Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy



Abstract
1. Introduction
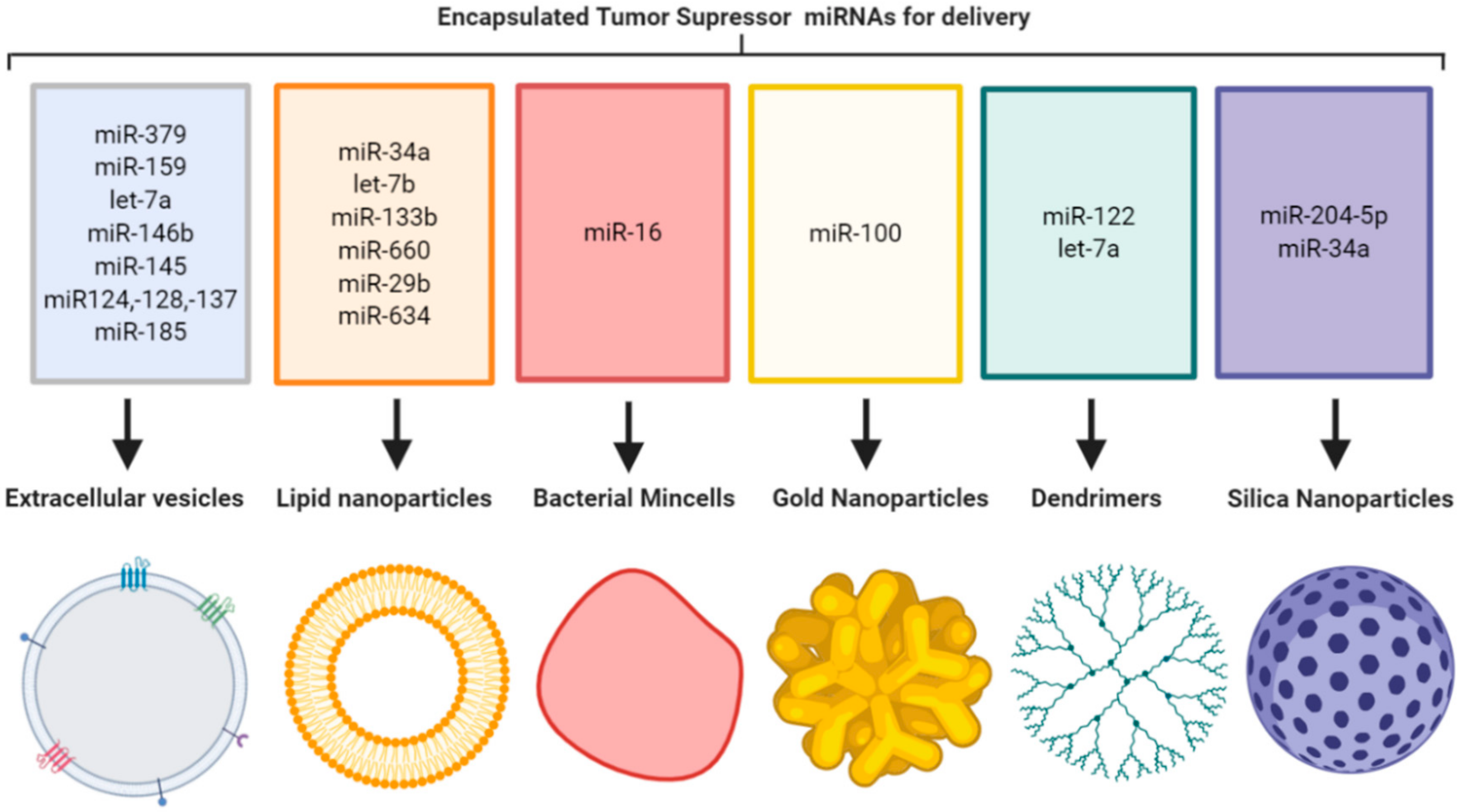
1.1. Organic Lipid Nanoparticles
1.2. Extracellular Vesicles
1.3. Bacterial Minicells
1.4. Inorganic Nanoparticles
2. Tumor Suppressor miRNA Delivery Via Lipid-Based Nanoparticles
3. Tumor Suppressive miRNA Delivery Via Extracellular Vesicles (EVs)
4. Inorganic-Based Nanoparticle Formulation
5. Clinical Trials
6. Discussion
Funding
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. Mirbase: From microrna sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.C.; Chung, H.Y.; Cho, D.Y.; Chan, T.M.; Liu, M.C.; Huang, H.M.; Li, T.Y.; Lin, J.Y.; Chou, P.C.; Fu, R.H.; et al. Therapeutic potential of microrna let-7: Tumor suppression or impeding normal stemness. Cell Transplant. 2014, 23, 459–469. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. Microrna-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.-A.; Murphy, P.R. Microrna: Biogenesis, function and role in cancer. Curr. Genomics 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed]
- Bail, S.; Swerdel, M.; Liu, H.; Jiao, X.; Goff, L.A.; Hart, R.P.; Kiledjian, M. Differential regulation of microrna stability. RNA 2010, 16, 1032–1039. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Lappalainen, K.; Jääskeläinen, I.; Syrjänen, K.; Urtti, A.; Syrjänen, S. Comparison of cell proliferation and toxicity assays using two cationic liposomes. Pharm. Res. 1994, 11, 1127–1131. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Jia, L.; Chu, F.; Zhou, Y.; He, Z.; Guo, M.; Chen, C.; Xu, L. Nanostructured lipid carriers for microrna delivery in tumor gene therapy. Cancer Cell Int. 2018, 18, 101. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.P.; Khan, S.; Gilligan, K.E.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (ev)-encapsulated microrna-379. Oncogene 2018, 37, 2137–2149. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Tian, J.; Wang, Z.; Gao, Y.; Wu, X.; Ding, X.; Qiang, L.; Li, G.; Han, Z.; Yuan, Y.; et al. Functional exosome-mediated co-delivery of doxorubicin and hydrophobically modified microrna 159 for triple-negative breast cancer therapy. J. Nanobiotechnology 2019, 17, 93. [Google Scholar] [CrossRef]
- Bhaskaran, V.; Nowicki, M.O.; Idriss, M.; Jimenez, M.A.; Lugli, G.; Hayes, J.L.; Mahmoud, A.B.; Zane, R.E.; Passaro, C.; Ligon, K.L.; et al. The functional synergism of microrna clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat. Commun. 2019, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Hu, W.; Chen, H.; Shou, X.; Ye, T.; Xu, Y. Cocktail strategy based on nk cell-derived exosomes and their biomimetic nanoparticles for dual tumor therapy. Cancers 2019, 11, 1560. [Google Scholar] [CrossRef] [PubMed]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.; Haasdyk, J.E.; Dickson, K.A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef]
- Jivrajani, M.; Shrivastava, N.; Nivsarkar, M. A combination approach for rapid and high yielding purification of bacterial minicells. J. Microbiol. Methods 2013, 92, 340–343. [Google Scholar] [CrossRef]
- Araújo, R.V.; Santos, S.D.S.; Igne Ferreira, E.; Giarolla, J. New advances in general biomedical applications of PAMAM dendrimers. Molecules 2018, 23, E2849. [Google Scholar] [CrossRef]
- Tivnan, A.; Orr, W.S.; Gubala, V.; Nooney, R.; Williams, D.E.; McDonagh, C.; Prenter, S.; Harvey, H.; Domingo-Fernández, R.; Bray, I.M.; et al. Inhibition of neuroblastoma tumor growth by targeted delivery of microrna-34a using anti-disialoganglioside gd2 coated nanoparticles. PLoS ONE 2012, 7, e38129. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, X.; Zhang, X.; Liu, B.; Huang, L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol. Ther. 2010, 18, 1650–1656. [Google Scholar] [CrossRef]
- Wu, Y.; Crawford, M.; Yu, B.; Mao, Y.; Nana-Sinkam, S.P.; Lee, L.J. Microrna delivery by cationic lipoplexes for lung cancer therapy. Mol. Pharm. 2011, 8, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.; Batte, K.; Yu, L.; Wu, X.; Nuovo, G.J.; Marsh, C.B.; Otterson, G.A.; Nana-Sinkam, S.P. MicroRNA 133b targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem. Biophys. Res. Commun. 2009, 388, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.-i.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microrna mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microrna-34. Cancer Res. 2010, 70, 5923–5930. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.; Di Paolo, D.; Milione, M.; Centonze, G.; Bornaghi, V.; Borzi, C.; Gandellini, P.; Perri, P.; Pastorino, U.; Ponzoni, M.; et al. Coated cationic lipid-nanoparticles entrapping mir-660 inhibit tumor growth in patient-derived xenografts lung cancer models. J. Control. Release 2019, 308, 44–56. [Google Scholar] [CrossRef]
- Wu, Y.; Crawford, M.; Mao, Y.; Lee, R.J.; Davis, I.C.; Elton, T.S.; Lee, L.J.; Nana-Sinkam, S.P. Therapeutic delivery of microRNA-29b by cationic lipoplexes for lung cancer. Mol. Ther. Nucleic Acids 2013, 2, e84. [Google Scholar] [CrossRef]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; de la Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnology 2019, 17, 85. [Google Scholar] [CrossRef]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing mir-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef]
- Sukumar, U.K.; Bose, R.J.C.; Malhotra, M.; Babikir, H.A.; Afjei, R.; Robinson, E.; Zeng, Y.; Chang, E.; Habte, F.; Sinclair, R.; et al. Intranasal delivery of targeted polyfunctional gold–iron oxide nanoparticles loaded with therapeutic micrornas for combined theranostic multimodality imaging and presensitization of glioblastoma to temozolomide. Biomaterials 2019, 218, 119342. [Google Scholar] [CrossRef] [PubMed]
- Oshima, G.; Guo, N.; He, C.; Stack, M.E.; Poon, C.; Uppal, A.; Wightman, S.C.; Parekh, A.; Skowron, K.B.; Posner, M.C.; et al. In vivo delivery and therapeutic effects of a microrna on colorectal liver metastases. Mol. Ther. 2017, 25, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, Y.; Qiu, Y.; Ding, M.; Zhang, Y. Mirna-204-5p and oxaliplatin-loaded silica nanoparticles for enhanced tumor suppression effect in cd44-overexpressed colon adenocarcinoma. Int. J. Pharm. 2019, 566, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Hu, Q.; Wang, M.; Shao, S.; Zhao, X.; Bai, H.; Huang, J.; Tang, G.; Liang, T. A pegylated megamer-based microrna delivery system activatable by stepwise microenvironment stimulation. Chem. Commun. (Camb.) 2019, 55, 9363–9366. [Google Scholar] [CrossRef] [PubMed]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic delivery of a Mir34a mimic as a potential therapeutic for liver cancer. Mol. Cancer Ther. 2014, 13, 2352. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, P.; Wang, J.; Wang, Y.; Sun, Z.; Zhou, Y.; Guan, X. Delivery of mesenchymal stem cells-derived extracellular vesicles with enriched mir-185 inhibits progression of opmd. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2481–2491. [Google Scholar] [CrossRef]
- Gokita, K.; Inoue, J.; Ishihara, H.; Kojima, K.; Inazawa, J. Therapeutic potential of lnp-mediated delivery of miR-634 for cancer therapy. Mol. Ther. Nucleic Acids 2019, 19, 330–338. [Google Scholar] [CrossRef]
- Yan, B.; Guo, Q.; Fu, F.-J.; Wang, Z.; Yin, Z.; Wei, Y.-B.; Yang, J.-R. The role of miR-29b in cancer: Regulation, function, and signaling. OncoTargets Ther. 2015, 8, 539–548. [Google Scholar]
- Fortunato, O.; Boeri, M.; Moro, M.; Verri, C.; Mensah, M.; Conte, D.; Caleca, L.; Roz, L.; Pastorino, U.; Sozzi, G. Mir-660 is downregulated in lung cancer patients and its replacement inhibits lung tumorigenesis by targeting mdm2-p53 interaction. Cell Death Dis. 2014, 5, e1564. [Google Scholar] [CrossRef]
- Gallardo, E.; Navarro, A.; Viñolas, N.; Marrades, R.M.; Diaz, T.; Gel, B.; Quera, A.; Bandres, E.; Garcia-Foncillas, J.; Ramirez, J.; et al. Mir-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis 2009, 30, 1903–1909. [Google Scholar] [CrossRef]
- Yamazaki, H.; Chijiwa, T.; Inoue, Y.; Abe, Y.; Suemizu, H.; Kawai, K.; Wakui, M.; Furukawa, D.; Mukai, M.; Kuwao, S.; et al. Overexpression of the mir-34 family suppresses invasive growth of malignant melanoma with the wild-type p53 gene. Exp. Ther. Med. 2012, 3, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.; Fox, J.; Ashton, B.; Middleton, J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 2007, 25, 2739–2749. [Google Scholar] [CrossRef]
- Baek, G.; Choi, H.; Kim, Y.; Lee, H.-C.; Choi, C. Mesenchymal stem cell-derived extracellular vesicles as therapeutics and as a drug delivery platform. Stem Cells Transl. Med. 2019, 8, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, K.E.; Dwyer, R.M. Extracellular vesicles for cancer therapy: Impact of host immune response. Cells 2020, 9, 224. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Yu, L.; Sun, C.; Cheng, D.; Yu, S.; Wang, Q.; Yan, Y.; Kang, C.; Jin, S.; et al. Mir-146b-5p inhibits glioma migration and invasion by targeting mmp16. Cancer Lett. 2013, 339, 260–269. [Google Scholar] [CrossRef]
- Liu, J.; Xu, J.; Li, H.; Sun, C.; Yu, L.; Li, Y.; Shi, C.; Zhou, X.; Bian, X.; Ping, Y.; et al. Mir-146b-5p functions as a tumor suppressor by targeting traf6 and predicts the prognosis of human gliomas. Oncotarget 2015, 6, 29129–29142. [Google Scholar] [CrossRef]
- Junichi, M.; Tessandra, S.; William, A.B.; Jing, Z. The transport mechanism of extracellular vesicles at the blood-brain barrier. Curr. Pharm. Des. 2017, 23, 6206–6214. [Google Scholar]
- Sun, C.-C.; Zhang, L.; Li, G.; Li, S.-J.; Chen, Z.-L.; Fu, Y.-F.; Gong, F.-Y.; Bai, T.; Zhang, D.-Y.; Wu, Q.-M.; et al. The lncrna pdia3p interacts with mir-185-5p to modulate oral squamous cell carcinoma progression by targeting cyclin d2. Mol. Ther. Nucleic Acids 2017, 9, 100–110. [Google Scholar] [CrossRef]
- Khan, S.; Brougham, C.L.; Ryan, J.; Sahrudin, A.; O’Neill, G.; Wall, D.; Curran, C.; Newell, J.; Kerin, M.J.; Dwyer, R.M. Mir-379 regulates cyclin b1 expression and is decreased in breast cancer. PLoS ONE 2013, 8, e68753. [Google Scholar] [CrossRef]
- Chin, A.R.; Fong, M.Y.; Somlo, G.; Wu, J.; Swiderski, P.; Wu, X.; Wang, S.E. Cross-kingdom inhibition of breast cancer growth by plant mir159. Cell Res. 2016, 26, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Thammaiah, C.K.; Jayaram, S. Role of let-7 family microrna in breast cancer. Noncoding RNA Res. 2016, 1, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and outcome of glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Liu, K.; Chen, H.; You, Q.; Ye, Q.; Wang, F.; Wang, S.; Zhang, S.; Yu, K.; Li, W.; Gu, M. Mir-145 inhibits human non-small-cell lung cancer growth by dual-targeting riok2 and nob1. Int. J. Oncol. 2018, 53, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Cheung, N.-K.V. Disialoganglioside gd2 as a therapeutic target for human diseases. Expert Opin. Ther. Targets 2015, 19, 349–362. [Google Scholar] [CrossRef]
- Comella, P.; Casaretti, R.; Sandomenico, C.; Avallone, A.; Franco, L. Role of oxaliplatin in the treatment of colorectal cancer. Ther. Clin. Risk Manag. 2009, 5, 229–238. [Google Scholar] [CrossRef]
- Alrfaei, B.M.; Vemuganti, R.; Kuo, J.S. Microrna mir-100 decreases glioblastoma growth by targeting smarca5 and erbb3 in tumor-initiating cells. bioRxiv 2019, 865105. [Google Scholar]
- Masoudi, M.S.; Mehrabian, E.; Mirzaei, H. Mir-21: A key player in glioblastoma pathogenesis. J. Cell. Biochem. 2018, 119, 1285–1290. [Google Scholar] [CrossRef]
- Kaur, K.; Kakkar, A.; Kumar, A.; Purkait, S.; Mallick, S.; Suri, V.; Sharma, M.C.; Julka, P.K.; Gupta, D.; Suri, A.; et al. Clinicopathological characteristics, molecular subgrouping, and expression of mir-379/mir-656 cluster (c14mc) in adult medulloblastomas. J. Neurooncol. 2016, 130, 423–430. [Google Scholar] [CrossRef]
- Kumar, A.; Nayak, S.; Pathak, P.; Purkait, S.; Malgulawar, P.B.; Sharma, M.C.; Suri, V.; Mukhopadhyay, A.; Suri, A.; Sarkar, C. Identification of mir-379/mir-656 (c14mc) cluster downregulation and associated epigenetic and transcription regulatory mechanism in oligodendrogliomas. J. Neurooncol. 2018, 139, 23–31. [Google Scholar] [CrossRef]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale coordination polymers for platinum-based anticancer drug delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase i study of mrx34, a liposomal mir-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microrna-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Zhu, X.; Badawi, M.; Pomeroy, S.; Sutaria, D.S.; Xie, Z.; Baek, A.; Jiang, J.; Elgamal, O.A.; Mo, X.; Perle, K.L.; et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from hek293t cells. J. Extracell. Vesicles 2017, 6, 1324730. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Cancer Type | miRNA(s) | Delivery Vehicle | Route | Study Outcome | Ref. |
|---|---|---|---|---|---|
| Breast cancer | miR-379 | MSC-EVs | IV | Significant reduction in tumor volume | [12] |
| miR-159, DOX | Monocyte-EVs with A15 | IV | Best therapeutic response after co-delivery of DOX and miR-159 | [13] | |
| let-7a | HEK-293-EVs with GE11 peptide | IV | Tumor growth suppression | [23] | |
| Lung cancer | miR-34a or let-7b | Neutral lipid emulsion | IV | Significantly lower tumor burden after treatment | [24] |
| miR-133b | Cationic Lipoplexes | IV | Significant increase in premiR-133b expression at lungs | [21] | |
| miR-34a | Neutral lipid reagent | IT, IV | Large areas of tumor necrosis | [25] | |
| miR-660 | CCL nanoparticles | IP, IV | Tumor growth significantly reduced | [26] | |
| miR-29b | Cationic lipoplexes | IV | Significantly smaller tumors after treatment | [27] | |
| miR-145 | EMNs | IP, RO | After IP injection EMN signal detected at tumor site | [28] | |
| miR-16 | Bacterial minicells | IV | Inhibition of tumor growth | [29] | |
| Glioblastoma | miR-146b | MSC-EVs | IT | Significant decrease in tumor volume | [30] |
| miR-124, -128, -137 (cluster 3) | Tumor derived EVs | IT | Significant increase in survival | [14] | |
| miR-100 and anti-miR-21 | Gold-iron oxide nanoparticles | Intranasal | Progressive accumulation of NPs in the prefrontal cortex and longer survival | [31] | |
| Neuroblastoma | miR-34a | Silica nanoparticles | IV | Significant reduction in tumor growth | [19] |
| let-7a | NN/NK-EV cocktail | IV | Significantly lower tumor bioluminescence signal | [15] | |
| Melanoma lung metastases | miR-34a and siRNAs | LPH nanoparticles with ScFv | IV | Co-delivery of miRNA and siRNA additively suppressed tumor growth | [20] |
| Colorectal liver metastases | miR-655-3p and OXL | NCPs | IP | Suppression of liver tumor development | [32] |
| Colorectal cancer | miR-204-5p and OXL | Silica nanoparticles | IV | Therapeutic agents combined had best therapeutic efficacy | [33] |
| Liver cancer | miR-122 | BOMB nanoparticles | IV | Lower tumor volumes in treatment group | [34] |
| miR-34a | Liposomal formulation | IV | Tumor growth inhibited | [35] | |
| OPMD | miR-185 | MSC-EVs | Topical | Reduced the incidence of transformation to OSCC | [36] |
| Pancreatic cancer | miR-634 | LNPs | IV | Significant reduction in tumor growth | [37] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neill, C.P.; Dwyer, R.M. Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells 2020, 9, 521. https://doi.org/10.3390/cells9020521
O’Neill CP, Dwyer RM. Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells. 2020; 9(2):521. https://doi.org/10.3390/cells9020521
Chicago/Turabian StyleO’Neill, Clodagh P., and Róisín M. Dwyer. 2020. "Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy" Cells 9, no. 2: 521. https://doi.org/10.3390/cells9020521
APA StyleO’Neill, C. P., & Dwyer, R. M. (2020). Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells, 9(2), 521. https://doi.org/10.3390/cells9020521