. Cell-type specific regulation of myostatin signaling. FASEB J. 2012, 26, 1462–1472. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Jen, Y.; Weintraub, H.; Benezra, R. Overexpression of Id protein inhibits the muscle differentiation program: In vivo association of Id with E2A proteins. Genes Dev. 1992, 6, 1466–1479. [Google Scholar] [CrossRef]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Muller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. Tgf-beta uses a novel mode of receptor activation to phosphorylate smad1/5 and induce epithelial-to-mesenchymal transition. Elife 2018, 7, e31756. [Google Scholar] [CrossRef]
- McFarlane, C.; Plummer, E.; Thomas, M.; Hennebry, A.; Ashby, M.; Ling, N.; Smith, H.; Sharma, M.; Kambadur, R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J. Cell. Physiol. 2006, 209, 501–514. [Google Scholar] [CrossRef]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NAPDH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Kang, J.S.; Derynck, R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004, 23, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Agarwal, M.; Kumar, A.; Kumar, P.; Saini, M.; Kardon, G.; Mathew, S.J. Myosin heavy chain-embryonic is a crucial regulator of skeletal muscle development and differentiation. bioRxiv 2018. [Google Scholar]
- Kadoguchi, T.; Shimada, K.; Koide, H.; Miyazaki, T.; Shiozawa, T.; Takahashi, S.; Aikawa, T.; Ouchi, S.; Kitamura, K.; Sugita, Y.; et al. Possible role of NAPDH oxidase 4 in angiotensin II-induced muscle wasting in mice. Front. Physiol. 2018, 9, 340. [Google Scholar] [CrossRef]
- Gundersen, K.; Merlie, J.P. Id-1 as a possible transcriptional mediator of muscle disuse atrophy. Proc. Natl. Acad. Sci. USA 1994, 91, 3647–3651. [Google Scholar] [CrossRef]
- Cencetti, F.; Bernacchioni, C.; Tonelli, F.; Roberts, E.; Donati, C.; Bruni, P. Tgfbeta1 evokes myoblast apoptotic response via a novel signaling pathway involving S1P4 transactivation upstream of Rho-kinase-2 activation. FASEB J. 2013, 27, 4532–4546. [Google Scholar] [CrossRef]
- Zammit, P.S.; Heslop, L.; Hudon, V.; Rosenblatt, J.D.; Tajbakhsh, S.; Buckingham, M.E.; Beauchamp, J.R.; Partridge, T.A. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp. Cell Res. 2002, 281, 39–49. [Google Scholar] [CrossRef]
- Mackey, A.L.; Magnan, M.; Chazaud, B.; Kjaer, M. Human skeletal muscle fibroblasts stimulate in vitro myogenesis and in vivo muscle regeneration. J. Physiol. 2017, 595, 5115–5127. [Google Scholar] [CrossRef]
- Frese, S.; Ruebner, M.; Suhr, F.; Konou, T.M.; Tappe, K.A.; Toigo, M.; Jung, H.H.; Henke, C.; Steigleder, R.; Strissel, P.L.; et al. Long-term endurance exercise in humans stimulates cell fusion of myoblasts along with fusogenic endogenous retroviral genes in vivo. PLoS ONE 2015, 10, e0132099. [Google Scholar] [CrossRef]
- Listrat, A.; Picard, B.; Geay, Y. Age-related changes and location of type I, III, IV, V and VI collagens during development of four foetal skeletal muscles of double-muscled and normal bovine animals. Tissue Cell 1999, 31, 17–27. [Google Scholar] [CrossRef]
- Light, N.; Champion, A.E. Characterization of muscle epimysium, perimysium and endomysium collagens. Biochem. J. 1984, 219, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Purslow, P.P. The structure and functional significance of variations in the connective tissue within muscle. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2002, 133, 947–966. [Google Scholar] [CrossRef]
- Huijing, P.A.; Jaspers, R.T. Adaptation of muscle size and myofascial force transmission: A review and some new experimental results. Scand J. Med. Sci. Sports 2005, 15, 349–380. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Engler, A.J.; Meyer, G.A. Extracellular matrix regulation in the muscle satellite cell niche. Connect. Tissue Res. 2015, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef]
- Boers, H.E.; Haroon, M.; Le Grand, F.; Bakker, A.D.; Klein-Nulend, J.; Jaspers, R.T. Mechanosensitivity of aged muscle stem cells. J. Orthop. Res. 2018, 36, 632–641. [Google Scholar]
- Romanazzo, S.; Forte, G.; Ebara, M.; Uto, K.; Pagliari, S.; Aoyagi, T.; Traversa, E.; Taniguchi, A. Substrate stiffness affects skeletal myoblast differentiation in vitro. Sci. Technol. Adv. Mater. 2012, 13, 064211. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Y.; Wu, X.; Peshavariya, H.M.; Dusting, G.J.; Zhang, M.; Jiang, F. Nox4 and redox signaling mediate tgf-beta-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 2014, 5, e1010. [Google Scholar] [CrossRef]
- Avin, K.G.; Chen, N.X.; Organ, J.M.; Zarse, C.; O’Neill, K.; Conway, R.G.; Konrad, R.J.; Bacallao, R.L.; Allen, M.R.; Moe, S.M. Skeletal muscle regeneration and oxidative stress are altered in chronic kidney disease. PLoS ONE 2016, 11, e0159411. [Google Scholar] [CrossRef]
- Lin, C.H.; Yu, M.C.; Tung, W.H.; Chen, T.T.; Yu, C.C.; Weng, C.M.; Tsai, Y.J.; Bai, K.J.; Hong, C.Y.; Chien, M.H.; et al. Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the Rac1/MLK3/JNK/AP-1 pathway. Biochim. Biophys. Acta 2013, 1833, 2823–2833. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, Z.; Liu, H.; Ren, Y.; Shao, D.; Zhang, W.; Lin, J.; Wolfram, J.; Wang, F.; Nie, S. Connective tissue growth factor stimulates the proliferation, migration and differentiation of lung fibroblasts during paraquat-induced pulmonary fibrosis. Mol. Med. Rep. 2015, 12, 1091–1097. [Google Scholar] [CrossRef]
- Ponticos, M.; Holmes, A.M.; Shi-wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009, 60, 2142–2155. [Google Scholar] [CrossRef]
- Ko, M.K.; Kay, E.P. Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4495–4503. [Google Scholar] [CrossRef]
- Park, D.S.; Park, J.C.; Lee, J.S.; Kim, T.W.; Kim, K.J.; Jung, B.J.; Shim, E.K.; Choi, E.Y.; Park, S.Y.; Cho, K.S.; et al. Effect of FGF-2 on collagen tissue regeneration by human vertebral bone marrow stem cells. Stem Cells Dev. 2015, 24, 228–243. [Google Scholar] [CrossRef]
- Yablonka-Reuveni, Z.; Rivera, A.J. Proliferative dynamics and the role of FGF2 during myogenesis of rat satellite cells on isolated fibers. Basic Appl. Myol. 1997, 7, 189–202. [Google Scholar]
- Liu, Y.; Schneider, M.F. FGF2 activates TRPC and Ca(2+) signaling leading to satellite cell activation. Front. Physiol. 2014, 5, 38. [Google Scholar] [CrossRef]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of tgf-beta family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar]
- Watt, K.I.; Jaspers, R.T.; Atherton, P.; Smith, K.; Rennie, M.J.; Ratkevicius, A.; Wackerhage, H. Sb431542 treatment promotes the hypertrophy of skeletal muscle fibers but decreases specific force. Muscle Nerve 2010, 41, 624–629. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| mGapdh-forward | TCCATGACAACTTTGGCATTG |
| mGapdh-reverse | TCACGCCACAGCTTTCCA |
| Myod1-forward | AGCACTACAGTGGCGACTCA |
| Myod1-reverse | GCTCCACTATGCTGGACAGG |
| Myog-forward | CCCAACCCAGGAGATCATTT |
| Myog-reverse | GTCTGGGAAGGCAACAGACA |
| Myh3-forward | CGCAGAATCGCAAGTCAATA |
| Myh3-reverse | CAGGAGGTCTTGCTCACTCC |
| Ctgf-forward | CCACCCGAGTTACCAATGAC |
| Ctgf-reverse | GCTTGGCGATTTTAGGTGTC |
| Fgf-2-forward | AAGCGGCTCTACTGCAAGAA |
| Fgf-2-reverse | GTAACACACTTAGAAGCCAGCAG |
| Col1a1-forward | ATGTTCAGCTTTGTGGACCT |
| Col1a1-reverse | CAGCTGACTTCAGGGATGT |
| Id1-forward | ACCCTGAACGGCGAGATCA |
| Id1-reverse | TCGTCGGCTGGAACACAT |
| Nox4-forward | CTTTTCATTGGGCGTCCTC |
| Nox4-reverse | GGGTCCACAGCAGAAAACTC |
| AB | Dilution | Experiment | Company |
|---|---|---|---|
| Phospho-SMAD2 (Ser465/467) Rabbit mAb | 1:1000 | WB | cell signaling/3108, Leiden, The Netherlands |
| SMAD2 Rabbit mAb | 1:1000, 1:200 | WB, IF | cell signaling/5339 |
| Phospho-SMAD3 (S423/425) Rabbit mAb | 1:1000 | WB | cell signaling/9520 |
| SMAD3 Rabbit mAb | 1:1000 | WB | cell signaling/9523 |
| Phospho-Akt (Ser473) Rabbit mAb | 1:2000 | WB | cell signaling/4060 |
| Akt (pan) Rabbit mAb | 1:1000 | WB | cell signaling/4691 |
| Phospho-ERK1/2 Rabbit mAb | 1:2000 | WB | cell signaling/4370 |
| ERK1/2 Rabbit mAb | 1:4000 | WB | cell signaling/4695 |
| Pan actin Rabbit mAb | 1:1000 | WB | cell signaling/8456 |
| Myosin, sarcomere (MHC) | 2.5 µg/mL | IF | DSHB/MF20-s, Iowa City, IA, USA |
| Anti-Rabbit IgG-POD (LumiLightPLUS Western Blotting Kit) | 1:2000 | WB | Roche/12015218001, Basel, Switzerland |
| Goat anti-Rabbit IgG (H + L), Alexa Fluor® 555 conjugate | 1:500 | IF | ThermoFisher Scientific/A21428, Waltham, MA, USA |
| Goat anti-Mouse IgG (H + L), Alexa Fluor® 488 conjugate | 1:250 | IF | ThermoFisher Scientific/A11001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillege, M.M.G.; Galli Caro, R.A.; Offringa, C.; de Wit, G.M.J.; Jaspers, R.T.; Hoogaars, W.M.H. TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression. Cells 2020, 9, 375. https://doi.org/10.3390/cells9020375
Hillege MMG, Galli Caro RA, Offringa C, de Wit GMJ, Jaspers RT, Hoogaars WMH. TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression. Cells. 2020; 9(2):375. https://doi.org/10.3390/cells9020375
Chicago/Turabian StyleHillege, Michèle M. G., Ricardo A. Galli Caro, Carla Offringa, Gerard M. J. de Wit, Richard T. Jaspers, and Willem M. H. Hoogaars. 2020. "TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression" Cells 9, no. 2: 375. https://doi.org/10.3390/cells9020375
APA StyleHillege, M. M. G., Galli Caro, R. A., Offringa, C., de Wit, G. M. J., Jaspers, R. T., & Hoogaars, W. M. H. (2020). TGF-β Regulates Collagen Type I Expression in Myoblasts and Myotubes via Transient Ctgf and Fgf-2 Expression. Cells, 9(2), 375. https://doi.org/10.3390/cells9020375