SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Treatments
2.2. Echocardiography
2.3. Cell Culture and Reagents
2.4. Cell death Assessment by Flow Cytometry
2.5. siRNA Transfection
2.6. RNA Isolation and Quantitative RT-PCR
2.7. Adult Cardiomyocytes Isolation
2.8. Evaluation of Adult Cardiomyocyte Death by Fluorescence Microscopy
2.9. Electron Microscopy
2.10. Western Blot Analysis
2.11. Autophagy Assessment
2.12. Enzyme Activity
2.13. Preparation of Isolated Mitochondria
2.14. Native and Denaturing Immunoprecipitations
2.15. Statistical Analysis
3. Results
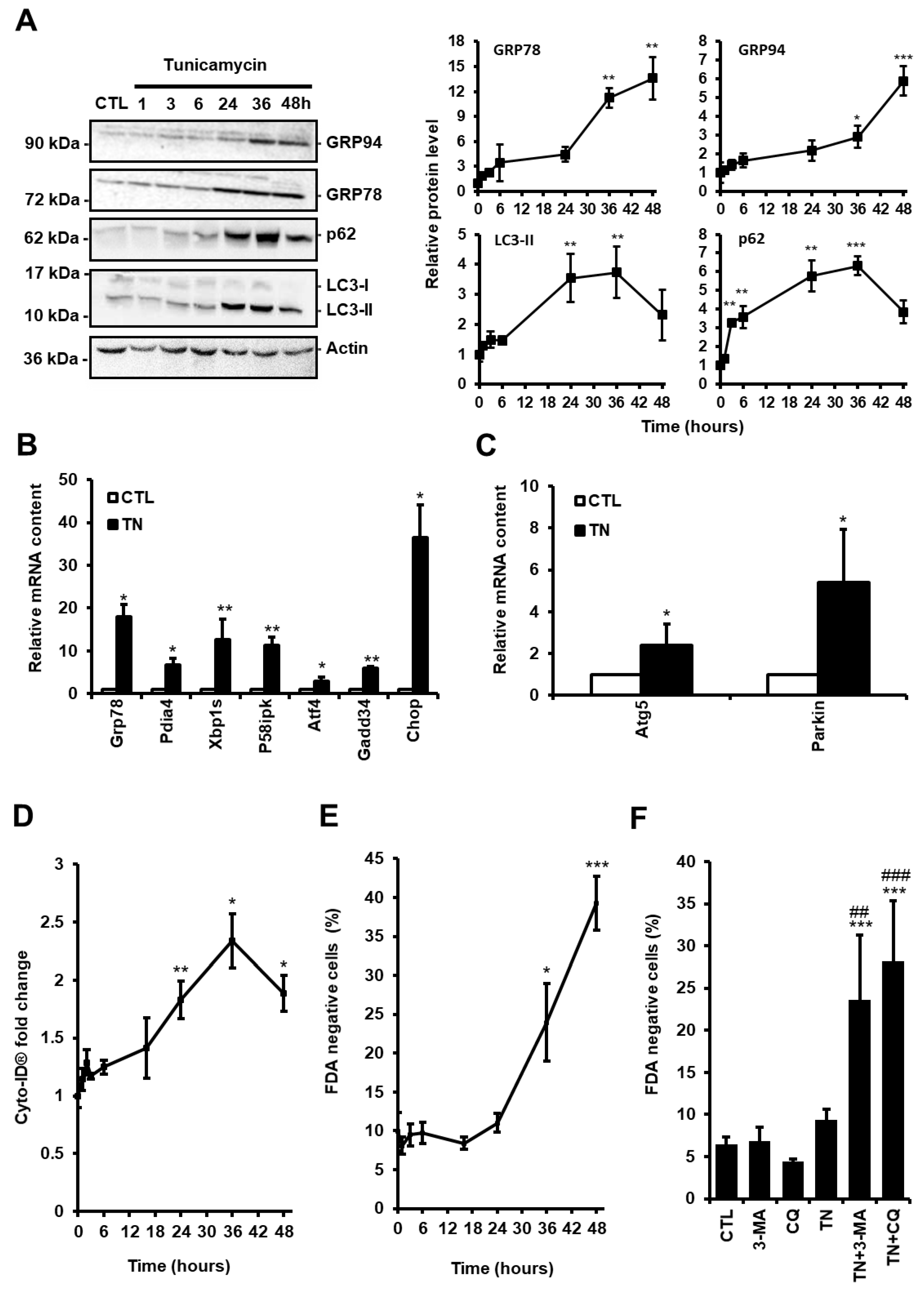
3.1. ER Stress Induces Protective Autophagy in Cardiac Cells
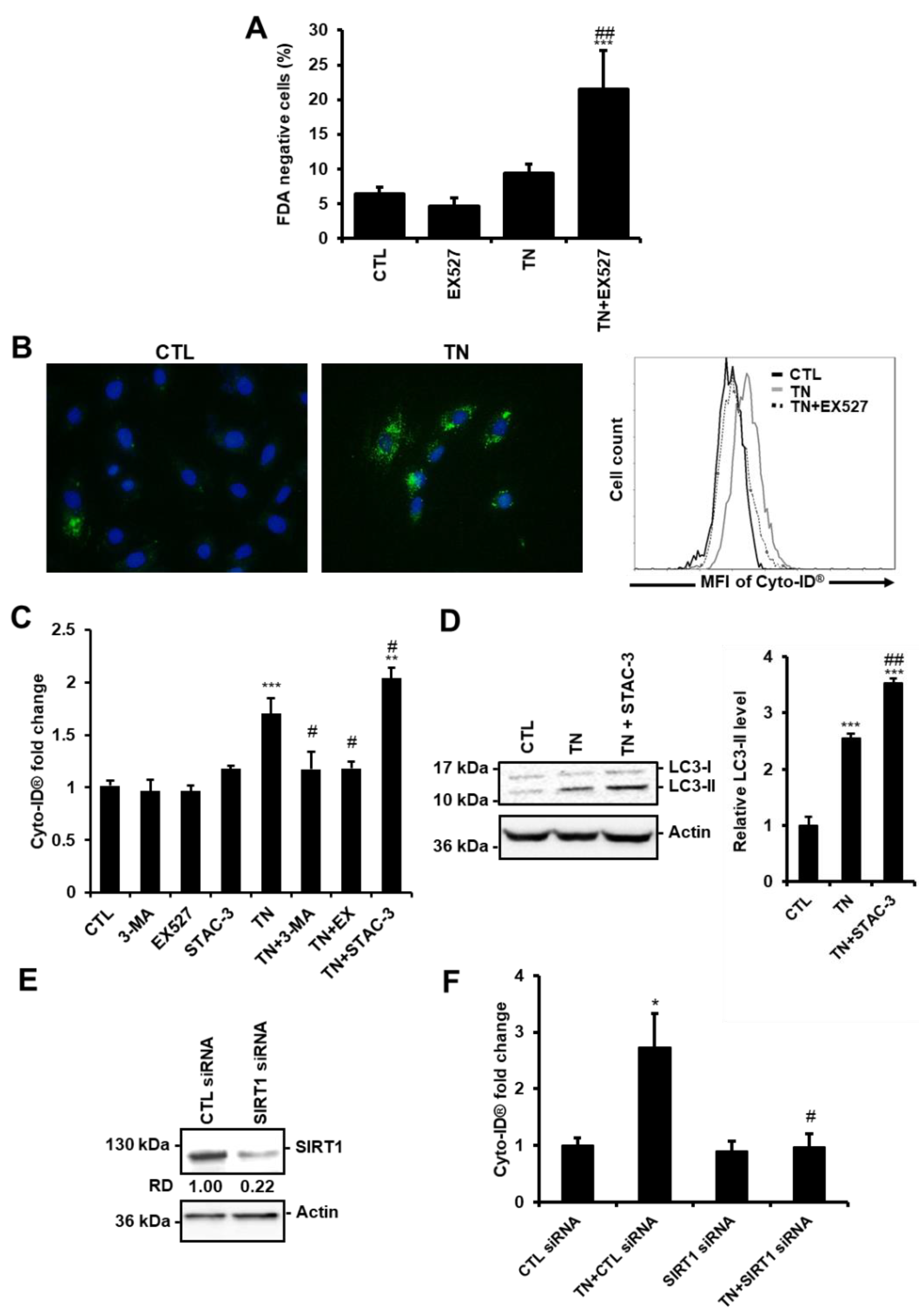
3.2. SIRT1 Regulates ER Stress-Induced Autophagy in Cardiac Cells
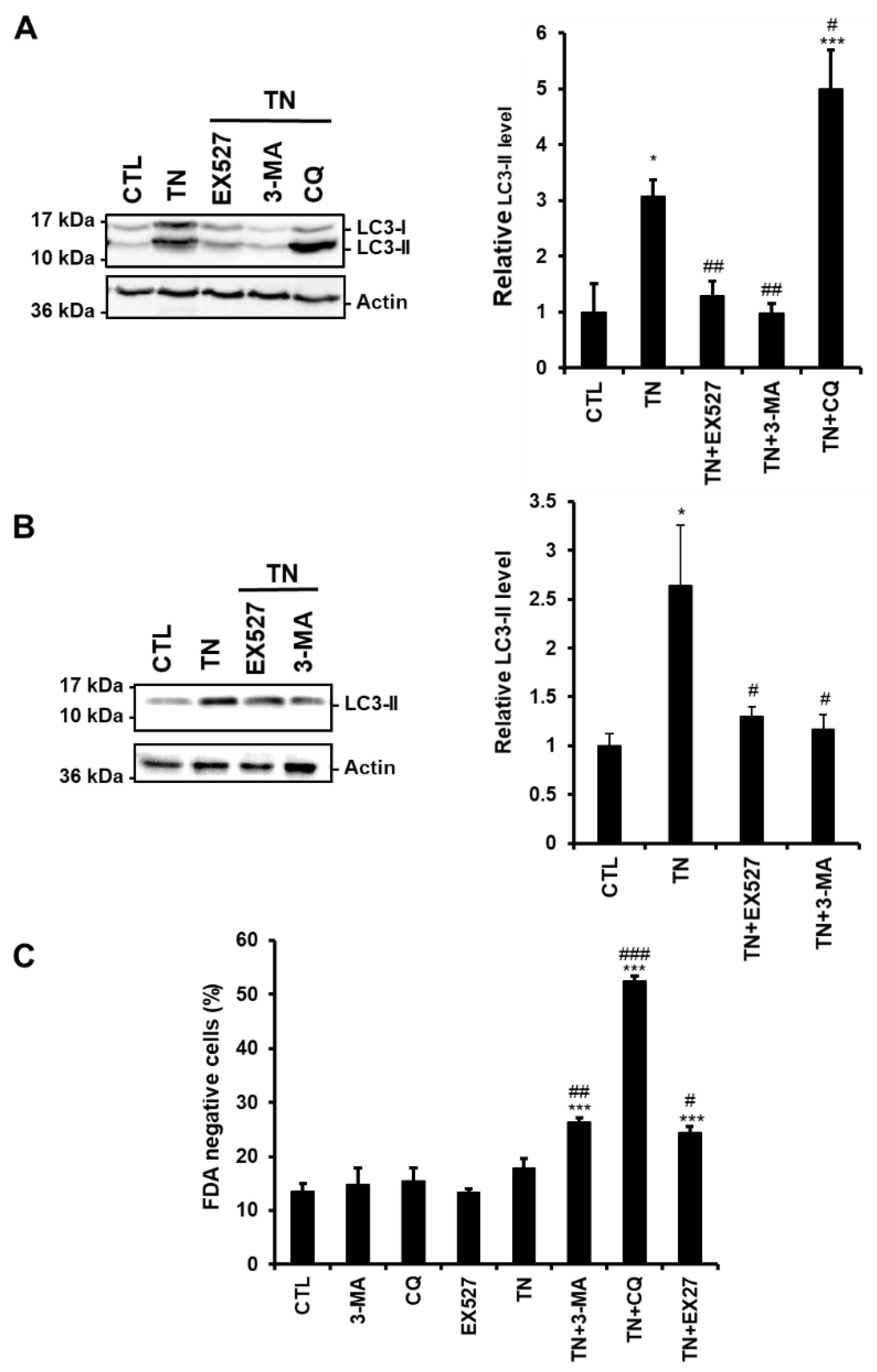
3.3. SIRT1 Inhibition Reduces the Accumulation of LC3-II Induced by ER Stress in Cardiac Cells
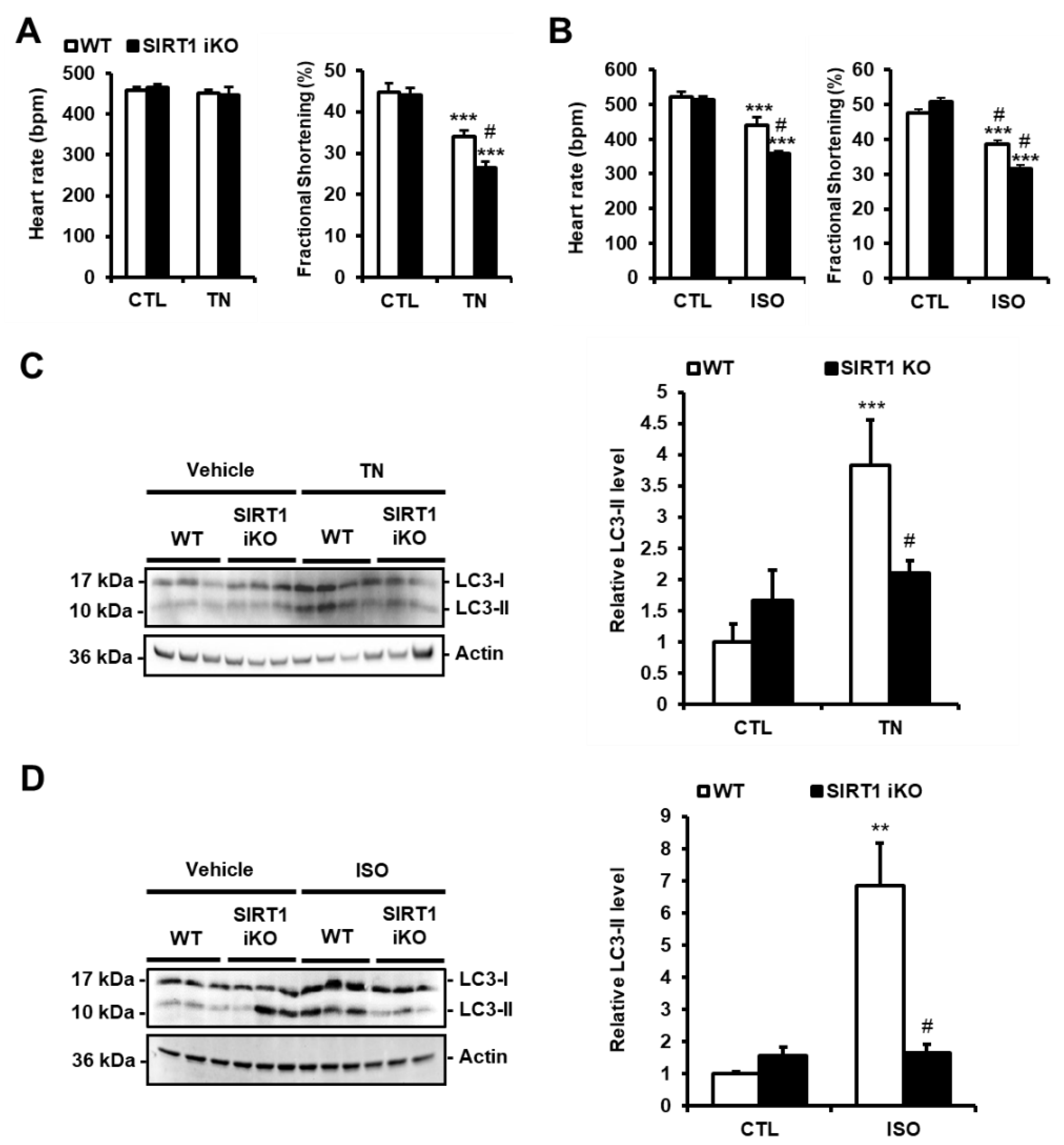
3.4. SIRT1 Deficiency Inhibits ER Stress-Induced Autophagy and Exacerbates Cardiac Injury
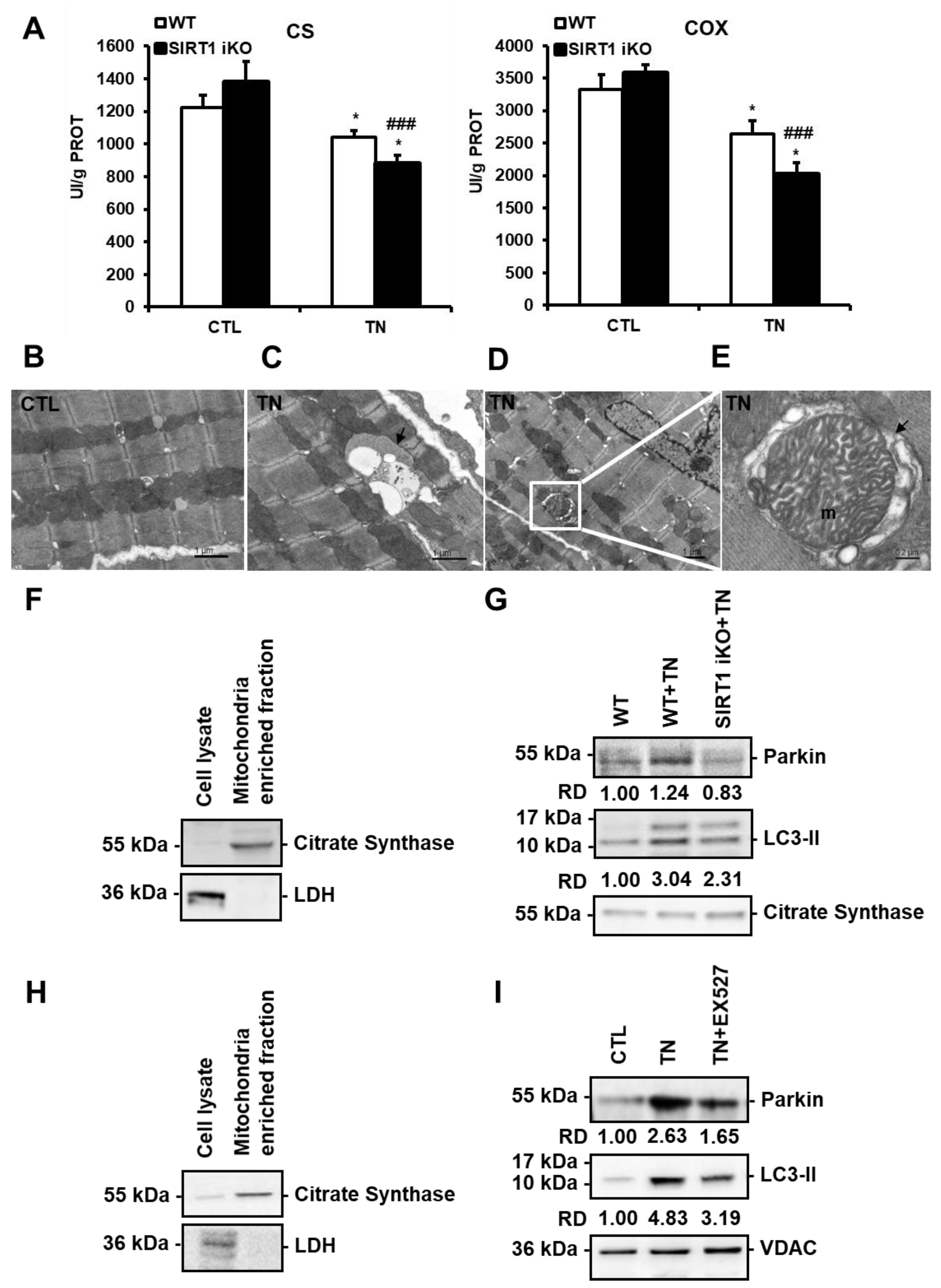
3.5. ER stress-Induced Cardiac Mitophagy Is Regulated by SIRT1
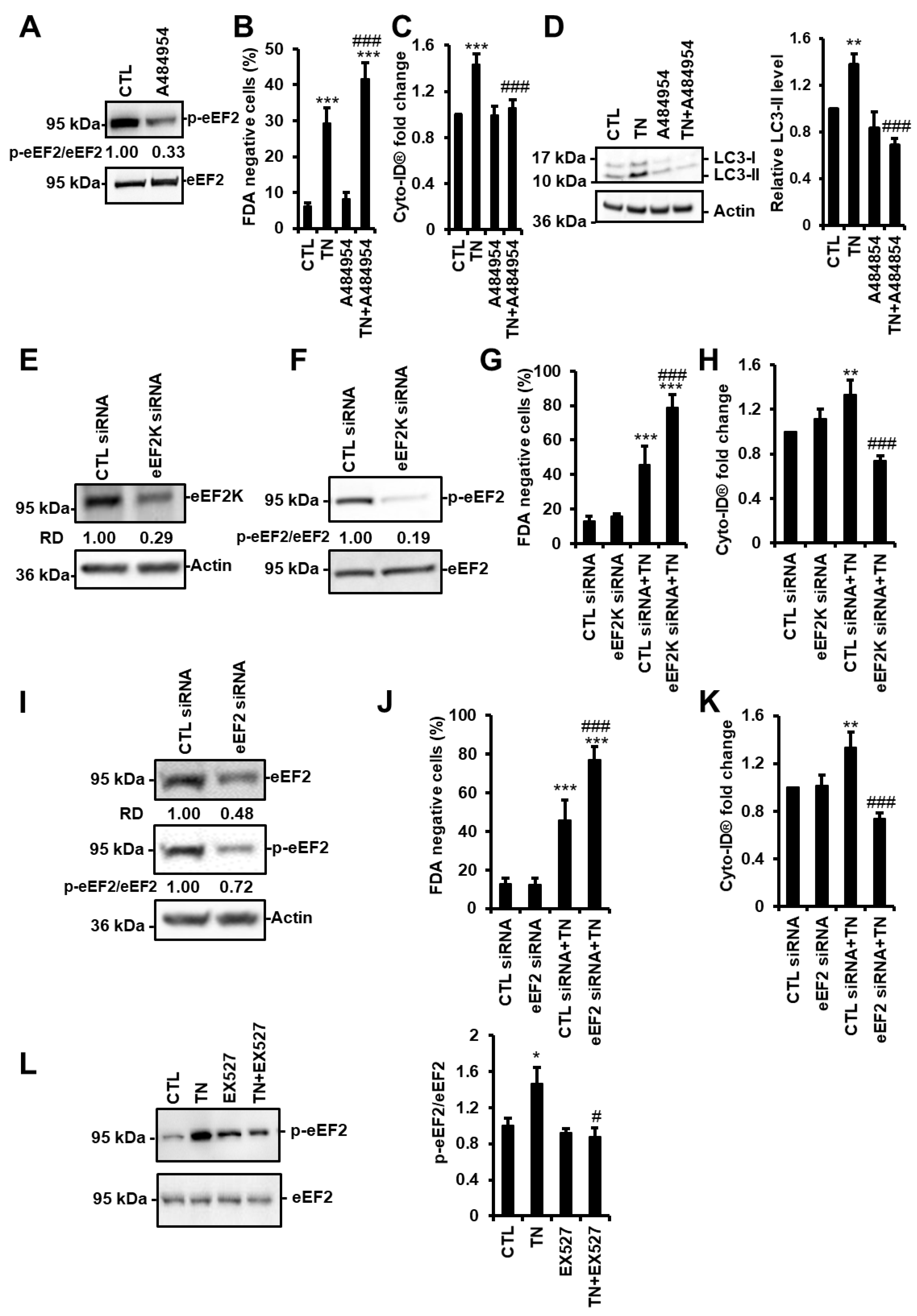
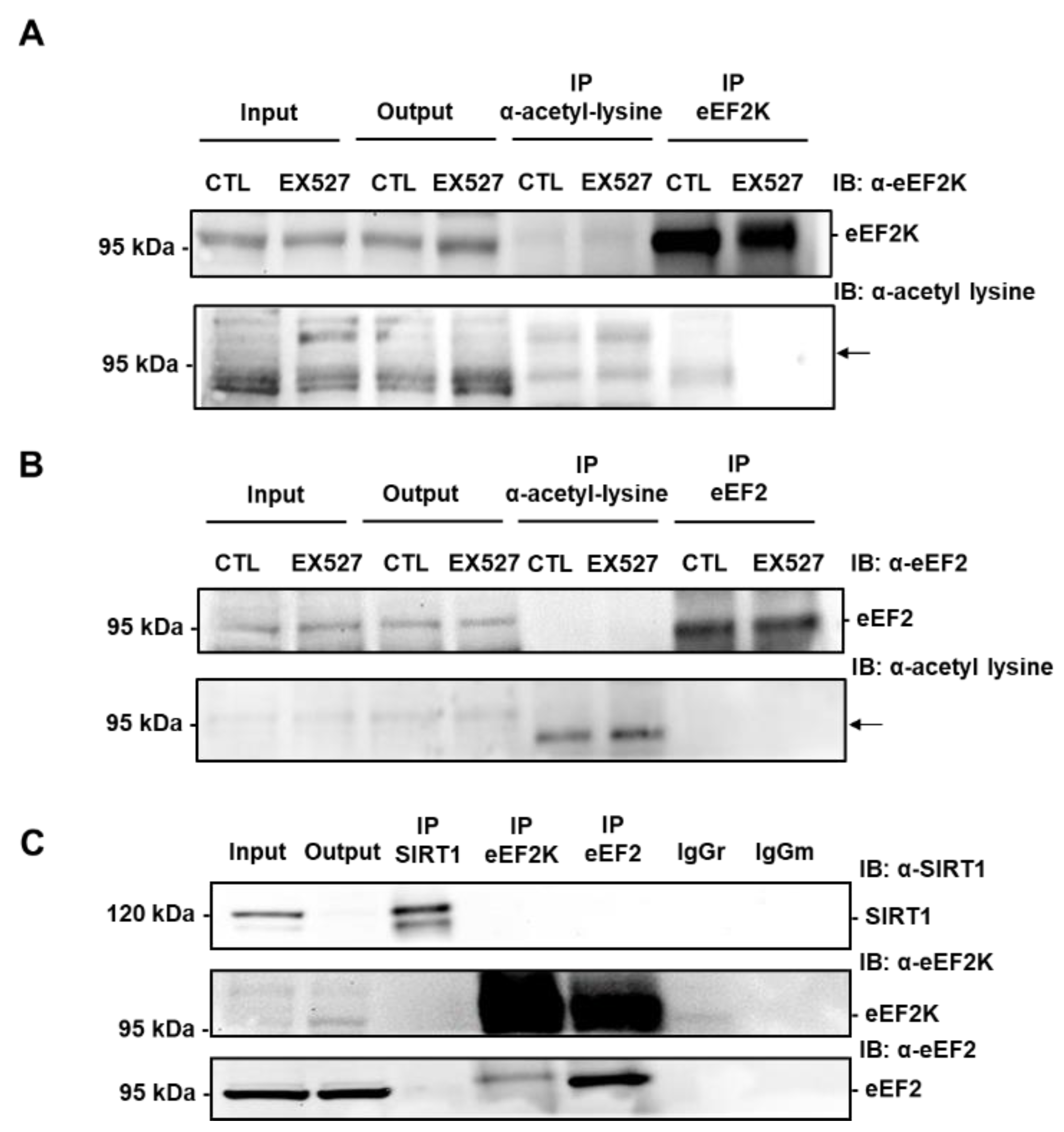
3.6. SIRT1 Activates eEF2K/eEF2 Pathway to Regulate Autophagy and Cell Death in Response to ER Stress
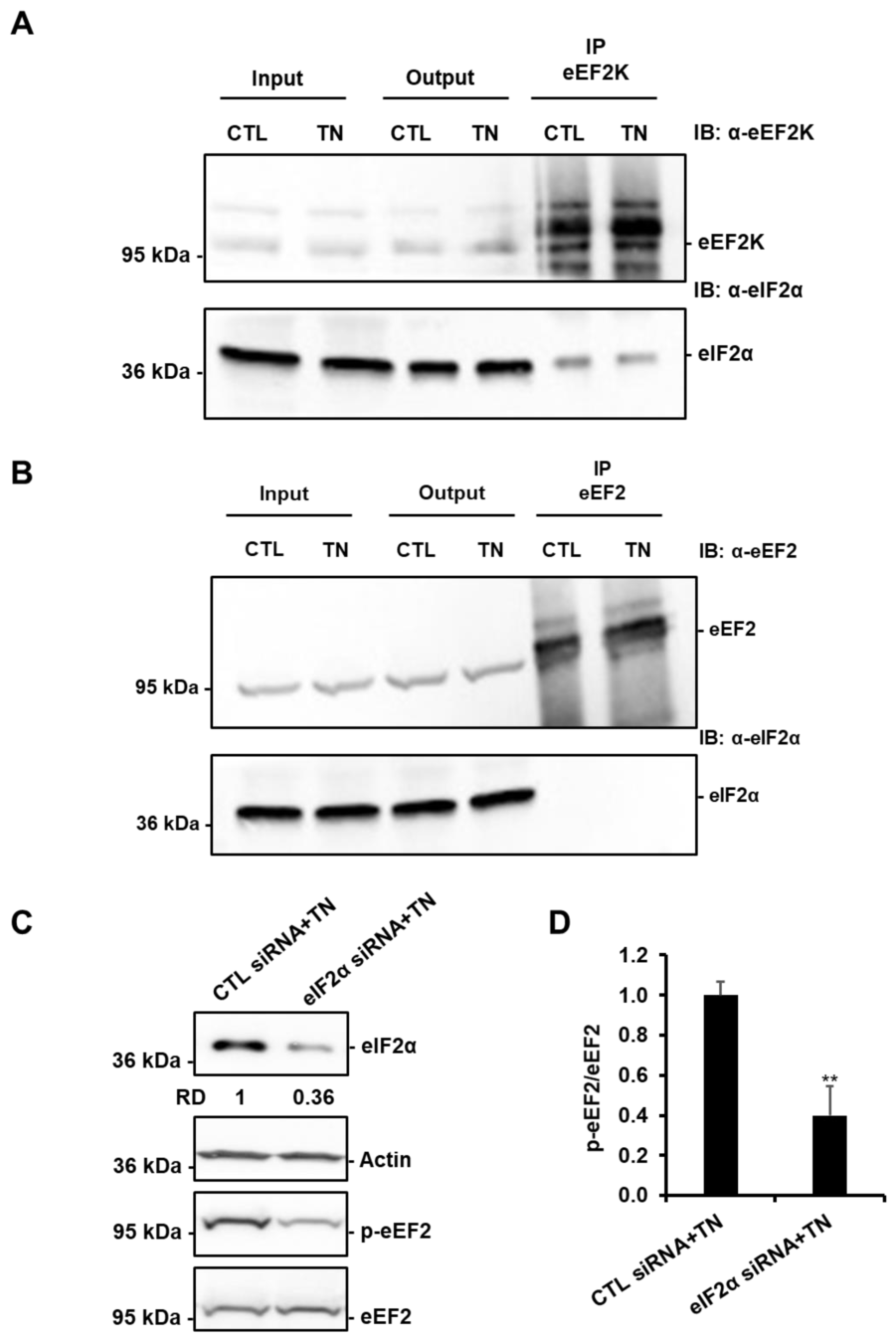
3.7. SIRT1-Mediated Activation of eEF2K/eEF2 Pathway Involves eIF2α
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Nakajima, M.; Uemura, K.; Yoshida, K. Ischemic preconditioning protects cardiomyocytes against ischemic injury by inducing GRP78. Biochem. Biophys. Res. Commun. 2006, 345, 1600–1605. [Google Scholar] [CrossRef]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Mialet-Perez, J.; Vindis, C. Autophagy in health and disease: Focus on the cardiovascular system. Essays Biochem. 2017, 61, 721–732. [Google Scholar]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of Autophagy and Apoptosis: Involvement of the Dual Role of Autophagy under ER Stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef]
- Cheng, Y.; Ren, X.; Zhang, Y.; Patel, R.; Sharma, A.; Wu, H.; Robertson, G.P.; Yan, L.; Rubin, E.; Yang, J.-M. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011, 71, 2654–2663. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y. SIRT1: Role in cardiovascular biology. Clin. Chim. Acta Int. J. Clin. Chem. 2014, 440C, 8–15. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Prola, A.; Pires Da Silva, J.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2α deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; Sook, Y.E.; Lamming, D.W.; et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef]
- Verde, I.; Vandecasteele, G.; Lezoualc’h, F.; Fischmeister, R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br. J. Pharmacol. 1999, 127, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Wiersma, M.; Meijering, R.A.M.; Qi, X.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B.J.J.M. Endoplasmic Reticulum Stress Is Associated with Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Heart Assoc. 2017, 6, e006458. [Google Scholar] [CrossRef]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef]
- Chan, L.L.-Y.; Shen, D.; Wilkinson, A.R.; Patton, W.; Lai, N.; Chan, E.; Kuksin, D.; Lin, B.; Qiu, J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012, 8, 1371–1382. [Google Scholar] [CrossRef]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.-F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Montenegro, J.; Vivar, R.; Letelier, A.; Urroz, P.A.; Copaja, M.; Pivet, D.; Humeres, C.; Troncoso, R.; Vicencio, J.M.; et al. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp. Mol. Pathol. 2012, 92, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Nah, J.; Miyamoto, S.; Sadoshima, J. Mitophagy as a Protective Mechanism against Myocardial Stress. Compr. Physiol. 2017, 7, 1407–1424. [Google Scholar]
- Zhu, H.; Yang, X.; Liu, J.; Zhou, L.; Zhang, C.; Xu, L.; Qin, Q.; Zhan, L.; Lu, J.; Cheng, H.; et al. Eukaryotic elongation factor 2 kinase confers tolerance to stress conditions in cancer cells. Cell Stress Chaperones 2015, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Castillero, E.; Akashi, H.; Pendrak, K.; Yerebakan, H.; Najjar, M.; Wang, C.; Naka, Y.; Mancini, D.M.; Sweeney, H.L.; D’Armiento, J.M.; et al. Attenuation of the Unfolded Protein Response and Endoplasmic Reticulum Stress after Mechanical Unloading in Dilated Cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Orogo, A.M.; Gustafsson, Å.B. Therapeutic Targeting of Autophagy: Potential and Concerns in Treating Cardiovascular Disease. Circ. Res. 2015, 116, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Nakaoka, M.; Iwai-Kanai, E.; Katamura, M.; Okawa, Y.; Mita, Y.; Matoba, S. An alpha-adrenergic agonist protects hearts by inducing Akt1-mediated autophagy. Biochem. Biophys. Res. Commun. 2015, 456, 250–256. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion: Roles of AMP-Activated Protein Kinase and Beclin 1 in Mediating Autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.-P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Ben Salem, I.; Boussabbeh, M.; Da Silva, J.P.; Guilbert, A.; Bacha, H.; Abid-Essefi, S.; Lemaire, C. SIRT1 protects cardiac cells against apoptosis induced by zearalenone or its metabolites α- and β-zearalenol through an autophagy-dependent pathway. Toxicol. Appl. Pharmacol. 2017, 314, 82–90. [Google Scholar] [CrossRef]
- Boyce, M.; Py, B.F.; Ryazanov, A.G.; Minden, J.S.; Long, K.; Ma, D.; Yuan, J. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 2008, 15, 589–599. [Google Scholar] [CrossRef]
- Py, B.F.; Boyce, M.; Yuan, J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy 2009, 5, 393–396. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2006, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Zhang, Y.; Shan, Y.; Huber-Keener, K.J.; Zhang, L.; Kimball, S.R.; Harvey, H.; Jefferson, L.S.; Yang, J.-M. Integrated regulation of autophagy and apoptosis by EEF2K controls cellular fate and modulates the efficacy of curcumin and velcade against tumor cells. Autophagy 2013, 9, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-O.; Seo, S.-K.; Woo, S.-H.; Kim, E.-S.; Lee, H.-C.; Yoo, D.-H.; An, S.; Choe, T.-B.; Lee, S.-J.; Hong, S.-I.; et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic. Biol. Med. 2009, 46, 1158–1167. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires Da Silva, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Piquereau, J.; Novotova, M.; Ventura-Clapier, R.; Garnier, A.; Lemaire, C. SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy. Cells 2020, 9, 426. https://doi.org/10.3390/cells9020426
Pires Da Silva J, Monceaux K, Guilbert A, Gressette M, Piquereau J, Novotova M, Ventura-Clapier R, Garnier A, Lemaire C. SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy. Cells. 2020; 9(2):426. https://doi.org/10.3390/cells9020426
Chicago/Turabian StylePires Da Silva, Julie, Kevin Monceaux, Arnaud Guilbert, Mélanie Gressette, Jérôme Piquereau, Marta Novotova, Renée Ventura-Clapier, Anne Garnier, and Christophe Lemaire. 2020. "SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy" Cells 9, no. 2: 426. https://doi.org/10.3390/cells9020426
APA StylePires Da Silva, J., Monceaux, K., Guilbert, A., Gressette, M., Piquereau, J., Novotova, M., Ventura-Clapier, R., Garnier, A., & Lemaire, C. (2020). SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy. Cells, 9(2), 426. https://doi.org/10.3390/cells9020426