The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Reagents
2.2. Cell Culture and Treatments
2.3. siRNA and Transfection
2.4. SDS-PAGE and Western Blotting
2.5. Quantitative Real-Time PCR (qPCR)
2.6. Immunofluorescence Imaging
2.7. Gelatin Degradation Assay
2.8. Measurements of Intracellular pH (pHi)
2.9. Single Cell Migration Analysis
2.10. Cell Invasion Analyses
2.11. Proliferation Assay
2.12. 3D spheroid Growth and CellTiter Glo Assay
2.13. Statistics
3. Results
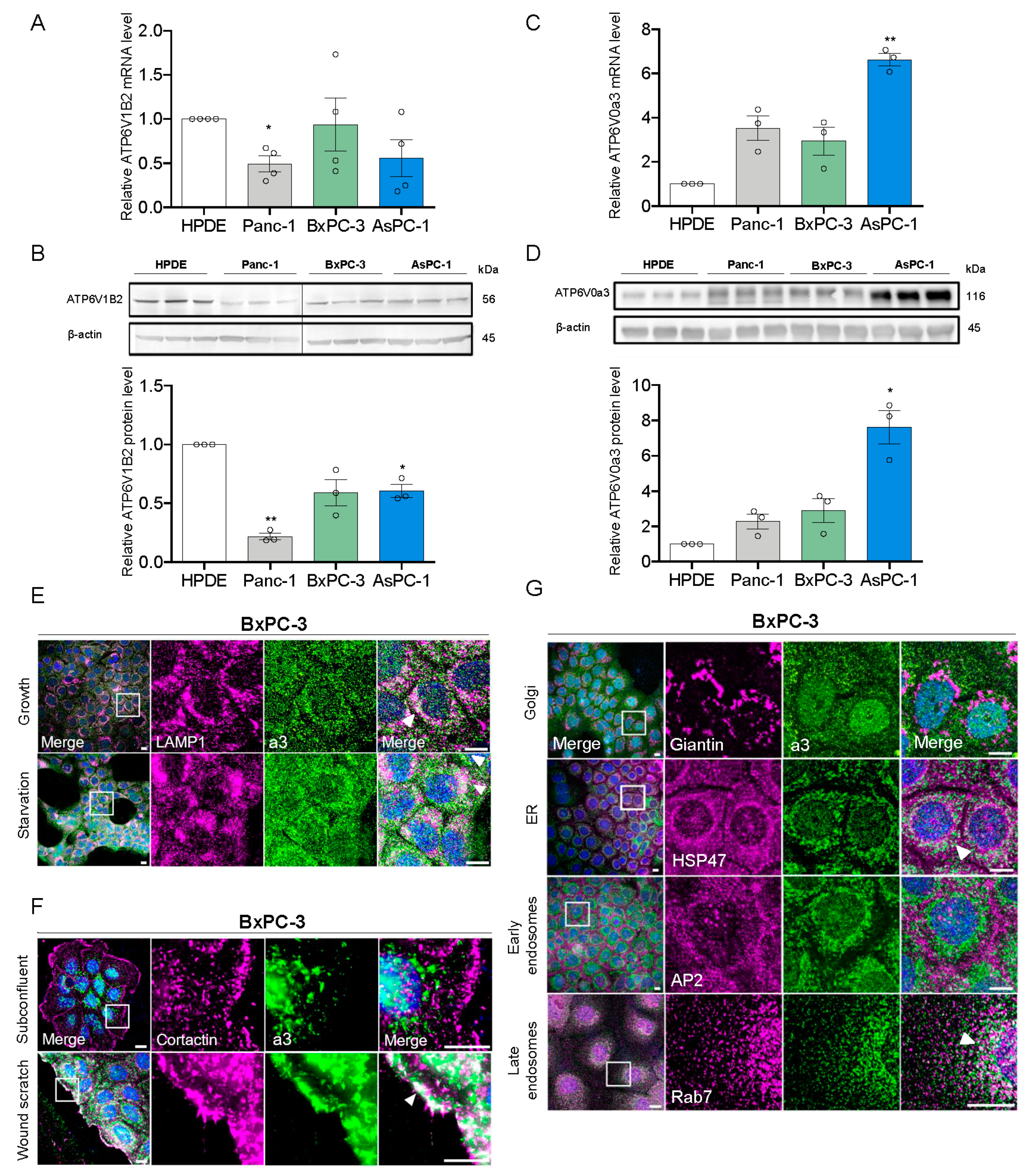
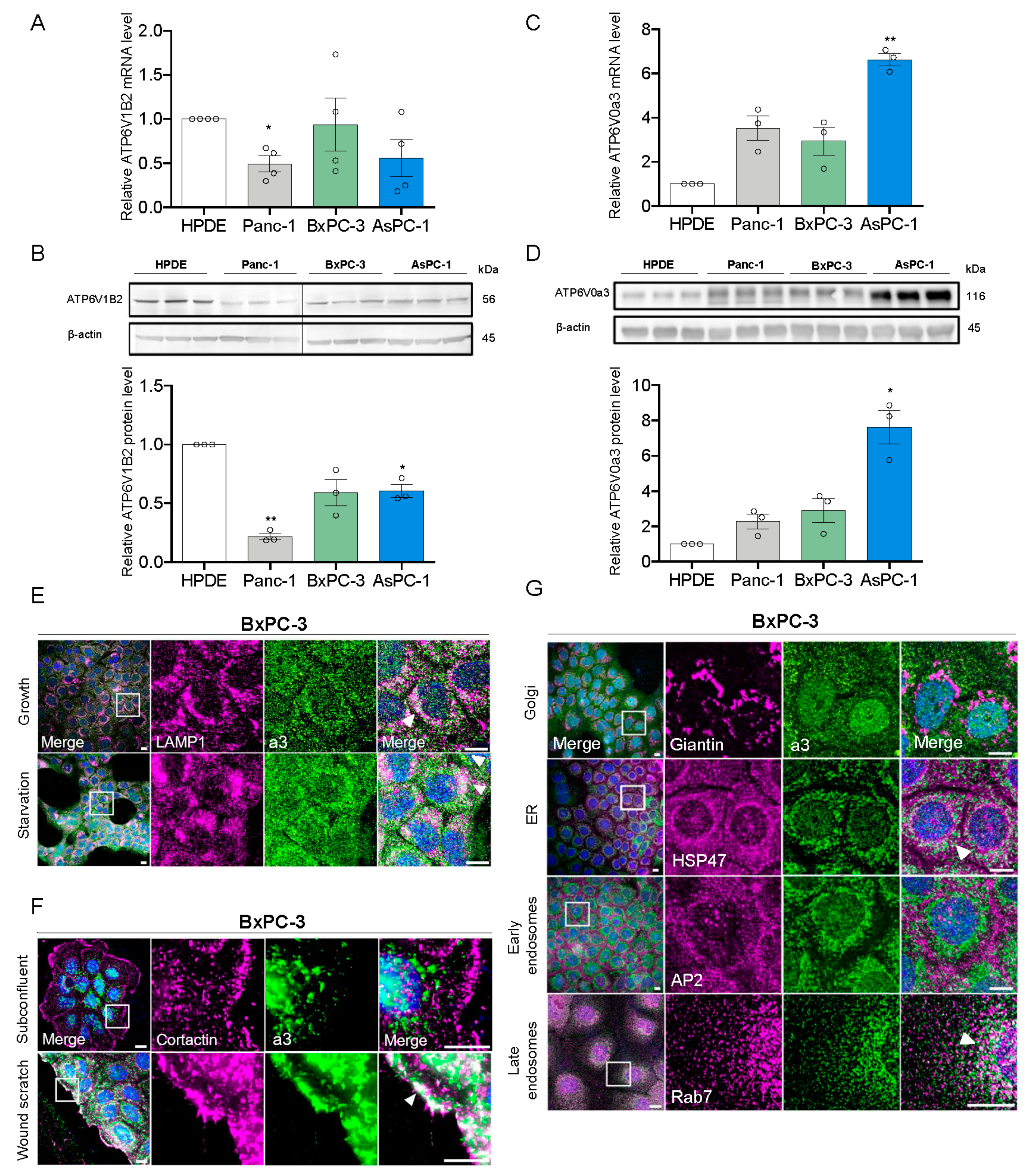
3.1. a3 is Upregulated in PDAC Cells and Mainly Localizes to Late Endosomes and Lysosomes
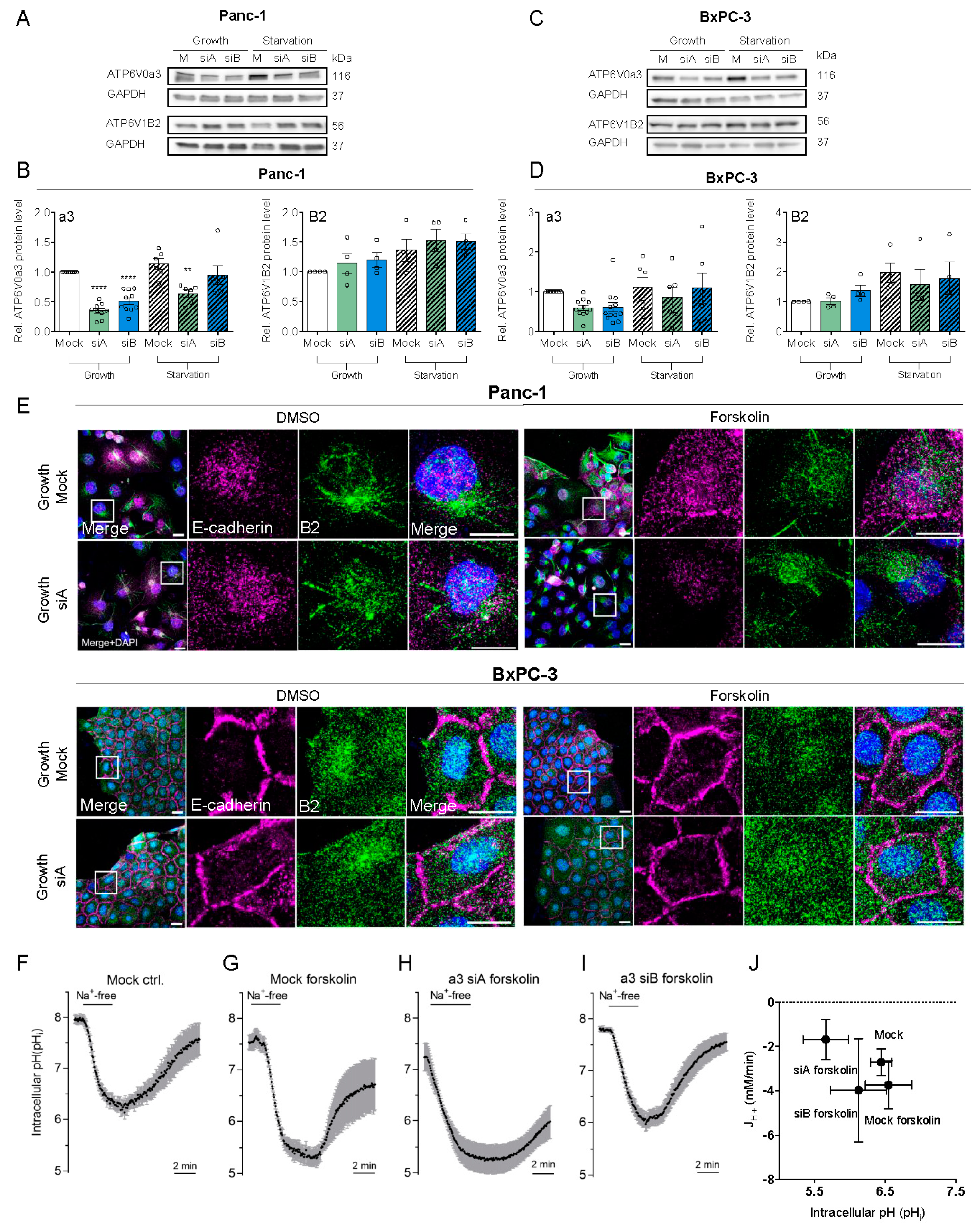
3.2. a3 Is Not. Upregulated by Starvation and Does Not. Contribute to pHi Recovery After Acidification
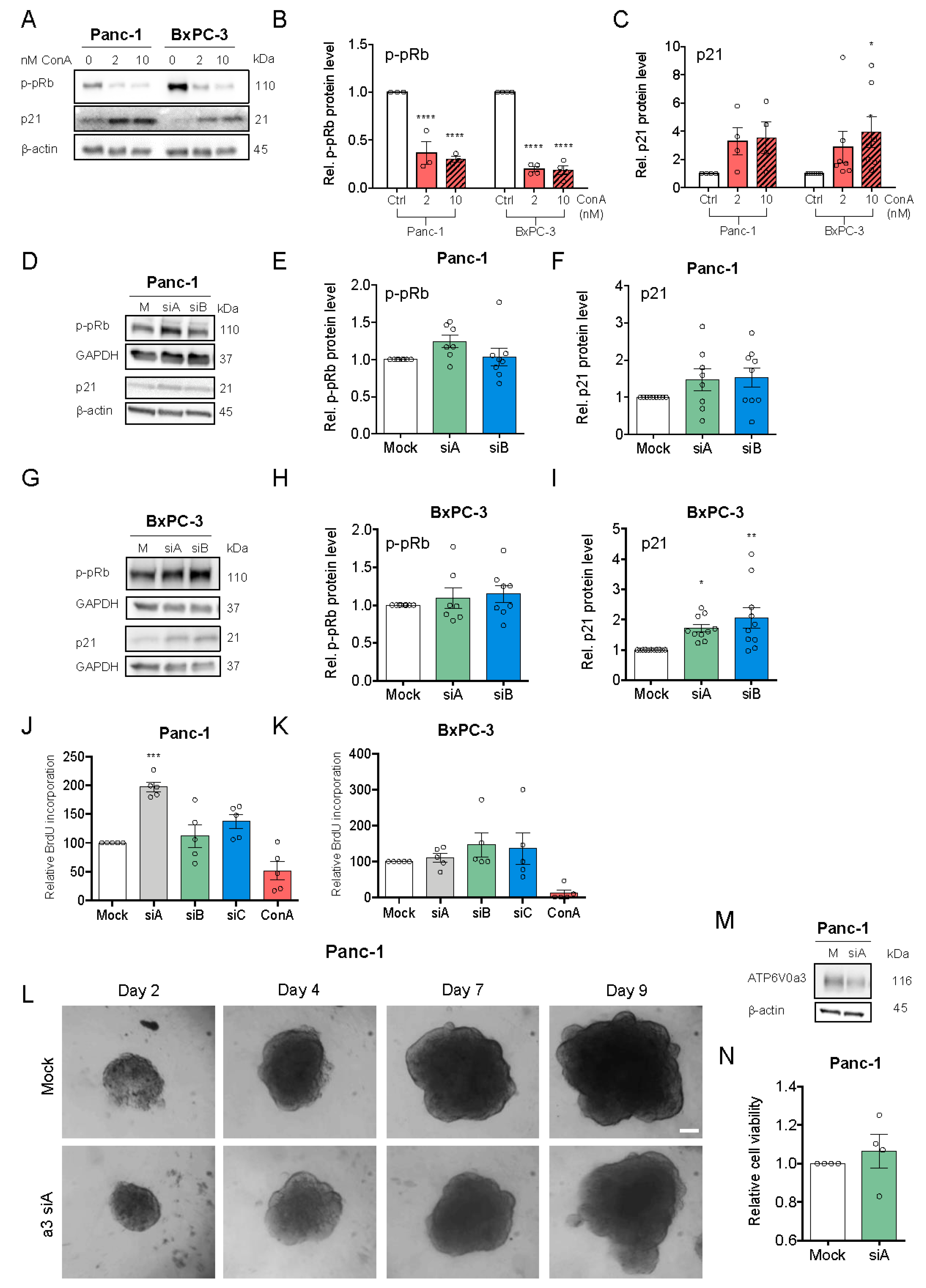
3.3. V-ATPase Inhibition, but not a3 KD, Reduces Proliferation and Increases Cell Death
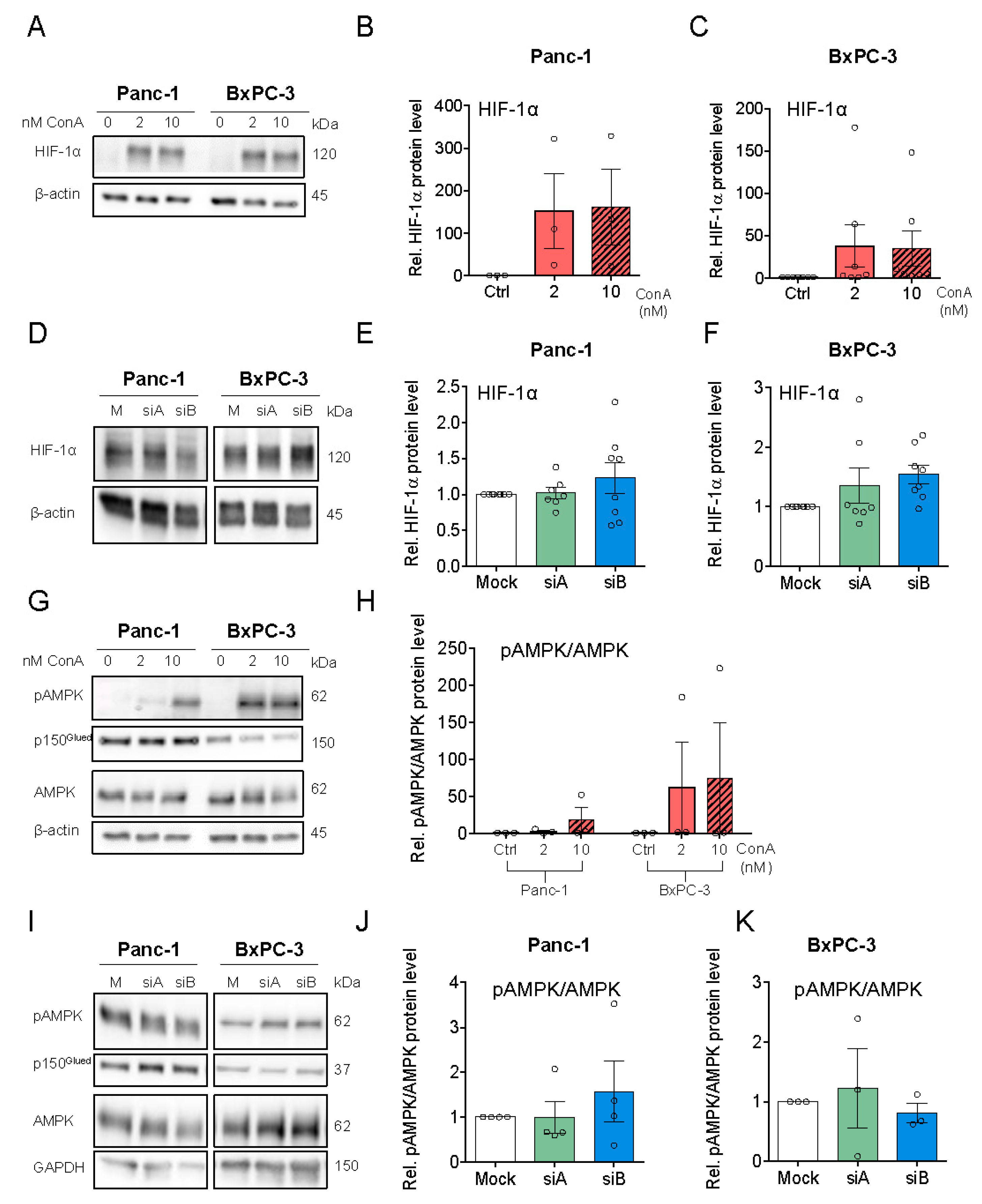
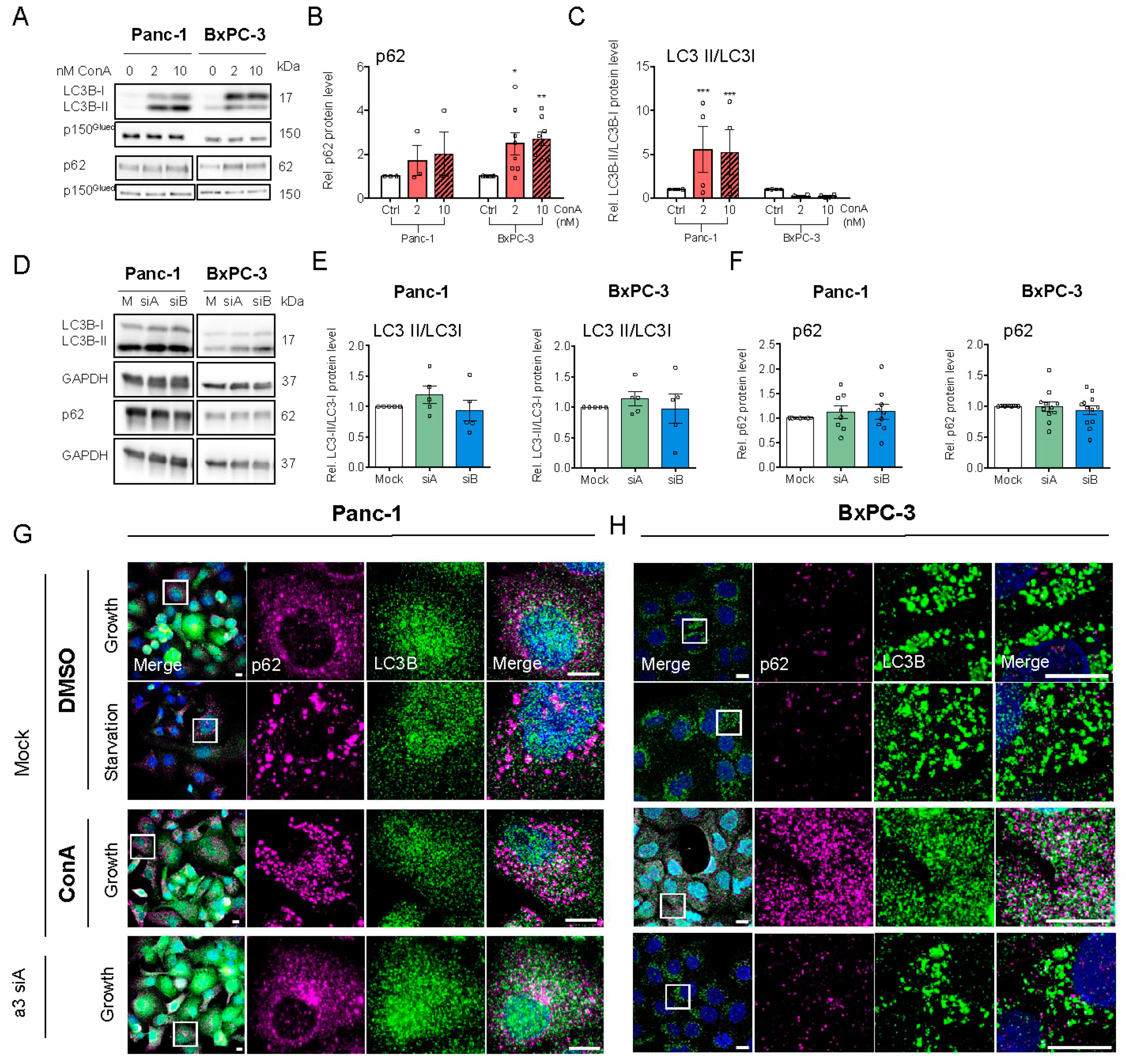
3.4. V-ATPase Inhibition Increases HIF-1α Protein Level, AMPK Activity and Alters Autophagic Flux
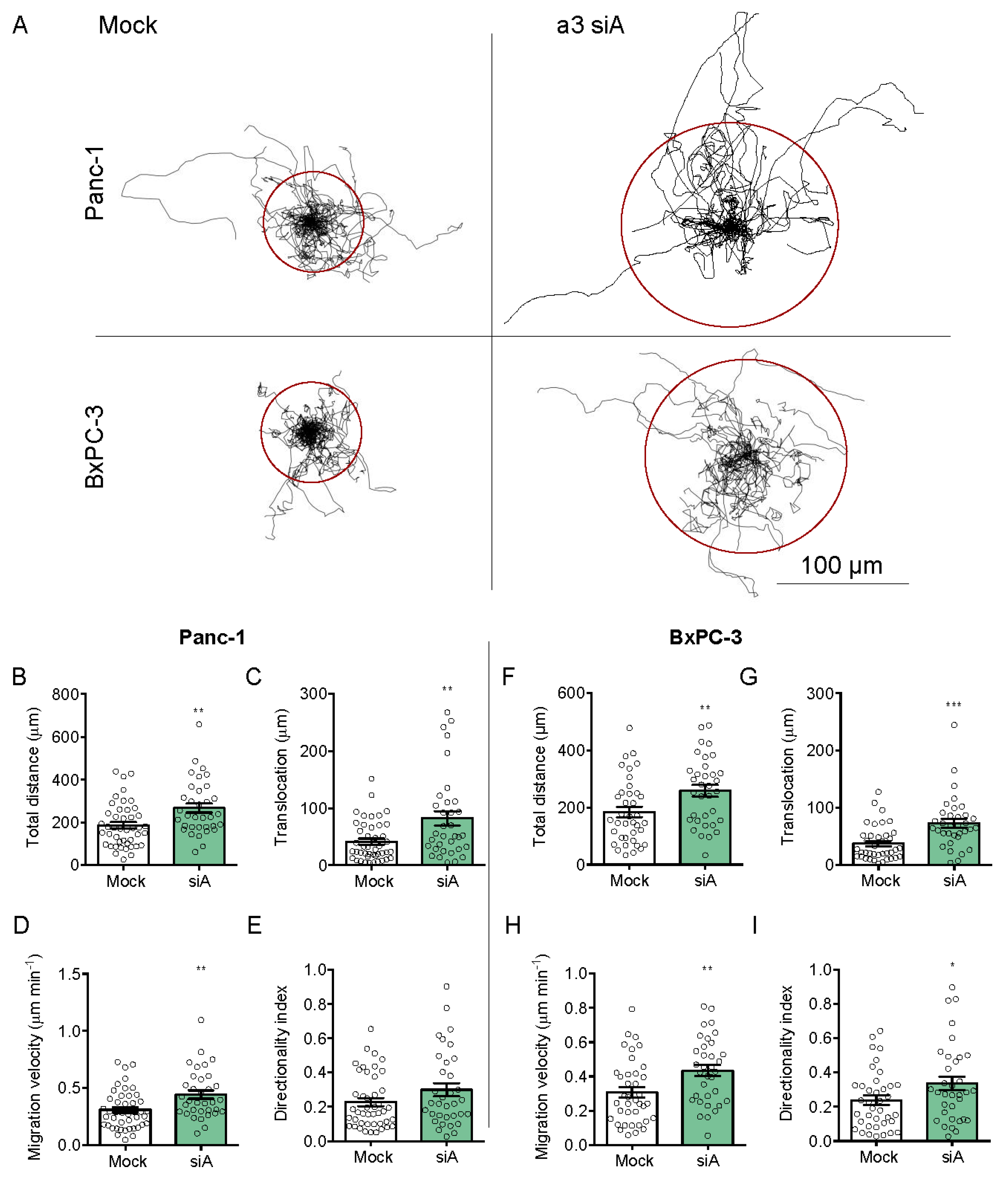
3.5. a3 Negatively Regulates Migration and Invasion of PDAC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gong, J.; Tuli, R.; Shinde, A.; Hendifar, A.E. Meta-analyses of treatment standards for pancreatic cancer. Mol. Clin. Oncol. 2016, 4, 315–325. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Pedersen, S.F. Roles of pH and the Na+/H+ exchanger NHE1 in cancer: From cell biology and animal models to an emerging translational perspective? Semin.Cancer Biol. 2017, 43, 5–16. [Google Scholar] [CrossRef]
- Boedtkjer, E.P.; Pedersen, S.F. The acidic tumor microenvironment as a driver of cancer. Ann. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Toyomura, T.; Murata, Y.; Yamamoto, A.; Oka, T.; Sun-Wada, G.H.; Wada, Y.; Futai, M. From lysosomes to the plasma membrane: Localization of vacuolar-type H+-ATPase with the a3 isoform during osteoclast differentiation. J. Biol. Chem. 2003, 278, 22023–22030. [Google Scholar] [CrossRef] [Green Version]
- Michel, V.; Licon-Munoz, Y.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 2013, 132, E1–E10. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.A.; Howell, G.J.; Phillips, C.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Extracellular and Luminal pH Regulation by Vacuolar H+-ATPase Isoform Expression and Targeting to the Plasma Membrane and Endosomes. J. Biol. Chem. 2016, 291, 8500–8515. [Google Scholar] [CrossRef] [Green Version]
- Sun-Wada, G.H.; Tabata, H.; Kawamura, N.; Aoyama, M.; Wada, Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J. Cell Sci. 2009, 122, 2504–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, H.; Kawamura, N.; Sun-Wada, G.H.; Wada, Y. Vacuolar-type H+-ATPase with the a3 isoform is the proton pump on premature melanosomes. Cell Tissue Res. 2008, 332, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Sun-Wada, G.H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J. Cell Sci. 2006, 119, 4531–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, C.; Hauser, A.D.; Vucic, E.A.; Bar-Sagi, D. Plasma membrane V-ATPase controls oncogenic RAS-induced macropinocytosis. Nature 2019, 576, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Sun-Wada, G.H.; Wada, Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim. Biophys. Acta 2015, 1847, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J. Biol. Chem. 2015, 290, 3680–3692. [Google Scholar] [CrossRef] [Green Version]
- Sennoune, S.R.; Bakunts, K.; Martinez, G.M.; Chua-Tuan, J.L.; Kebir, Y.; Attaya, M.N.; Martinez-Zaguilan, R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: Distribution and functional activity. Am. J. Physiol. Cell Physiol. 2004, 286, C1443–C1452. [Google Scholar] [CrossRef]
- Nishisho, T.; Hata, K.; Nakanishi, M.; Morita, Y.; Sun-Wada, G.H.; Wada, Y.; Yasui, N.; Yoneda, T. The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol. Cancer Res. 2011, 9, 845–855. [Google Scholar] [CrossRef] [Green Version]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capecci, J.; Forgac, M. The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 2013, 288, 32731–32741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, C.M.; Collins, M.P.; Sun-Wada, G.; Wada, Y.; Forgac, M. Isoform-specific gene disruptions reveal a role for the V-ATPase subunit a4 isoform in the invasiveness of 4T1-12B breast cancer cells. J. Biol. Chem. 2019, 294, 11248–11258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Chung, C.; Mader, C.C.; Schmitz, J.C.; Atladottir, J.; Fitchev, P.; Cornwell, M.L.; Koleske, A.J.; Crawford, S.E.; Gorelick, F. The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab. Investig. 2011, 91, 732–743. [Google Scholar] [CrossRef] [Green Version]
- Ohta, T.; Numata, M.; Yagishita, H.; Futagami, F.; Tsukioka, Y.; Kitagawa, H.; Kayahara, M.; Nagakawa, T.; Miyazaki, I.; Yamamoto, M.; et al. Expression of 16 kDa proteolipid of vacuolar-type H(+)-ATPase in human pancreatic cancer. Br. J. Cancer 1996, 73, 1511–1517. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, B.K.; Belinsky, G.S.; Einwachter, H.; Rhim, A.D.; Schmid, R.; Chung, C. Polarization of the vacuolar adenosine triphosphatase delineates a transition to high-grade pancreatic intraepithelial neoplasm lesions. Pancreas 2014, 43, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, T.; Hosotani, R.; Wada, M.; Miyamoto, Y.; Fujimoto, K.; Lee, J.U.; Doi, R.; Arii, S.; Imamura, M. Involvement of matrix metalloproteinase-2 activity in invasion and metastasis of pancreatic carcinoma. Cancer 1998, 82, 642–650. [Google Scholar] [CrossRef]
- Knapinska, A.M.; Estrada, C.A.; Fields, G.B. The Roles of Matrix Metalloproteinases in Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2017, 148, 339–354. [Google Scholar] [PubMed]
- Maatta, M.; Soini, Y.; Liakka, A.; Autio-Harmainen, H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: Implications for tumor progression and clinical prognosis. Clin. Cancer Res. 2000, 6, 2726–2734. [Google Scholar] [PubMed]
- Imamura, T.; Ohshio, G.; Mise, M.; Harada, T.; Suwa, H.; Okada, N.; Wang, Z.; Yoshitomi, S.; Tanaka, T.; Sato, H.; et al. Expression of membrane-type matrix metalloproteinase-1 in human pancreatic adenocarcinomas. J. Cancer Res. Clin. Oncol. 1998, 124, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Duguid, W.P.; Rosenberg, L.; Viallet, J.; Galloway, D.A.; Tsao, M.S. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 1996, 148, 1763–1770. [Google Scholar]
- Ouyang, H.; Mou, L.; Luk, C.; Liu, N.; Karaskova, J.; Squire, J.; Tsao, M.S. Immortal human pancreatic duct epithelial cell lines with near normal genotype and phenotype. Am. J. Pathol. 2000, 157, 1623–1631. [Google Scholar] [CrossRef] [Green Version]
- Andersen, A.P.; Samsoe-Petersen, J.; Oernbo, E.K.; Boedtkjer, E.; Moreira, J.M.A.; Kveiborg, M.; Pedersen, S.F. The net acid extruders NHE1, NBCn1 and MCT4 promote mammary tumor growth through distinct but overlapping mechanisms. Int. J. Cancer 2018, 142, 2529–2542. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- McGuire, C.M.; Forgac, M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018, 293, 9113–9123. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Sekiya, M.; Tohyama, K.; Ishiyama-Matsuura, E.; Sun-Wada, G.H.; Wada, Y.; Futai, M.; Nakanishi-Matsui, M. Essential Role of the a3 Isoform of V-ATPase in Secretory Lysosome Trafficking via Rab7 Recruitment. Sci. Rep. 2018, 8, 6701. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Soler, N.M.; Hallows, K.R.; Smolak, C.; Gong, F.; Brown, D.; Breton, S. Alkaline pH- and cAMP-induced V-ATPase membrane accumulation is mediated by protein kinase A in epididymal clear cells. Am. J. Physiol. Cell Physiol. 2008, 294, C488–C494. [Google Scholar] [CrossRef] [Green Version]
- Malinda, R.R.Z.; Sharki, P.C.; Ludwig, M.Q.; Pedersen, L.B.; Christensen, S.T.; Pedersen, S.F. TGFβ signaling increases net acid extrusion, proliferation and invasion in Panc-1 pancreatic cancer cells: SMAD4 dependence and link to Merlin/NF2 signaling. Front. Oncol. 2019. in review. [Google Scholar]
- Lim, J.H.; Park, J.W.; Kim, M.S.; Park, S.K.; Johnson, R.S.; Chun, Y.S. Bafilomycin induces the p21-mediated growth inhibition of cancer cells under hypoxic conditions by expressing hypoxia-inducible factor-1alpha. Mol. Pharmacol. 2006, 70, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, M.L.; Pellegrini, P.; Di, L.G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hagg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Xia, H.G.; Yuan, J. Pharmacologic agents targeting autophagy. J. Clin. Investig. 2015, 125, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Liu, Y.; Hong, L.; Yang, Z.; Cai, X.; Chen, X.; Fu, Y.; Lin, Y.; Wen, W.; Li, S.; et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 2016, 7, e2330. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, D.; Blauer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, J.; Roether, I.; Kern, H.F. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas 1987, 2, 14–24. [Google Scholar] [CrossRef]
- Toth, M.; Chvyrkova, I.; Bernardo, M.M.; Hernandez-Barrantes, S.; Fridman, R. Pro-MMP-9 activation by the MT1-MMP/MMP-2 axis and MMP-3: Role of TIMP-2 and plasma membranes. Biochem. Biophys. Res. Commun. 2003, 308, 386–395. [Google Scholar] [CrossRef]
- Von Schwarzenberg, K.; Lajtos, T.; Simon, L.; Muller, R.; Vereb, G.; Vollmar, A.M. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol. Oncol. 2014, 8, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Villanger, O.; Veel, T.; Raeder, M.G. Secretin causes H+/HCO3- Secretion From Pig Pancreatic Ductules by Vacuolar-Type H+-Adenosine Triphosphatase. Gastroenterology 1995, 108, 850–859. [Google Scholar] [CrossRef]
- Hulikova, A.; Harris, A.L.; Vaughan-Jones, R.D.; Swietach, P. Regulation of intracellular pH in cancer cell lines under normoxia and hypoxia 2. J. Cell Physiol. 2013, 228, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, G.; Jensen, M.B.; Boedtkjer, E.; Dybboe, R.; Aalkjaer, C.; Nylandsted, J.; Pedersen, S.F. NBCn1 and NHE1 expression and activity in DeltaNErbB2 receptor-expressing MCF-7 breast cancer cells: Contributions to pHi regulation and chemotherapy resistance. Exp. Cell Res. 2010, 316, 2538–2553. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.C.; Wu, W.K.; Li, Y.; Yu, L.; Li, Z.J.; Wong, C.C.; Li, H.T.; Sung, J.J.; Cho, C.H. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem. Biophys. Res. Commun. 2009, 382, 451–456. [Google Scholar] [CrossRef]
- Manabe, T.; Yoshimori, T.; Henomatsu, N.; Tashiro, Y. Inhibitors of vacuolar-type H+-ATPase suppresses proliferation of cultured cells. J. Cell Physiol. 1993, 157, 445–452. [Google Scholar] [CrossRef]
- Von Schwarzenberg, K.; Wiedmann, R.M.; Oak, P.; Schulz, S.; Zischka, H.; Wanner, G.; Efferth, T.; Trauner, D.; Vollmar, A.M. Mode of cell death induction by pharmacological vacuolar H+-ATPase (V-ATPase) inhibition. J. Biol. Chem. 2013, 288, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Park, J.W.; Kim, S.J.; Kim, M.S.; Park, S.K.; Johnson, R.S.; Chun, Y.S. ATP6V0C competes with von Hippel-Lindau protein in hypoxia-inducible factor 1alpha (HIF-1alpha) binding and mediates HIF-1alpha expression by bafilomycin A1. Mol. Pharmacol. 2007, 71, 942–948. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Hu, H.; Kshitiz; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, L.S.; von, S.K.; Lehr, T.; Ulrich, M.; Kubisch-Dohmen, R.; Liebl, J.; Trauner, D.; Menche, D.; Vollmar, A.M. Vacuolar-ATPase Inhibition Blocks Iron Metabolism to Mediate Therapeutic Effects in Breast Cancer. Cancer Res. 2015, 75, 2863–2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissing, S.; Hermsen, C.; Repnik, U.; Nesset, C.K.; von, B.K.; Griffiths, G.; Ichihara, A.; Lee, B.S.; Schwake, M.; De, B.J.; et al. Vacuolar ATPase in phagosome-lysosome fusion. J. Biol. Chem. 2015, 290, 14166–14180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvezin, C.; Nagy, P.; Juhasz, G.; Neufeld, T.P. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef] [Green Version]
- Maquoi, E.; Peyrollier, K.; Noel, A.; Foidart, J.M.; Frankenne, F. Regulation of membrane-type 1 matrix metalloproteinase activity by vacuolar H+-ATPases. Biochem. J. 2003, 373, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Pratt, J.; Roy, R.; Annabi, B. Concanavalin-A-induced autophagy biomarkers requires membrane type-1 matrix metalloproteinase intracellular signaling in glioblastoma cells. Glycobiology 2012, 22, 1245–1255. [Google Scholar] [CrossRef] [Green Version]
- Vogelmann, R.; Nguyen-Tat, M.D.; Giehl, K.; Adler, G.; Wedlich, D.; Menke, A. TGFbeta-induced downregulation of E-cadherin-based cell-cell adhesion depends on PI3-kinase and PTEN. J. Cell Sci. 2005, 118, 4901–4912. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wang, Z.; Zhang, W.; Qian, K.; Li, H.; Kong, D.; Li, Y.; Tang, Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett. 2015, 356, 613–627. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flinck, M.; Hagelund, S.; Gorbatenko, A.; Severin, M.; Pedraz-Cuesta, E.; Novak, I.; Stock, C.; Pedersen, S.F. The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells. Cells 2020, 9, 465. https://doi.org/10.3390/cells9020465
Flinck M, Hagelund S, Gorbatenko A, Severin M, Pedraz-Cuesta E, Novak I, Stock C, Pedersen SF. The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells. Cells. 2020; 9(2):465. https://doi.org/10.3390/cells9020465
Chicago/Turabian StyleFlinck, Mette, Sofie Hagelund, Andrej Gorbatenko, Marc Severin, Elena Pedraz-Cuesta, Ivana Novak, Christian Stock, and Stine Falsig Pedersen. 2020. "The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells" Cells 9, no. 2: 465. https://doi.org/10.3390/cells9020465
APA StyleFlinck, M., Hagelund, S., Gorbatenko, A., Severin, M., Pedraz-Cuesta, E., Novak, I., Stock, C., & Pedersen, S. F. (2020). The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells. Cells, 9(2), 465. https://doi.org/10.3390/cells9020465