The Timing and Extent of Motor Neuron Vulnerability in ALS Correlates with Accumulation of Misfolded SOD1 Protein in the Cortex and in the Spinal Cord



Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Histology
2.3. Immunocytochemistry and Cellular Staining
2.4. Imaging
2.5. Data Collection and Analysis
3. Results
3.1. Misfolded SOD1 Is Expressed mainly in Layer 5 of the Motor Cortex and Colocalizes with Diseased CSMN
3.2. Misfolded SOD1 Protein Is Detected Primarily in Vulnerable and Degenerating SMN
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef]
- Lalancette-Hebert, M.; Sharma, A.; Lyashchenko, A.K.; Shneider, N.A. Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc. Natl. Acad. Sci. USA 2016, 113, E8316–E8325. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef]
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008, 586, 3337–3351. [Google Scholar] [CrossRef]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef] [Green Version]
- Rochat, C.; Bernard-Marissal, N.; Schneider, B.L. Selective Vulnerability of Neuronal Subtypes in ALS: A Fertile Ground for the Identification of Therapeutic Targets. Update Amyotroph. Lateral Scler. 2016, 165–194. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Wells, C.; Brennan, S.E.; Keon, M.; Saksena, N.K. Prionoid Proteins in the Pathogenesis of Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 12, 271. [Google Scholar] [CrossRef]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Gros-Louis, F.; Soucy, G.; Lariviere, R.; Julien, J.P. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J. Neurochem. 2010, 113, 1188–1199. [Google Scholar] [CrossRef]
- Saxena, S.; Roselli, F.; Singh, K.; Leptien, K.; Julien, J.P.; Gros-Louis, F.; Caroni, P. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 2013, 80, 80–96. [Google Scholar] [CrossRef] [Green Version]
- Leyton-Jaimes, M.F.; Kahn, J.; Israelson, A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 2019, 116, 14755–14760. [Google Scholar] [CrossRef] [Green Version]
- Dutta, K.; Patel, P.; Julien, J.P. Protective effects of Withania somnifera extract in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2018, 309, 193–204. [Google Scholar] [CrossRef]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef]
- Jara, J.H.; Genc, B.; Klessner, J.L.; Ozdinler, P.H. Retrograde labeling, transduction, and genetic targeting allow cellular analysis of corticospinal motor neurons: Implications in health and disease. Front. Neuroanat. 2014, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Genc, B.; Gozutok, O.; Ozdinler, P.H. Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons. Int. J. Mol. Sci. 2019, 20, 3848. [Google Scholar] [CrossRef] [Green Version]
- Commisso, B.; Ding, L.; Varadi, K.; Gorges, M.; Bayer, D.; Boeckers, T.M.; Ludolph, A.C.; Kassubek, J.; Muller, O.J.; Roselli, F. Stage-dependent remodeling of projections to motor cortex in ALS mouse model revealed by a new variant retrograde-AAV9. eLife 2018, 7. [Google Scholar] [CrossRef]
- Yasvoina, M.V.; Genc, B.; Jara, J.H.; Sheets, P.L.; Quinlan, K.A.; Milosevic, A.; Shepherd, G.M.; Heckman, C.J.; Ozdinler, P.H. eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 7890–7904. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Hernandez, M.; Tadokoro, T.; Navarro, M.R.; Platoshyn, O.; Kobayashi, Y.; Marsala, S.; Miyanohara, A.; Juhas, S.; Juhasova, J.; Skalnikova, H.; et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Ozdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef]
- Brown, R.H., Jr.; Robberecht, W. Amyotrophic lateral sclerosis: Pathogenesis. Semin. Neurol. 2001, 21, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef]
- Ravits, J.; Paul, P.; Jorg, C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef]
- Genc, B.; Jara, J.H.; Lagrimas, A.K.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Ozdinler, P.H. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef] [Green Version]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Ozdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Ozdinler, P.H. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum. Mol. Genet. 2016, 25, 1074–1087. [Google Scholar] [CrossRef] [Green Version]
- Jara, J.H.; Genc, B.; Cox, G.A.; Bohn, M.C.; Roos, R.P.; Macklis, J.D.; Ulupinar, E.; Ozdinler, P.H. Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function. Cereb Cortex 2015, 25, 4259–4272. [Google Scholar] [CrossRef] [Green Version]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2017, 26, 686–701. [Google Scholar] [CrossRef]
- Joyce, P.I.; McGoldrick, P.; Saccon, R.A.; Weber, W.; Fratta, P.; West, S.J.; Zhu, N.; Carter, S.; Phatak, V.; Stewart, M.; et al. A novel SOD1-ALS mutation separates central and peripheral effects of mutant SOD1 toxicity. Hum. Mol. Genet. 2015, 24, 1883–1897. [Google Scholar] [CrossRef] [Green Version]
- Atlasi, R.S.; Malik, R.; Corrales, C.I.; Tzeplaeff, L.; Whitelegge, J.P.; Cashman, N.R.; Bitan, G. Investigation of Anti-SOD1 Antibodies Yields New Structural Insight into SOD1 Misfolding and Surprising Behavior of the Antibodies Themselves. ACS Chem. Biol. 2018, 13, 2794–2807. [Google Scholar] [CrossRef]
- Pickles, S.; Vande Velde, C. Misfolded SOD1 and ALS: Zeroing in on mitochondria. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2012, 13, 333–340. [Google Scholar] [CrossRef]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Rotunno, M.S.; Bosco, D.A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 2013, 7, 253. [Google Scholar] [CrossRef] [Green Version]
- Pare, B.; Lehmann, M.; Beaudin, M.; Nordstrom, U.; Saikali, S.; Julien, J.P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz, S.; Bui, A.; Saberi, S.; Lee, S.K.; Stauffer, J.; McAlonis-Downes, M.; Schulte, D.; Pizzo, D.P.; Parone, P.A.; Cleveland, D.W.; et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017, 134, 97–111. [Google Scholar] [CrossRef]
- Hayashi, Y.; Homma, K.; Ichijo, H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv. Biol. Regul. 2016, 60, 95–104. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Sunyach, C.; Marissal, T.; Raoul, C.; Pettmann, B. Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice. Neurobiol. Dis. 2015, 73, 130–136. [Google Scholar] [CrossRef]
- Fogarty, M.J.; Mu, E.W.H.; Lavidis, N.A.; Noakes, P.G.; Bellingham, M.C. Size-Dependent Vulnerability of Lumbar Motor Neuron Dendritic Degeneration in SOD1(G93A) Mice. Anat. Rec. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ruegsegger, C.; Maharjan, N.; Goswami, A.; Filezac de L’Etang, A.; Weis, J.; Troost, D.; Heller, M.; Gut, H.; Saxena, S. Aberrant association of misfolded SOD1 with Na(+)/K(+)ATPase-alpha3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol. 2016, 131, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Semmler, S.; Gagné, M.; Garg, P.; Pickles, S.R.; Baudouin, C.; Hamon-Keromen, E.; Destroismaisons, L.; Khalfallah, Y.; Chaineau, M.; Caron, E.; et al. The E3 ubiquitin ligase TRAF6 is a novel interacting protein of amyotrophic lateral sclerosis-linked misfolded SOD1. bioRxiv 2019, 780460. [Google Scholar] [CrossRef]
- Filezac de L’Etang, A.; Maharjan, N.; Cordeiro Brana, M.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J.; et al. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 2015, 18, 227–238. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Moumen, A.; Sunyach, C.; Pellegrino, C.; Dudley, K.; Henderson, C.E.; Raoul, C.; Pettmann, B. Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 4901–4912. [Google Scholar] [CrossRef]
- Tung, Y.T.; Peng, K.C.; Chen, Y.C.; Yen, Y.P.; Chang, M.; Thams, S.; Chen, J.A. Mir-17 approximately 92 Confers Motor Neuron Subtype Differential Resistance to ALS-Associated Degeneration. Cell Stem Cell 2019, 25, 193–209.e7. [Google Scholar] [CrossRef]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Han, J.S.; McAlonis-Downes, M.; Vetto, A.P.; Lee, S.K.; Tseng, E.; Cleveland, D.W. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 4657–4671. [Google Scholar] [CrossRef]
- Irvin, C.W.; Kim, R.B.; Mitchell, C.S. Seeking homeostasis: Temporal trends in respiration, oxidation, and calcium in SOD1 G93A Amyotrophic Lateral Sclerosis mice. Front. Cell. Neurosci. 2015, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.; Spiller, K.J.; Towne, C.; Kanning, K.C.; Choe, G.T.; Geber, A.; Akay, T.; Aebischer, P.; Henderson, C.E. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron 2014, 81, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Morisaki, Y.; Niikura, M.; Watanabe, M.; Onishi, K.; Tanabe, S.; Moriwaki, Y.; Okuda, T.; Ohara, S.; Murayama, S.; Takao, M.; et al. Selective Expression of Osteopontin in ALS-resistant Motor Neurons is a Critical Determinant of Late Phase Neurodegeneration Mediated by Matrix Metalloproteinase-9. Sci. Rep. 2016, 6, 27354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirren, E.; Aebischer, J.; Rochat, C.; Towne, C.; Schneider, B.L.; Aebischer, P. SOD1 silencing in motoneurons or glia rescues neuromuscular function in ALS mice. Ann. Clin. Transl. Neurol. 2015, 2, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannitti, T.; Scarrott, J.M.; Likhite, S.; Coldicott, I.R.P.; Lewis, K.E.; Heath, P.R.; Higginbottom, A.; Myszczynska, M.A.; Milo, M.; Hautbergue, G.M.; et al. Translating SOD1 Gene Silencing toward the Clinic: A Highly Efficacious, Off-Target-free, and Biomarker-Supported Strategy for fALS. Mol. Nucleic Acids 2018, 12, 75–88. [Google Scholar] [CrossRef] [PubMed]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Ulzi, G.; Ramirez, A.; Rizzuti, M.; Bordoni, A.; Bucchia, M.; Gatti, S.; Bresolin, N.; et al. Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype. Sci. Rep. 2016, 6, 21301. [Google Scholar] [CrossRef] [Green Version]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.; Wong, L.F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef]
- Raoul, C.; Abbas-Terki, T.; Bensadoun, J.C.; Guillot, S.; Haase, G.; Szulc, J.; Henderson, C.E.; Aebischer, P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005, 11, 423–428. [Google Scholar] [CrossRef]
- van Zundert, B.; Brown, R.H., Jr. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci. Lett. 2017, 636, 32–39. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Qiu, L.; Yang, C.; Kramer, J.; Su, Q.; Guo, Y.; Brown, R.H., Jr.; Gao, G.; Xu, Z. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene 2019. [Google Scholar] [CrossRef] [PubMed]





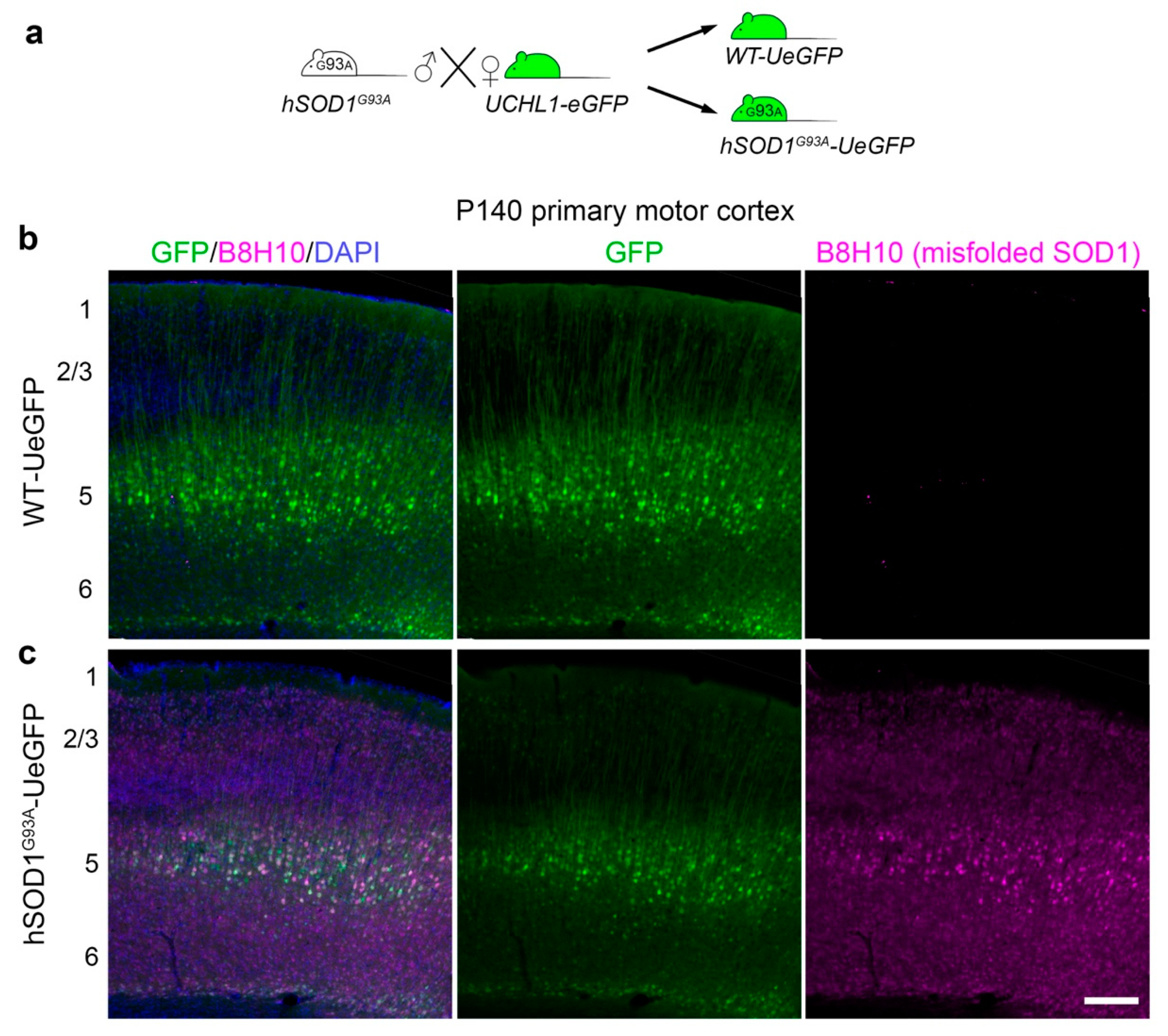{kind=link}
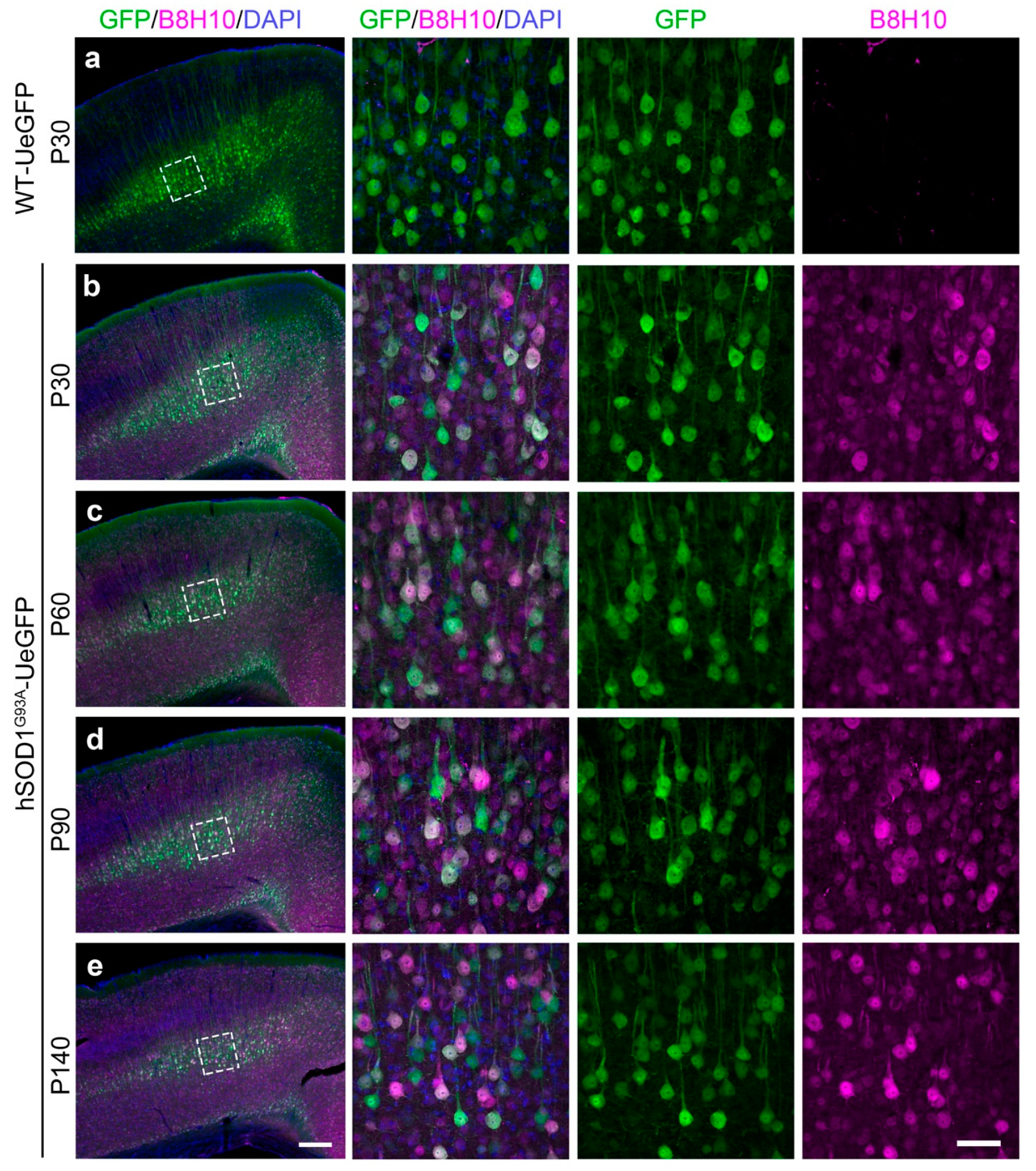{kind=link}
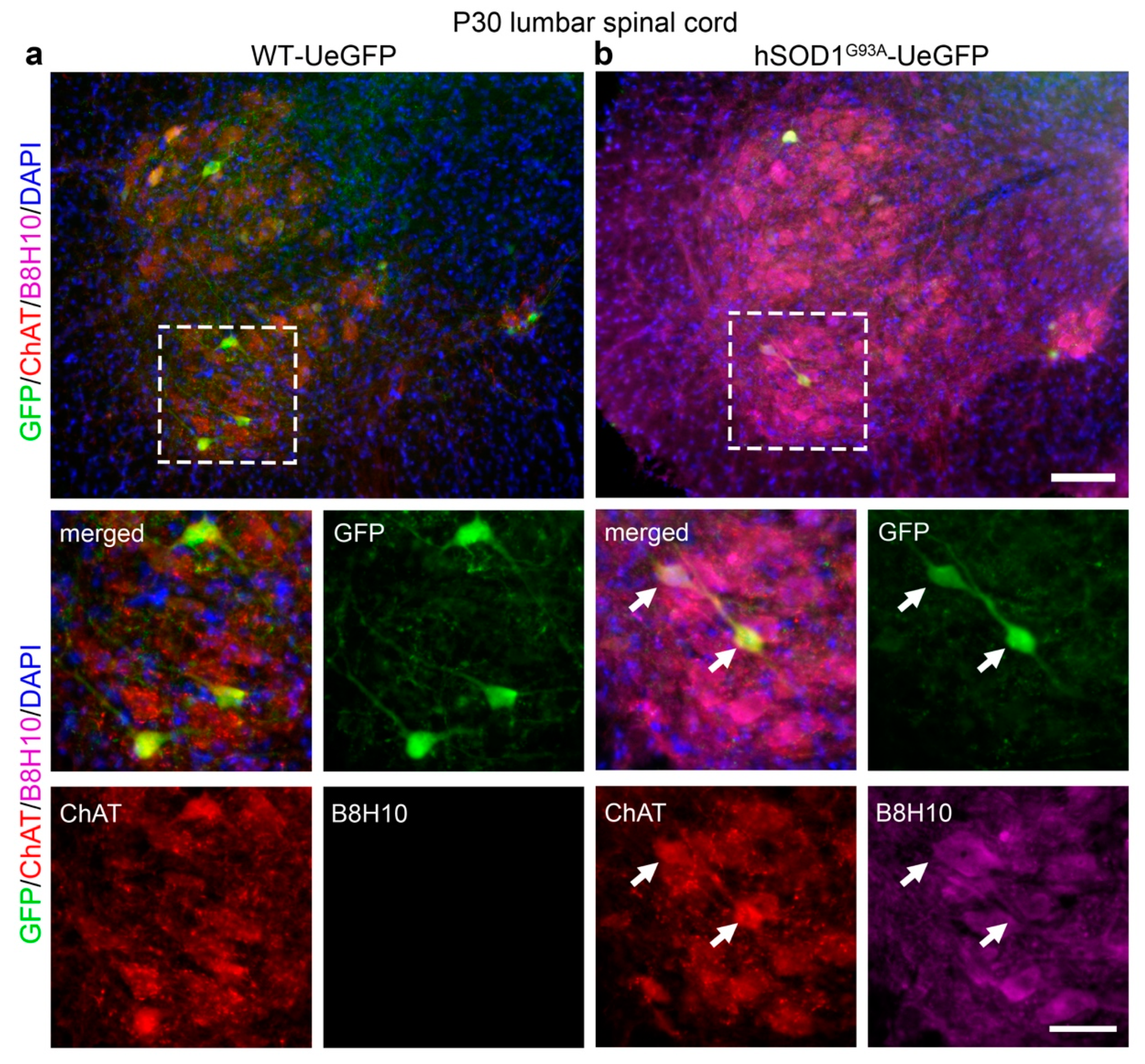{kind=link}
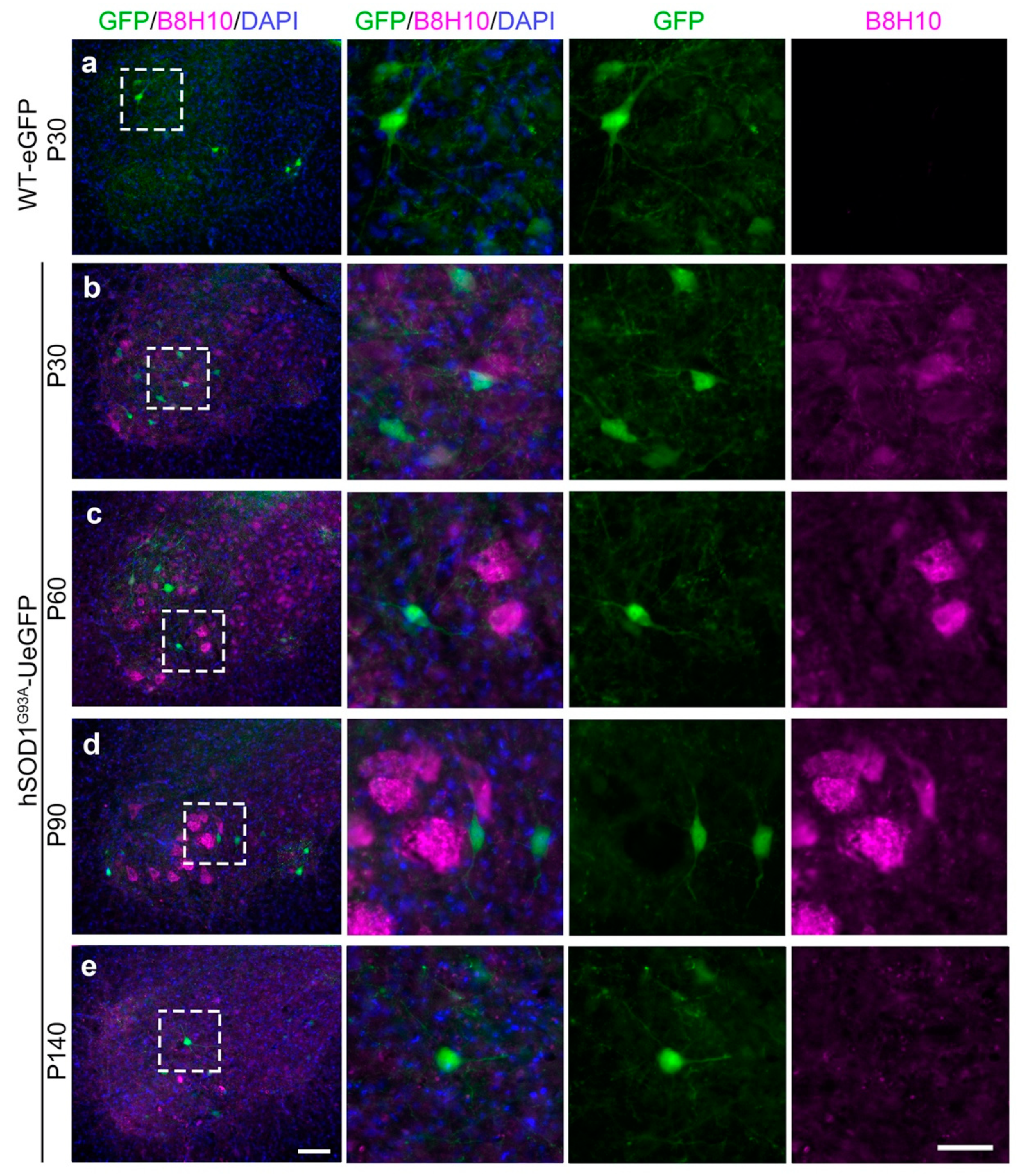{kind=link}
| Age | Number of Mice | Total Number of Neurons | Average % of Non-CSMN with Misfolded SOD1 | S.E.M. | Average % of CSMN with Misfolded SOD1 | S.E.M. |
|---|---|---|---|---|---|---|
| P30 | 3 | 452 | 64.68% | 2.98% | 78.52% | 5.40% |
| P60 | 4 | 485 | 58.81% | 1.10% | 90.29% | 1.05% |
| P90 | 4 | 579 | 60.08% | 2.36% | 86.06% | 2.74% |
| P140 | 4 | 575 | 52.41% | 2.40% | 86.37% | 6.74% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genc, B.; Gozutok, O.; Kocak, N.; Ozdinler, P.H. The Timing and Extent of Motor Neuron Vulnerability in ALS Correlates with Accumulation of Misfolded SOD1 Protein in the Cortex and in the Spinal Cord. Cells 2020, 9, 502. https://doi.org/10.3390/cells9020502
Genc B, Gozutok O, Kocak N, Ozdinler PH. The Timing and Extent of Motor Neuron Vulnerability in ALS Correlates with Accumulation of Misfolded SOD1 Protein in the Cortex and in the Spinal Cord. Cells. 2020; 9(2):502. https://doi.org/10.3390/cells9020502
Chicago/Turabian StyleGenc, Baris, Oge Gozutok, Nuran Kocak, and P. Hande Ozdinler. 2020. "The Timing and Extent of Motor Neuron Vulnerability in ALS Correlates with Accumulation of Misfolded SOD1 Protein in the Cortex and in the Spinal Cord" Cells 9, no. 2: 502. https://doi.org/10.3390/cells9020502
APA StyleGenc, B., Gozutok, O., Kocak, N., & Ozdinler, P. H. (2020). The Timing and Extent of Motor Neuron Vulnerability in ALS Correlates with Accumulation of Misfolded SOD1 Protein in the Cortex and in the Spinal Cord. Cells, 9(2), 502. https://doi.org/10.3390/cells9020502