mTOR Regulation of Metabolism in Hematologic Malignancies



Abstract
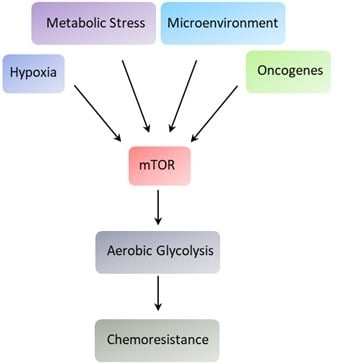
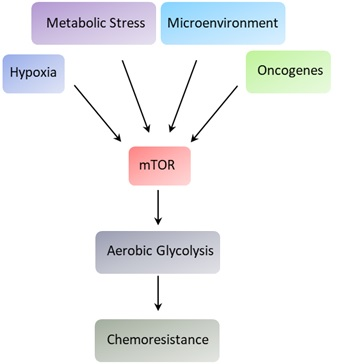
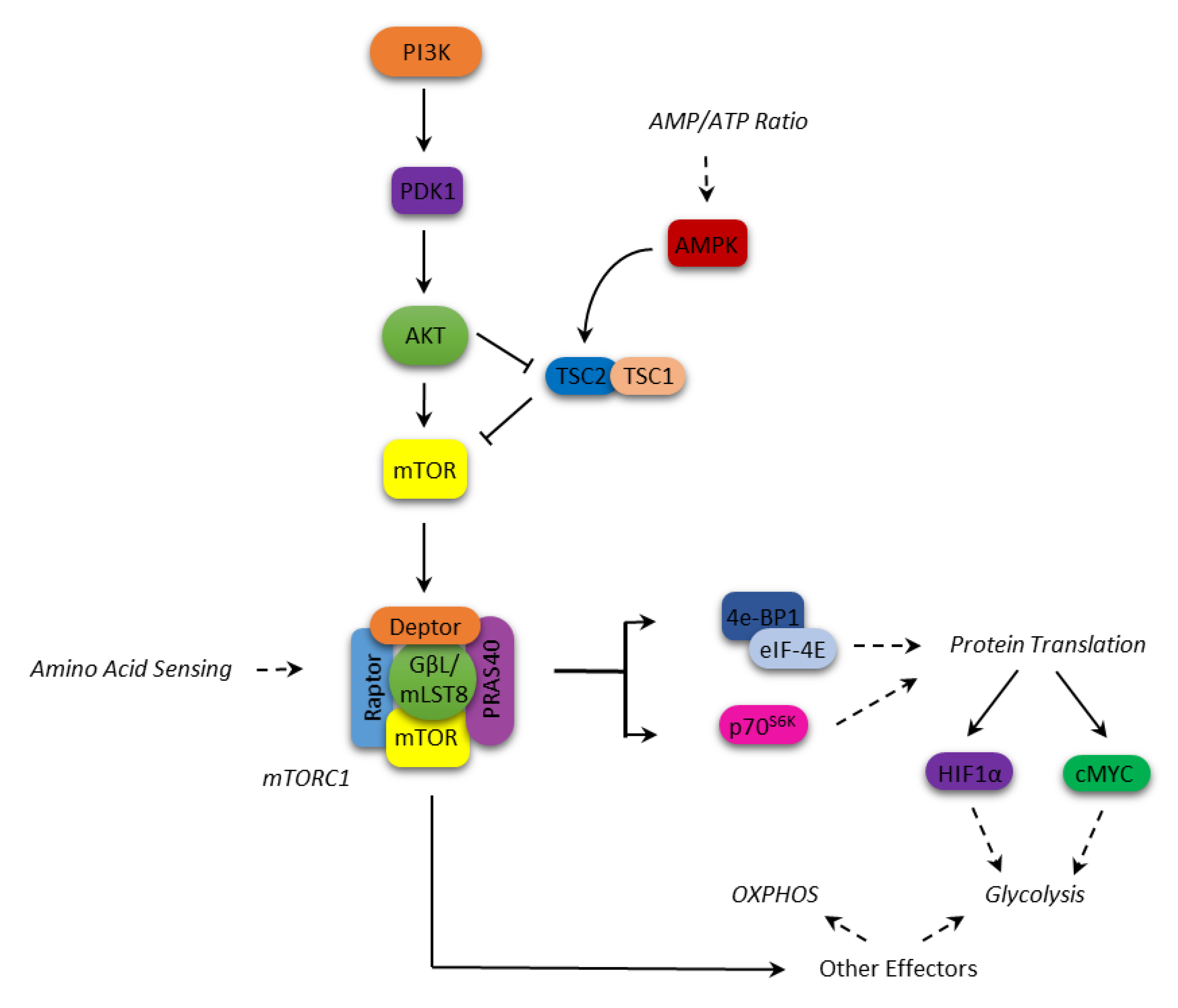
1. mTOR Structure and Function
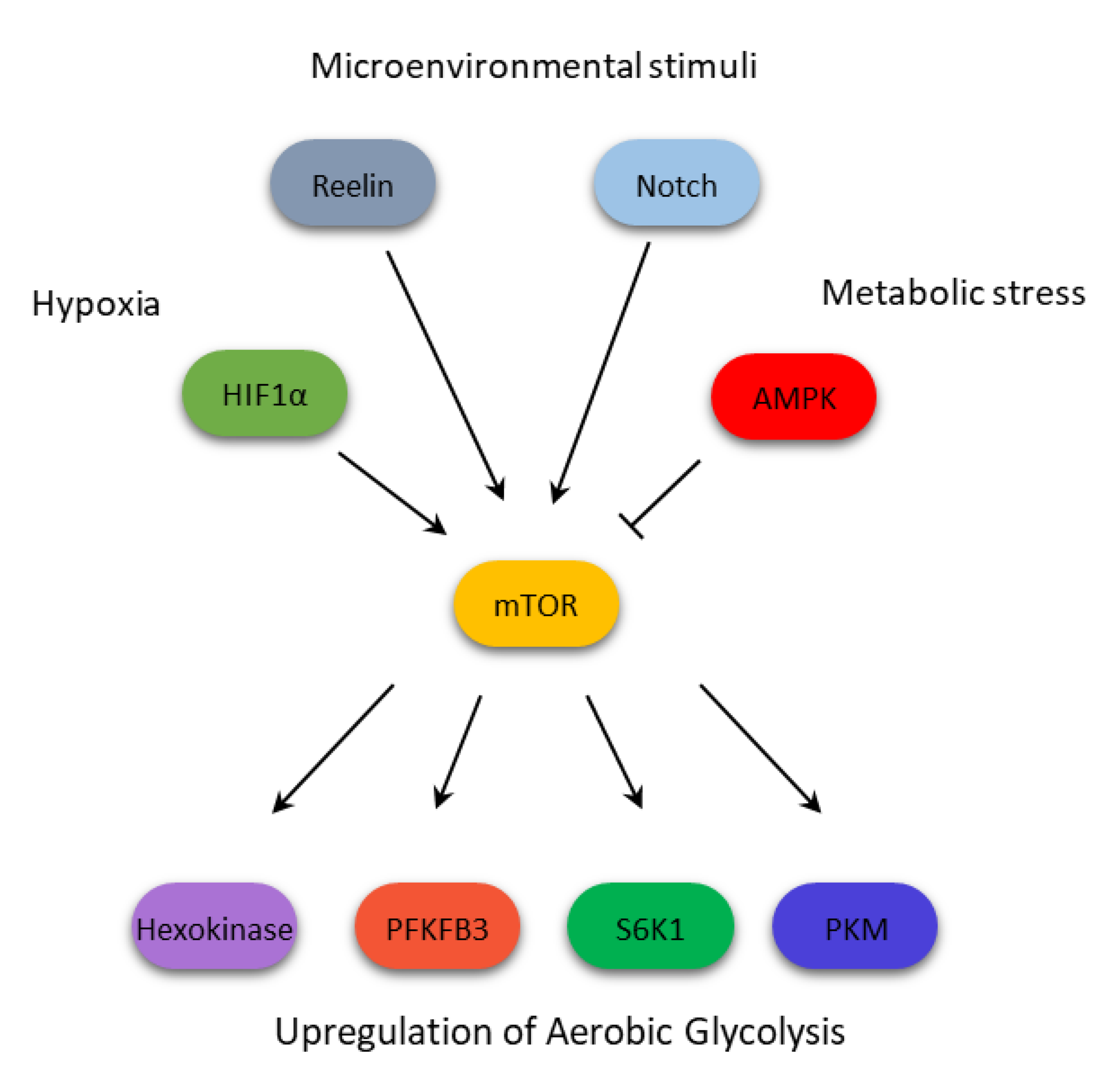
2. Cancer Metabolism and Cell Signaling
3. mTOR and the Metabolism of Hematologic Malignancies
3.1. Acute Myeloid Leukemia
3.2. Chronic Myeloid Leukemia
3.3. Acute Lymphoblastic Leukemia
3.4. Chronic Lymphocytic Leukemia
3.5. Multiple Myeloma
3.6. Lymphomas
4. Summary and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Faenza, I.; Billi, A.M.; Manzoli, L.; Evangelisti, C.; Falà, F.; Cocco, L. Intranuclear 3-phosphoinositide metabolism and Akt signaling: New mechanisms for tumori genesis and protection against apoptosis? Cell Signal 2006, 18, 1101–1107. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef]
- Sarbassov, S.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Zhang, H.; Cicchetti, G.; Onda, H.; Koon, H.B.; Asrican, K.; Bajraszewski, N.; Vazquez, F.; Carpenter, C.L.; Kwiatkowski, D.J. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin. Investig. 2003, 112, 1223–1233. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the mTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Boil. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Meyuhas, O.; Ruvinsky, I. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Esnault, S.; Shen, Z.-J.; Malter, J.S. Protein Translation and Signaling in Human Eosinophils. Front. Med. 2017, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.; Tee, A. Mammalian target of rapamycin complex 1: Signalling inputs, substrates and feedback mechanisms. Cell. Signal. 2009, 21, 827–835. [Google Scholar] [CrossRef]
- Mamane, Y.; Petroulakis, E.; LeBacquer, O.; Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 2006, 25, 6416–6422. [Google Scholar] [CrossRef]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef]
- Howell, J.J.; Ricoult, S.J.; Manning, B.D.; Ben-Sahra, I. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; León-Annicchiarico, C.L.; Munoz-Pinedo, C. Regulation of Cancer Metabolism by Oncogenes and Tumor Suppressors. Methods Enzymol. 2014, 542, 59–80. [Google Scholar] [PubMed]
- Jacquel, A.; Luciano, F.; Robert, G.; Auberger, P. Implication and Regulation of AMPK during Physiological and Pathological Myeloid Differentiation. Int. J. Mol. Sci. 2018, 19, 2991. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; Phillips, D.; McCoy, J.P.; Aponte, A.M.; Shen, R.-F.; Balaban, R.S.; Finkel, T. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J. Boil. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Boil. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sarbassov, S.D.; Ali, S.M.; Kim, D.-H.; A Guertin, D.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr. Boil. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Niles, B.J.; Powers, T. TOR complex 2–Ypk1 signaling regulates actin polarization via reactive oxygen species. Mol. Boil. Cell 2014, 25, 3962–3972. [Google Scholar] [CrossRef]
- Zoncu, R.; Sabatini, D.M.; Efeyan, A. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Kamada, Y.; Fujioka, Y.; Suzuki, N.N.; Inagaki, F.; Wullschleger, S.; Loewith, R.; Hall, M.N.; Ohsumi, Y. Tor2 Directly Phosphorylates the AGC Kinase Ypk2 To Regulate Actin Polarization†. Mol. Cell. Boil. 2005, 25, 7239–7248. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bickle, M.; Beck, T.; Hall, M.N. The Yeast Phosphatidylinositol Kinase Homolog TOR2 Activates RHO1 and RHO2 via the Exchange Factor ROM2. Cell 1997, 88, 531–542. [Google Scholar] [CrossRef]
- Dada, S.; Demartines, N.; Dormond, O. mTORC2 regulates PGE2-mediated endothelial cell survival and migration. Biochem. Biophys. Res. Commun. 2008, 372, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by Association with the Ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, e3249. [Google Scholar] [CrossRef]
- Moloughney, J.G.; Kim, P.K.; Vega-Cotto, N.M.; Wu, C.-C.; Zhang, S.; Adlam, M.; Lynch, T.; Chou, P.-C.; Rabinowitz, J.D.; Werlen, G.; et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol. Cell 2016, 63, 811–826. [Google Scholar] [CrossRef]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR Signaling Pathways in Cancer: New Data on Targeted Therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Kohli, L.; Passegué, E. Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Boil. 2014, 24, 479–487. [Google Scholar] [CrossRef]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, U.S.; et al. Inhibition of Pyruvate Kinase M2 by Reactive Oxygen Species Contributes to Cellular Antioxidant Responses. Sci. 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Boil. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet. Med. 2008, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically ‘fertilize’ the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
- Yuneva, M.O.; Fan, T.W.M.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Matés, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The Metabolic Profile of Tumors Depends on Both the Responsible Genetic Lesion and Tissue Type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Sci. 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Li, B.; Simon, M.C. Molecular Pathways: Targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin. Cancer Res. 2013, 19, 5835–5841. [Google Scholar] [CrossRef]
- Jiang, Y.; Nakada, D. Cell intrinsic and extrinsic regulation of leukemia cell metabolism. Int. J. Hematol. 2016, 103, 607–616. [Google Scholar] [CrossRef]
- Krejčí, A. Metabolic sensors and their interplay with cell signalling and transcription. Biochem. Soc. Trans. 2012, 40, 311–323. [Google Scholar] [CrossRef]
- Metallo, C.M.; Heiden, M.G.V. Metabolism strikes back: metabolic flux regulates cell signaling. Genome Res. 2010, 24, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Israelsen, W.J.; Lee, N.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Heiden, M.G.V.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Petronini, P.G.; Urbani, S.; Alfieri, R.; Borghetti, A.F.; Guidotti, G.G. Cell susceptibility to apoptosis by glutamine deprivation and rescue: Survival and apoptotic death in cultured lymphoma-leukemia cell lines. J. Cell. Physiol. 1996, 169, 175–185. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Saitoh, K.; Yang, H.; Sekihara, K.; Yamatani, K.; Ruvolo, V.; Taka, H.; Kaga, N.; Kikkawa, M.; Arai, H.; et al. Inhibition of FAO in AML co-cultured with BM adipocytes: mechanisms of survival and chemosensitization to cytarabine. Sci. Rep. 2018, 8, 16837. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Liu, L.-L.; Long, Z.-J.; Wang, L.-X.; Zheng, F.-M.; Fang, Z.-G.; Yan, M.; Xu, D.-F.; Chen, J.-J.; Wang, S.-W.; Lin, N.-J.; et al. Inhibition of mTOR Pathway Sensitizes Acute Myeloid Leukemia Cells to Aurora Inhibitors by Suppression of Glycolytic Metabolism. Mol. Cancer Res. 2013, 11, 1326–1336. [Google Scholar] [CrossRef]
- Allegretti, M.; Ricciardi, M.R.; Licchetta, R.; Mirabilii, S.; Orecchioni, S.; Reggiani, F.; Talarico, G.; Foà, R.; Bertolini, F.; Amadori, S.; et al. The pan-class I phosphatidyl-inositol-3 kinase inhibitor NVP-BKM120 demonstrates anti-leukemic activity in acute myeloid leukemia. Sci. Rep. 2015, 5, 18137. [Google Scholar] [CrossRef]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leuk. 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Pradelli, L.A.; Bénéteau, M.; Chauvin, C.; Jacquin, M.A.; Marchetti, S.; Muñoz-Pinedo, C.; Auberger, P.; Pende, M.; Ricci, J.E. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translations. Oncogene 2010, 29, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Estañ, M.C.; Calviño, E.; De Blas, E.; Boyano-Adánez, M.D.C.; Mena, M.L.; Gómez-Gómez, M.; Rial, E.; Aller, P. 2-Deoxy-d-glucose cooperates with arsenic trioxide to induce apoptosis in leukemia cells: Involvement of IGF-1R-regulated Akt/mTOR, MEK/ERK and LKB-1/AMPK signaling pathways. Biochem. Pharmacol. 2012, 84, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girão, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell. Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef]
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903. [Google Scholar] [CrossRef]
- Braun, M.; Qorraj, M.; Büttner, M.; A Klein, F.; Saul, D.; Aigner, M.; Huber, W.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leuk. 2016, 30, 1788–1792. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M. Role of Microenvironment in Resistance to Therapy in AML. Curr. Hematol. Malign- Rep. 2015, 10, 96–103. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. The mTOR Pathway in the Control of Protein Synthesis. Physiol. 2006, 21, 362–369. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Nepstad, I.; Reikvam, H.; Brenner, A.K.; Bruserud, Ø.; Hatfield, K.J. Resistance to the Antiproliferative In Vitro Effect of PI3K-Akt-mTOR Inhibition in Primary Human Acute Myeloid Leukemia Cells Is Associated with Altered Cell Metabolism. Int. J. Mol. Sci. 2018, 19, 382. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, S. Imatinib (STI571)-Mediated Changes in Glucose Metabolism in Human Leukemia BCR-ABL-Positive Cells. Clin. Cancer Res. 2004, 10, 6661–6668. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Klawitter, J.; Brown, J.L.; Boros, L.G.; Melo, J.V.; Eckhardt, S.G.; Serkova, N.J. Abnormalities in Glucose Uptake and Metabolism in Imatinib-Resistant Human BCR-ABL-Positive Cells. Clin. Cancer Res. 2009, 15, 3442–3450. [Google Scholar] [CrossRef]
- De Rosa, V.; Monti, M.; Terlizzi, C.; Fonti, R.; Del Vecchio, S.; Iommelli, F. Coordinate Modulation of Glycolytic Enzymes and OXPHOS by Imatinib in BCR-ABL Driven Chronic Myelogenous Leukemia Cells. Int. J. Mol. Sci. 2019, 20, 3134. [Google Scholar] [CrossRef] [PubMed]
- Bentley, J.; Walker, I.; McIntosh, E.; Whetton, A.; Owen-Lynch, P.J.; Baldwin, S.A. Glucose transport regulation by p210 Bcr-Abl in a chronic myeloid leukaemia model. Br. J. Haematol. 2001, 112, 212–215. [Google Scholar] [CrossRef]
- Sontakke, P.; Koczula, K.M.; Jaques, J.; Wierenga, A.T.; Brouwers-Vos, A.Z.; Pruis, M.; Günther, U.L.; Vellenga, E.; Schuringa, J.J. Hypoxia-Like Signatures Induced by BCR-ABL Potentially Alter the Glutamine Uptake for Maintaining Oxidative Phosphorylation. PLoS ONE 2016, 11, e0153226. [Google Scholar] [CrossRef]
- Zhao, F.; Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1a-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar] [CrossRef]
- Noel, B.M.; Ouellette, S.B.; Marholz, L.; Dickey, D.; Navis, C.; Yang, T.-Y.; Nguyen, V.; Parker, S.J.; Bernlohr, D.; Sachs, Z.; et al. Multiomic Profiling of Tyrosine Kinase Inhibitor-Resistant K562 Cells Suggests Metabolic Reprogramming To Promote Cell Survival. J. Proteome Res. 2019, 18, 1842–1856. [Google Scholar] [CrossRef]
- Flis, K.; Irvine, D.; Copland, M.; Bhatia, R.; Skorski, T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk. Lymphoma 2012, 53, 2474–2478. [Google Scholar] [CrossRef][Green Version]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemic stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.; He, J.; Agarwal, P.; Shah, M.; Welner, R.; Darley-Usmar, V.M.; et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investig. 2019, 129, 2685–2701. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Barger, J.F.; Gallo, C.A.; Tandon, P.; Liu, H.; Sullivan, A.; Grimes, H.L.; Plas, D.R. S6K1 determines the metabolic requirements for BCR-ABL survival. Oncogene 2013, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Taniguchi, K.; Kumazaki, M.; Yamada, N.; Ito, Y.; Otsuki, Y.; Uno, B.; Hayakawa, F.; Minami, Y.; Naoe, T.; et al. Anti-cancer fatty-acid derivative induces autophagic cell death through modulation of PKM isoform expression profile mediated by bcr-abl in chronic myeloid leukemia. Cancer Lett. 2015, 360, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Deng, X.; Liu, Y.; Liu, Y.; Sun, L.; Chen, F. PKM2, function and expression and regulation. Cell Biosci. 2019, 9, 52. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Ai, Z.; Zhang, Z.; Lin, L.; Wei, H. Targeting glycometabolic reprogramming to restore the sensitivity of leukemia drug-resistant K562/ADM cells to adriamycin. Life Sci. 2018, 215, 1–10. [Google Scholar] [CrossRef]
- Boag, J.M.; Beesley, A.H.; Firth, M.J.; Freitas, J.R.; Ford, J.; Hoffmann, K.; Cummings, A.J.; De Klerk, N.H.; Kees, U.R. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leuk. 2006, 20, 1731–1737. [Google Scholar] [CrossRef]
- Liu, T.; Kishton, R.J.; Macintyre, A.N.; Gerriets, V.A.; Xiang, H.; Liu, X.; Abel, E.D.; Rizzieri, D.; Locasale, J.W.; Rathmell, J.C. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis 2014, 5, e1470. [Google Scholar] [CrossRef]
- Stäubert, C.; Bhuiyan, H.; Lindahl, A.; Broom, O.J.; Zhu, Y.; Islam, S.; Linnarsson, S.; Lehtiö, J.; Nordström, A. Rewired Metabolism in Drug-resistant Leukemia Cells: A metabolic switch hallmarked by reduced dependence on exogenous glutamine. J. Biol. Chem. 2015, 290, 8348–8359. [Google Scholar] [CrossRef]
- Aoki, S.; Morita, M.; Hirao, T.; Yamaguchi, M.; Shiratori, R.; Kikuya, M.; Chibana, H.; Ito, K. Shift in energy metabolism caused by glucocorticoids enhances the effect of cytotoxic anti-cancer drugs against acute lymphoblastic leukemia cells. Oncotarget 2017, 8, 94271–94285. [Google Scholar] [CrossRef][Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; MacIntyre, A.N.; Goraksha-Hicks, P.; De Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A. The role of NOTCH1 signaling in T-ALL. Hematol. 2009, 2009, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sanchez-Martin, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Boil. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, G.J.; DeSalvo, J.; Du, J.; Gao, N.; Leclerc, G.M.; Lehrman, M.A.; Lampidis, T.J.; Barredo, J.C. Mcl-1 downregulation leads to the heightened sensitivity exhibited by BCR-ABL positive ALL to induction of energy and ER-stress. Leuk. Res. 2015, 39, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Miriyala, S.; Keaton, M.A.; Jordan, C.T.; Wiedl, C.; Clair, D.K.S.; Moscow, J.A. Metabolic Effects of Acute Thiamine Depletion Are Reversed by Rapamycin in Breast and Leukemia Cells. PLoS ONE 2014, 9, e85702. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.A.; Marchetti-Laurent, C.; Poindessous, V.; Antonio, S.; Laurent-Puig, P.; Bortoli, S.; Loriot, M.-A.; Pallet, N. 6-mercaptopurine promotes energetic failure in proliferating T cells. Oncotarget 2017, 8, 43048–43060. [Google Scholar] [CrossRef]
- Clapham, C.E.; Pettitt, A.R.; Slupsky, J.R. Targeting Cell Metabolism In Chronic Lymphocytic Leukaemia (CLL); A Viable Therapeutic Approach? J. Hematol. Oncol. Res. 2014, 1, 7–23. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Aloyz, R. Metabolic rewiring beyond Warburg in chronic lymphocytic leukemia: How much do we actually know? Crit. Rev. Oncol. 2019, 134, 65–70. [Google Scholar] [CrossRef]
- Koczula, K.M.; Ludwig, C.; Hayden, R.; Cronin, L.; Pratt, G.; Parry, H.; Tennant, D.; Drayson, M.; Bunce, C.M.; Khanim, F.L.; et al. Metabolic plasticity in CLL: Adaptation to the hypoxic niche. Leukemia 2016, 30, 65–73. [Google Scholar] [CrossRef]
- Vangapandu, H.V.; Havranek, O.; Ayres, M.L.; Kaipparettu, B.A.; Balakrishnan, K.; Wierda, W.G.; Keating, M.J.; Davis, R.E.; Stellrecht, C.M.; Gandhi, V. B-cell Receptor Signaling Regulates Metabolism in Chronic Lymphocytic Leukemia. Mol. Cancer Res. 2017, 15, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Braun, M.; Qorraj, M.; Saul, D.; Le Blanc, K.; Zenz, T.; Mougiakakos, D. Stromal cell–mediated glycolytic switch in CLL cells involves Notch-c-Myc signaling. Blood 2015, 125, 3432–3436. [Google Scholar] [CrossRef] [PubMed]
- Vangapandu, H.V.; Ayres, M.L.; Bristow, C.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Stellrecht, C.M.; Gandhi, V. The Stromal Microenvironment Modulates Mitochondrial Oxidative Phosphorylation in Chronic Lymphocytic Leukemia Cells. Neoplasia 2017, 19, 762–771. [Google Scholar] [CrossRef]
- Martinez-Marignac, V.; Smith, S.; Toban, N.; Bazile, M.; Aloyz, R. Resistance to Dasatinib in primary chronic lymphocytic leukemia lymphocytes involves AMPK-mediated energetic re-programming. Oncotarget 2013, 4, 2550–2566. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Janocha, A.J.; Hill, B.T.; Smith, M.R.; Erzurum, S.C.; Almasan, A. Targeting mTORC1-mediated metabolic addiction overcomes fludarabine resistance in malignant B cells. Mol. Cancer Res. 2014, 12, 1205–1215. [Google Scholar] [CrossRef]
- Siska, P.J.; Van Der Windt, G.J.W.; Kishton, R.J.; Cohen, S.; Eisner, W.; Maciver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef]
- McBrayer, S.K.; Cheng, J.C.; Singhal, S.; Krett, N.L.; Rosen, S.T.; Shanmugam, M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood 2012, 119, 4686–4697. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1β regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595. [Google Scholar] [CrossRef]
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015, 75, 2071–2082. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Ramakrishnan, V.; Hitosugi, T.; Ghosh, T.; Jevremovic, D.; Dutta, T.; Sakrikar, D.; Petterson, X.-M.; Wellik, L.; Kumar, S.K.; et al. Glutamine-derived 2-hydroxyglutarate is associated with disease progression in plasma cell malignancies. JCI Insight 2018, 3, 94543. [Google Scholar] [CrossRef]
- Hresko, R.C.; Hruz, P.W. HIV Protease Inhibitors Act as Competitive Inhibitors of the Cytoplasmic Glucose Binding Site of GLUTs with Differing Affinities for GLUT1 and GLUT4. PLoS ONE 2011, 6, e25237. [Google Scholar] [CrossRef] [PubMed]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Shanmugam, M.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; et al. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Demel, H.-R.; Feuerecker, B.; Piontek, G.; Seidl, C.; Blechert, B.; Pickhard, A.; Essler, M. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. Am. J. Cancer Res. 2015, 5, 1649–1664. [Google Scholar] [PubMed]
- Lis, P.; Dyląg, M.; Niedźwiecka, K.; Ko, Y.H.; Pedersen, P.L.; Goffeau, A.; Ułaszewski, S. The HK2 Dependent “Warburg Effect” and Mitochondrial Oxidative Phosphorylation in Cancer: Targets for Effective Therapy with 3-Bromopyruvate. Mol. 2016, 21, 1730. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Miki, H.; Nakamura, S.; Harada, T.; Oda, A.; Amou, H.; Fujii, S.; Kagawa, K.; Takeuchi, K.; Ozaki, S.; et al. Up-regulation of hexokinaseII in myeloma cells: targeting myeloma cells with 3-bromopyruvate. J. Bioenerg. Biomembr. 2012, 44, 31–38. [Google Scholar] [CrossRef]
- Khialeeva, E.; Carpenter, E.M. Nonneuronal roles for the reelin signaling pathway. Dev. Dyn. 2017, 246, 217–226. [Google Scholar] [CrossRef]
- Qin, X.; Lin, L.; Cao, L.; Zhang, X.; Song, X.; Hao, J.; Zhang, Y.; Wei, R.; Huang, X.; Lu, J.; et al. Extracellular matrix protein Reelin promotes myeloma progression by facilitating tumor cell proliferation and glycolysis. Sci. Rep. 2017, 7, 45305. [Google Scholar] [CrossRef]
- Zhang, D.X.; Zhang, J.P.; Hu, J.Y.; Huang, Y.S. The potential regulatory roles of NAD(+) and its metabolism in autophagy. Metabolism 2016, 65, 454–462. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef]
- Mikkilineni, L.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Sprandio, J.; Avena, P.; Cotzia, P.; Dulau-Florea, A.; Gong, J.; Uppal, G.; Zhan, T.; et al. Hodgkin lymphoma: A complex metabolic ecosystem with glycolytic reprogramming of the tumor microenvironment. Semin. Oncol. 2017, 44, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, N.; Aguiar, R.C.T.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef]
- Lee, S.C.; Marzec, M.; Liu, X.; Wehrli, S.; Kantekure, K.; Ragunath, P.N.; Nelson, D.S.; Delikatny, E.J.; Glickson, J.D.; Wasik, M.A. Decreased lactate concentration and glycolytic enzyme expression reflect inhibition of mTOR signal. NMR Biomed. 2013, 26, 106–114. [Google Scholar] [CrossRef]
- Mediani, L.; Gibellini, F.; Bertacchini, J.; Frasson, C.; Bosco, R.; Accordi, B.; Basso, G.; Bonora, M.; Calabrò, M.L.; Mattiolo, A.; et al. Reversal of the glycolytic phenotype of primary effusion lymphoma cells by combined targeting of cellular metabolism and PI3K/Akt/mTOR signaling. Oncotarget 2016, 7, 5521–5537. [Google Scholar] [CrossRef]
- Akhenblit, P.J.; Hanke, N.T.; Gill, A.; Persky, D.O.; Howison, C.M.; Pagel, M.D.; Baker, A.F. Assessing Metabolic Changes in Response to mTOR Inhibition in a Mantle Cell Lymphoma Xenograft Model Using AcidoCEST MRI. Mol. Imaging 2016, 15, 1536012116645439. [Google Scholar] [CrossRef]
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017, 8, 34552–34564. [Google Scholar] [CrossRef]
- Coloff, J.L.; MacIntyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef]
- Chiche, J.; Reverso-Meinietti, J.; Mouchotte, A.; Rubio-Patiño, C.; Mhaidly, R.; Villa, E.; Bossowski, J.P.; Proics, E.; Grima-Reyes, M.; Paquet, A.; et al. GAPDH Expression Predicts the Response to R-CHOP, the Tumor Metabolic Status, and the Response of DLBCL Patients to Metabolic Inhibitor. Cell Metab. 2019, 29, 1243–1257. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Effect (Ref) |
|---|---|---|
| rapamycin | mTORC1 | Decreased Glucose uptake (59); |
| Shift from Glycolysis to OXPHOS (61); | ||
| Decreased glycolysis through PFKB3 downregulation (65) | ||
| BKM-120 | PI3K | Decreased Glycolysis and OXPHOS |
| 2DG | Hexokinase | mTOR inactivation (62); |
| mTOR activation (63) | ||
| L-asparaginases | Asparagine and glutamine degradation | mTOR inactivation (70) |
| Type | Compound | Target | Effect (Ref) |
|---|---|---|---|
| B-ALL | everolimus | mTORC1 | Decreased glycolysis and lactate generation (94) |
| rapamycin | mTORC1 | Decrease of glycolysis and increase of OXPHOS, reversion of thiaminase effects (96) | |
| T-ALL | 6-mercaptopurine | mTOR through AMPK activation | Decreased glucose and glutamine consumption (97) |
| Type | Compound | Target | Effect (Ref) |
|---|---|---|---|
| PEL | LY294002 | PI3K | Decreased glycolysis and FAS (125) |
| PF04691502 | PI3K/mTOR | Reduction of lactate production (126) | |
| Akti 1/2 | Akt | Reduction of lactate production (126) | |
| MCL | Everolimus | mTORC1 | Reduction of lactate production (127) |
| AZD-2014 | mTORC1/2 | Downregulation of glycolytic enzymes (128) | |
| DBLCL | 2DG | hexokinase | Inactivation of Akt/mTOR and decreased expression of Mcl-1 (129) |
| rapamycin | mTORC1 | Reduction of OXPHOS and increase of glycolytic ATP in the oxidative DBLCL subset (130) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirabilii, S.; Ricciardi, M.R.; Tafuri, A. mTOR Regulation of Metabolism in Hematologic Malignancies. Cells 2020, 9, 404. https://doi.org/10.3390/cells9020404
Mirabilii S, Ricciardi MR, Tafuri A. mTOR Regulation of Metabolism in Hematologic Malignancies. Cells. 2020; 9(2):404. https://doi.org/10.3390/cells9020404
Chicago/Turabian StyleMirabilii, Simone, Maria Rosaria Ricciardi, and Agostino Tafuri. 2020. "mTOR Regulation of Metabolism in Hematologic Malignancies" Cells 9, no. 2: 404. https://doi.org/10.3390/cells9020404
APA StyleMirabilii, S., Ricciardi, M. R., & Tafuri, A. (2020). mTOR Regulation of Metabolism in Hematologic Malignancies. Cells, 9(2), 404. https://doi.org/10.3390/cells9020404