1. Introduction
Receptor tyrosine kinases (RTKs) are crucial regulators of key cellular processes such as cell growth, differentiation, neovascularization, and tissue repair. Hepatocyte growth factor receptor (MET or c-MET) is an RTK produced predominantly in cells of epithelial origin [
1]. Its only known high-affinity ligand is the hepatocyte growth factor (HGF) [
2] and both are essential to embryonic development and organ regeneration. The binding of MET and HGF activates the kinase activity of MET and several pathways, such as the mitogen-activated protein kinase (MAPK) cascade, the dylinositol 3-kinase pathway (PIK3K-Akt), the signal transducer and activator of transcription (STAT) pathway, and the IκBα–NF-κB complex [
3]. These pathways can activate cell proliferation, survival, migration, motility, invasion, angiogenesis, apoptosis, and epithelial-to-mesenchymal transition [
3,
4]. In addition, MET can interact with other cell membrane receptors, such as integrins, CD44, class B Plexins, and other tyrosine kinase receptors (e.g., HER2, AXL, EGFR, and VEGF) [
1,
2].
Deregulation of the MET pathway has been associated with cancer growth and metastasis in several types of tumors (lung, head and neck, gastric, and colorectal, among others) [
1,
3], taking place through several mechanisms including overexpression, amplification, autocrine signaling, and mutational activation [
5]. In addition to its role as an oncogenic driver, MET alterations have been described as a mechanism of resistance to different therapies [
1,
6,
7]. Therefore, several clinical trials have assessed the efficacy of selective and broad-spectrum MET inhibitors in an extensive panel of cancers [
8], generating interest in MET activity as a promising target for anticancer therapy.
MET protein overexpression and/or amplification are frequently found in different cancers. In fact,
MET amplification has been also described in diverse tumor types, such as non-small cell lung cancer (NSCLC), gastric cancer, esophageal cancer, colorectal cancer, medulloblastoma, and glioblastoma, and has been associated with bad prognosis and poor survival [
1,
9,
10,
11,
12]. Furthermore, this alteration is more frequent in metastatic patients and has been specifically associated with the development of resistance to anti-EGFR therapy [
1,
9]. On the other hand, MET overexpression is known to activate the MET-signaling pathway, promoting tumor cell growth, survival, migration, and invasion. This alteration has been associated with bad prognosis and the generation of metastasis in different tumor types, such as NSCLC, hepatocellular carcinoma, kidney cancer, head and neck cancer, colorectal cancer, gastric cancer, nasopharyngeal carcinoma, and glioblastomas [
2,
3,
6,
11,
12,
13,
14]. In addition, MET overexpression has been associated with chemotherapy and radiotherapy resistance in breast cancer [
1,
6,
7].
All these data indicate that
MET amplification and/or MET overexpression may be a potential biomarker for the evaluation of patients who will benefit from treatment with MET inhibitors [
14]. However, there are no standardized methods at present to confirm these molecular alterations. In fact, variable
MET amplification rates can be detected, depending on the detection techniques [
14], for example, fluorescence in situ hybridization (FISH), single-nucleotide polymorphism (SNP) genotyping, or quantitative polymerase chain reaction (qPCR), in which different scoring criteria may define high amplification.
In the same line, MET protein levels can be dependent on the antibodies employed, which can recognize different MET epitopes and domains, showing different membrane and/or cytoplasmic staining intensities by immunohistochemistry (IHC) [
14]. Moreover,
MET genomic changes have mostly been associated with metastatic patients and normally appear with the progression of disease [
5]. Therefore, tissue re-biopsy constitutes the best alternative for such molecular analysis in tissue samples; however, re-biopsy is often not possible, making the validation of liquid biopsy strategies to address MET status essential. In fact, circulating tumor cells (CTCs) and circulating free DNA (cfDNA) represent an accessible and noninvasive alternative for detecting MET alterations in patient’s blood, particularly in patients for whom tissue biopsies are inaccessible or problematic to carry out or repeat [
15]. Therefore, the purpose of the present study was to investigate the utility of a liquid biopsy-based strategy to assess MET alterations in cancer patients. For this aim, we evaluated the
MET copy number (CN) status in cfDNA and the MET expression in CTCs from a cohort of cancer patients with different tumor types and evaluated its clinical potential to detect the appearance of resistances and to guide the treatment with anti-MET drugs in a noninvasive manner.
2. Materials and Methods
2.1. Cell Lines
The MET Genetic Alteration Cell Panel was purchased from the American Type Culture Collection (ATCC, TCP-1036). The panel was composed of five human tumor cell lines (SNU-5, Hs746T, C32, AU565, and NCI-N87) showing various degrees of MET gene CN changes and expression levels. We also included the MET-negative prostate cancer cell line LNCaP and the cancer cell lines A549 (lung, NSCLC) and PC3 (prostate), purchased from the ATCC, in the study. The cell lines were routinely cultured in ATCC’s recommended growth medium at 37 °C, 5% CO2, and 95% humidity.
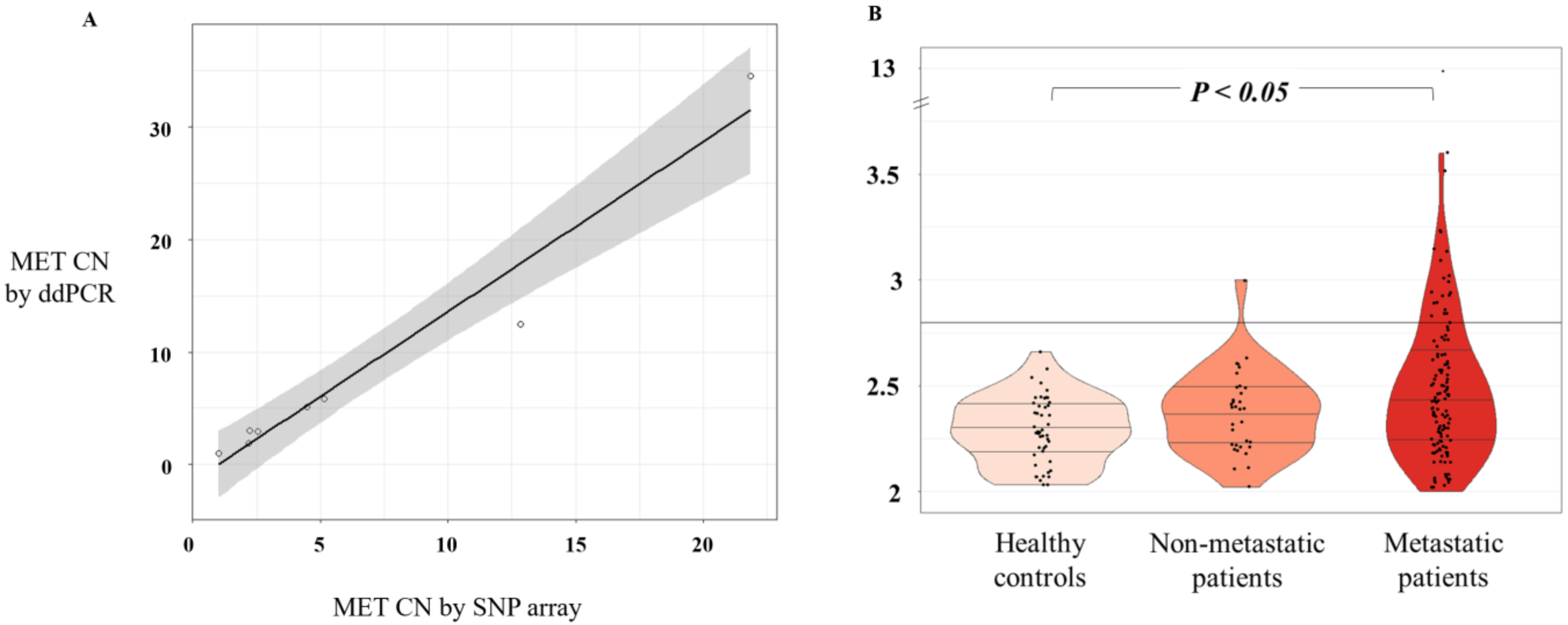
MET CN data of the eight cell lines were obtained from the Cancer Cell Line Encyclopedia (CCLE) database (
http://www.broadinstitute.org/ccle/home), determined using the Affymetrix Genome-Wide Human SNP Array 6.0 platform. Next, genomic DNA (gDNA) was extracted from the same cancer cell lines with a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s suggested protocol. DNA quantity was assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.2. Patients and Samples
The study design for testing the utility of blood samples to assess the MET status in cancer patients is shown in
Supplementary Figure S1. We designed a prospective study to analyze
MET amplification, including 112 patients diagnosed with diverse metastatic cancers (such as NSCLC, colorectal cancer, head and neck, or melanoma, among others) who had progressed to at least one line of therapy and also 34 patients with non-metastatic tumors. They were recruited between August 2017 and October 2019 at the Medical Oncology Service of Complexo Hospitalario Universitario de Santiago de Compostela and Hospital HM La Rosaleda of Santiago de Compostela. We also included 28 stage IV NSCLC EGFR mutated patients treated with tyrosine kinase inhibitors at the Medical Oncology Service of Hospital Universitario Son Espases (Mallorca, Spain) (
Table 1,
Supplementary Table S1). Frozen plasma samples from this last cohort, collected between September 2016 and November 2019, were retrospectively assessed for
MET CN. These patients had a progression to first-line tyrosine kinase inhibitors (TKIs) and some of them even to osimertinib in second-line (irreversible EGFR TKI). In addition, we recruited 49 healthy volunteers as controls for testing the specificity of the
MET CN analyses. Among the patients recruited, those diagnosed with lung or head and neck cancers starting with anti-EGFR treatment (
n = 30), were monitored by means of cfDNA and CTCs analysis before the initiation of therapy (baseline), at 12 and 24 weeks after the onset of therapy, and at disease progression in order to evaluate the dynamics of
MET amplification and overexpression within tumor evolution.
All individuals gave written informed consent approved by Santiago de Compostela and Lugo, and Islas Baleares Ethics Committees (Ref: 2017/538 and IB2584/15; respectively) prior to enrolling in the study. The protocol approved by the Ethics Committee was conducted according to the Declaration of Helsinki.
2.3. Circulating Free DNA Isolation from Plasma Samples
Ten milliliters of peripheral whole blood from controls and patients were obtained by direct venipuncture directly with Streck Cell-Free DNA blood collection tubes (Streck, La Vista, NE, USA). Plasma was separated within 96 h after blood collection as a result of two sequential centrifugation steps (10 min at 1600 g, and 10 min at 6000 g; both at room temperature), and then stored at -80 °C for further processing. cfDNA was extracted from 3 mL of plasma using a QIAmp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. cfDNA yields were assessed using a Qubit 1X dsDNA Hs Assay Kit, according to the manufacturer’s instructions (Thermo Fisher Scientific, USA).
2.4. Droplet Digital PCR to Detect MET CN
Genomic DNA extracted from the cancer cell lines (n = 8) and cfDNA isolated from plasma (n = 140 metastatic patients, n = 34 non-metastatic patients, and n = 49 healthy controls) was amplified using ddPCR to analyze MET CN. The ddPCR experiments were performed according to the manufacturer’s protocol (Bio-Rad, Hercules, CA, USA). Each assay contained 20 μL reaction mixture of 10 μL ddPCR Supermix for Probes (Bio-Rad, CA, USA), 1 μL PrimePCR ddPCR CN assay for MET with a FAM fluorophore (dHsaCP2500321, Bio-Rad, CA, USA), 1 μL PrimePCR ddPCR CN assay for the reference gene with a HEX fluorophore (EIF2C1, dHsaCPE2500349, Bio-Rad, CA, USA), and 8 μL DNA template and nuclease-free water. In the case of gDNA from cancer cell lines, we added the HaeIII enzyme (Takara Bio, USA) to the mix, according to the manufacturer’s instructions. Droplets were generated using a QX200 AutoDG Droplet Digital PCR System (Bio-Rad, CA, USA) with Droplet Generation Oil for Probes (Bio-Rad, CA, USA) and transferred to a 96-well plate. PCR amplification was performed with the following conditions: 95 °C for 10 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 60 s, and 10 min incubation at 98 °C. The plates were read on a Bio-Rad QX200 droplet reader (Bio-Rad, CA, USA) and we calculated MET CN using the QuantaSoft Pro software (Bio-Rad, CA, USA).
2.5. Fluorescent In Situ Hybridization (FISH) Analysis
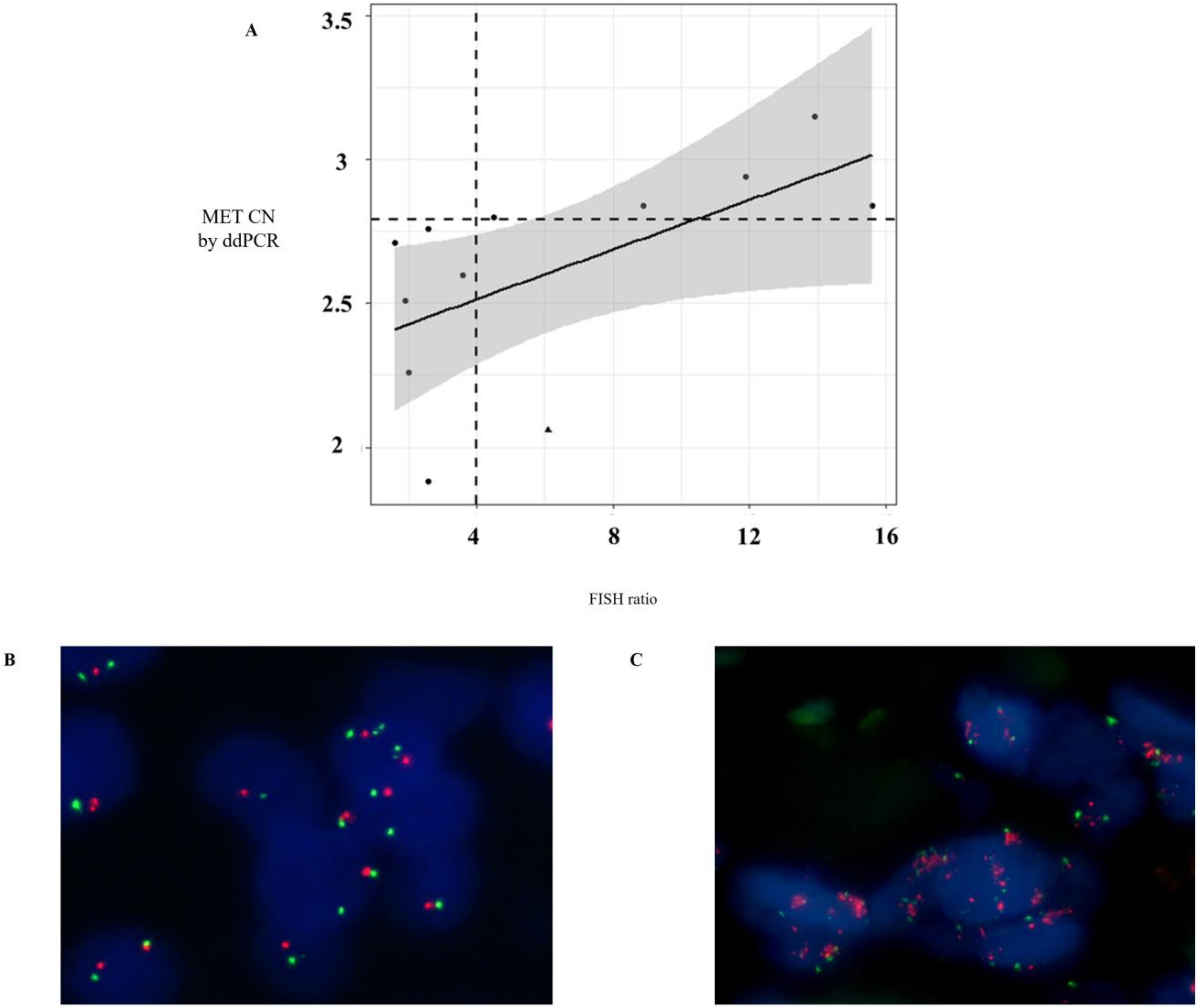
Fluorescent In Situ Hybridization (FISH) analysis was conducted on formalin-fixed paraffin-embedded (FFPE) tissues in a subgroup of patients who had a blood sample taken within 2 months after tissue biopsy (
n = 12). The tissue sections were hybridized with MET (SpectrumRed)/CEN7 (SpectrumGreen) Dual Color FISH Probe (Vysis, Abbott Molecular) following the manufacturer’s protocol. In each case, 60 non-overlapping nuclei were examined by a fluorescence microscope (BX51, Olympus Corporation, Barcelona, Spain). FISH positivity was estimated using the standard Colorado criteria (gene amplification; 2 >
MET gene (red)/CEP7q (green) per cell plus high polysomy; 5 copies > mean
MET per cell) [
16].
2.6. Spiked Experiments
The assays to evaluate MET expression on CTCs were tested using cancer cell lines spiked in whole blood from the healthy volunteers recruited. A total of 7.5 mL of blood from healthy donors was collected in CellSave tubes (Menarini, Silicon Biosystems, Bologna, Italy). Cells were spiked at a final concentration of 30 cells per mL of blood. Concurrently, we employed samples from three healthy donors per each cell line. These samples were analyzed using the CellSearch® and Parsortix Systems, running identical blood samples with both technologies. All spiked samples were enriched within 96 h of collection.
2.7. Analysis of MET Expression on CTCs Isolated Using Cell Search®
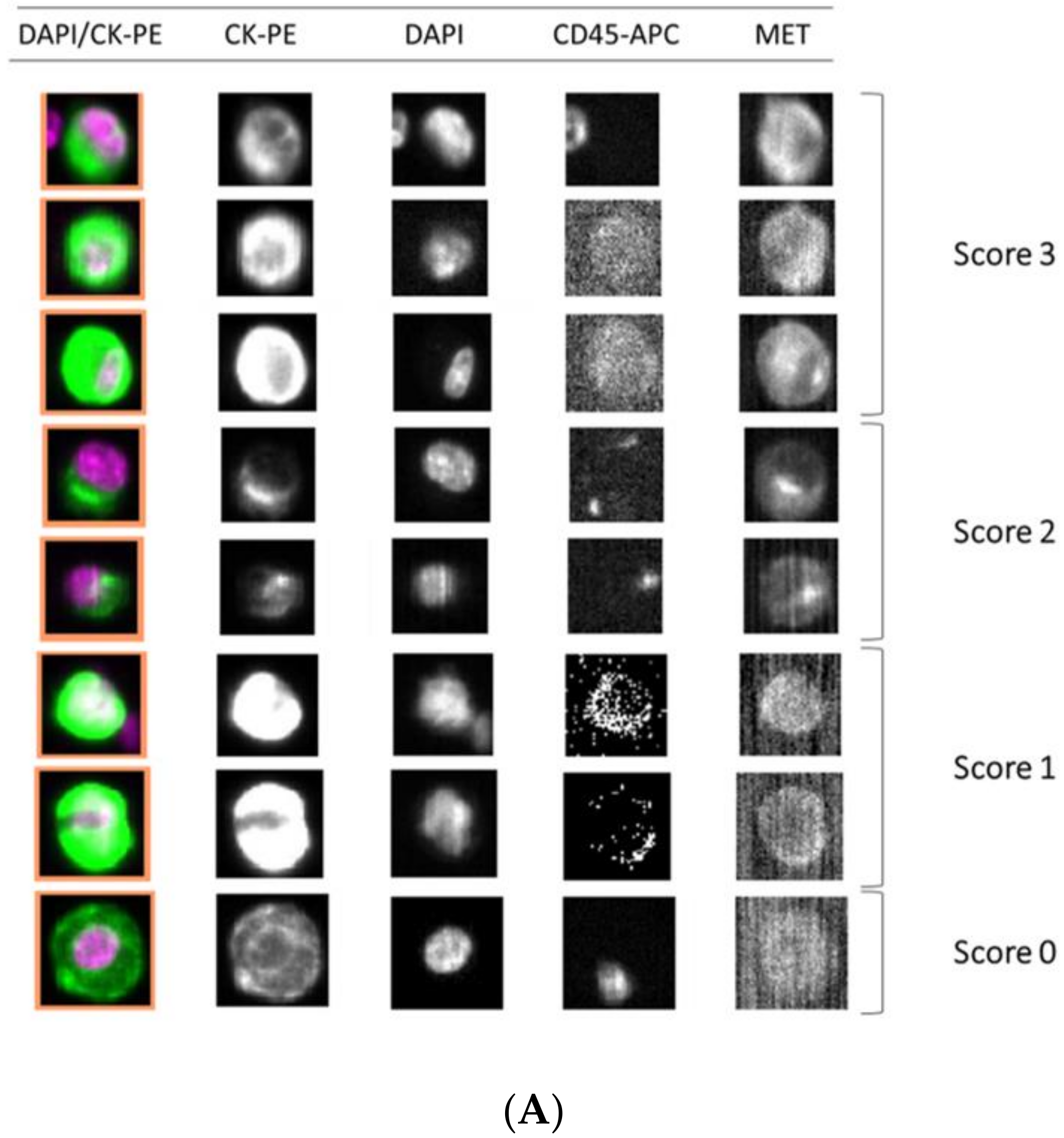
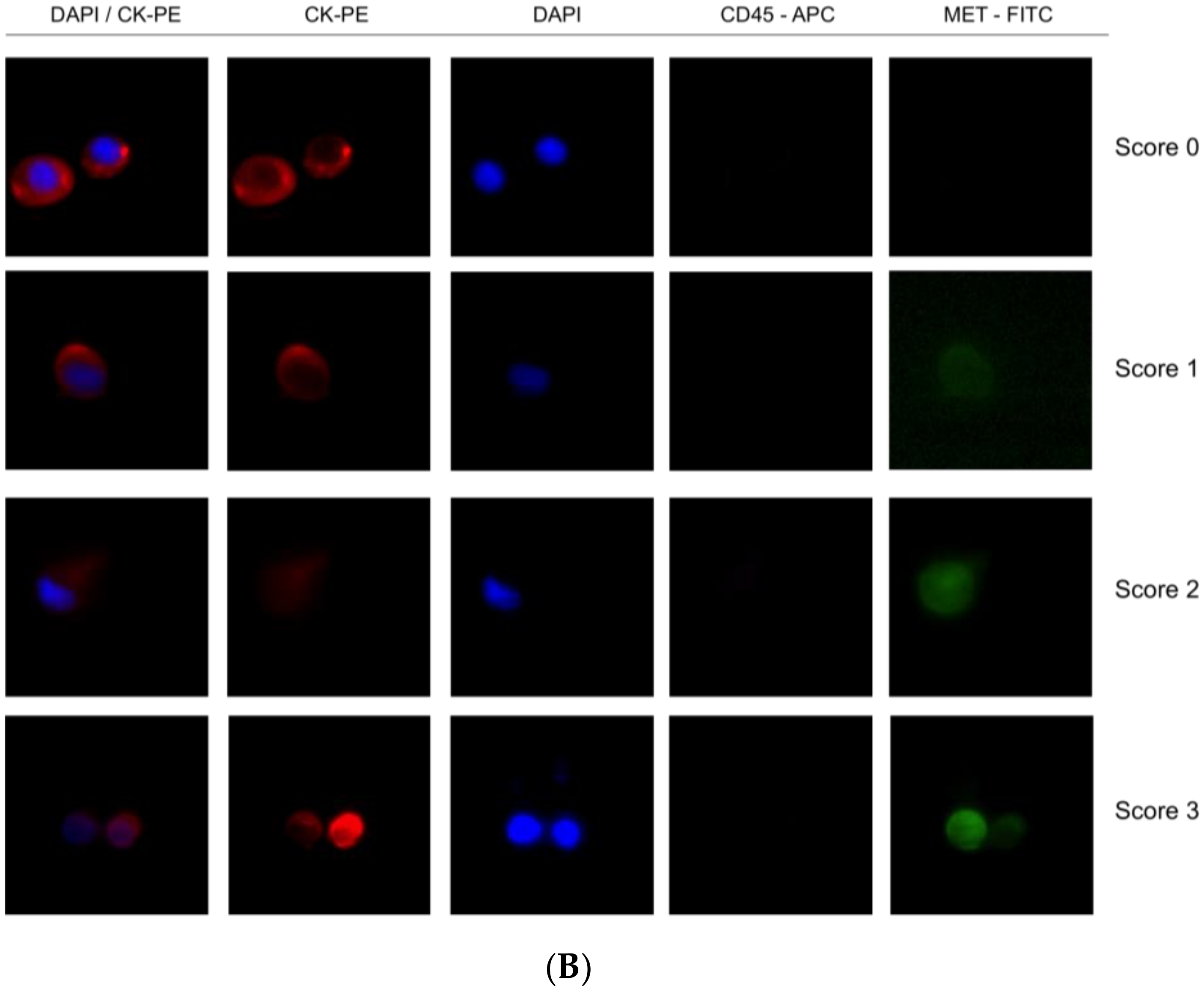
A total of 7.5 mL of peripheral whole blood samples were collected in CellSave tubes (Menarini, Silicon Biosystems, Bologna, Italy) for CTCs enumeration using the FDA-cleared CellSearch® system (Menarini, Silicon Biosystems, Bologna, Italy). CellSearch® Circulating Tumor Cell Kit (Menarini, Silicon Biosystems, Bologna, Italy) was used for these specific experiments, including ferrofluids coated with epithelial cell-specific anti-Epithelial cell adhesion molecule (EpCAM) antibodies to immunomagnetically enrich epithelial cells; a mixture of antibodies directed to cytokeratins (CKs) 8, 18, and 19 conjugated to phycoerythrin (PE); anti-CD45 mAb conjugated to allophycocyanin (APC); and nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) to fluorescently label the cells. The C32 cells and human melanoma samples were analyzed using a CellSearch® Circulating Melanoma Cell Kit (Menarini, Silicon Biosystems, Bologna, Italy), which included ferrofluid coated with antibodies targeting the CD146 antigen to capture circulating melanoma cells (CMCs) and fluorescent reagents against anti-high molecular weight melanoma-associated antigen (HMW-MAA-PE (MEL-PE)), anti-CD34-APC for endothelial cells, anti-CD45-APC for leukocytes, and DAPI for staining cell nuclei. The open 4th antibody position of the CellSearch® system was used to evaluate MET expression. We employed the anti-MET fluorescein (FITC)-conjugated monoclonal antibody (D-4, sc-514148, Santa Cruz, CA, USA) at a final concentration of 20 μg/mL. CTCs were identified as EpCAM+, CK+, CD45−, and DAPI+, and the level of MET expression was recorded for each CTC, on a 0–3 scale, by comparison to the MET expression levels of the different cell lines reference.
2.8. Analysis of MET Expression on CTCs Isolated Using Parsortix System
A total of 7.5 mL of peripheral whole blood was collected in CellSave tubes (Menarini, Silicon Biosystems, Bologna, Italy) and loaded into the Parsortix microfluidic device (Angle Inc, Guildford, UK). CTCs were then enriched from blood samples in disposable Parsortix cassettes (GEN3D6.5, Angle Inc, Guildford, UK), according to the manufacturer’s guidelines. CTCs were trapped in the Parsortix cassette due to their large size and lower compressibility than the remaining blood cells. After separation, we fixed the sample and carried out immunofluorescence staining. CTCs were subjected to on-cassette-staining with selected antibodies, according to the manufacturer’s guidelines, followed by fluorescence microscopy detection (Leica DMI8, Leica Microsystems, Germany). The selected antibodies included Alexa Fluor (AF)546-conjugated pan-Cytokeratin (C11, sc-8018, Santa Cruz, CA, USA), AF647-conjugated CD45 (35-Z6, sc-1178, Santa Cruz, CA, USA), anti-MET FITC-conjugated (D-4, sc-514148, Santa Cruz, CA, USA), and DAPI to fluorescently label the cells. CTCs were identified as CK+, CD45−, and DAPI+, and the level of MET expression on CTCs was determined, on a scale of 0–3 scale, depending on the MET antibody intensity, according to the results obtained in cell-line controls.
2.9. Statistical Analysis
Statistical analyses were performed using R version 3.4.0. Pearson’s correlation and linear regression were used to evaluate the relationship between ddPCR and SNP6.0 or FISH results. The Mann–Whitney–Wilcoxon U-Test was used to compare the MET CN between cancer patients and healthy controls. Two-tailed P values of 0.05 or less were considered significant. The cut-off value for the MET CN test to define the presence of MET amplification was determined using the mean plus 3 standard deviations of healthy controls. The Kappa test was used to determine the concordance between MET amplification based on FISH and ddPCR analyses. Paired Student t tests were used to compare the CTCs enrichment potential between the Parsortix and the CellSearch® systems.
4. Discussion
This study was designed to investigate the utility of liquid biopsy in assessing MET protein expression and MET CN, defined as MET status, in cancer patients. We used ddPCR to quantify MET CN in cfDNA and in a complementary way, we evaluated two approaches to detect MET presence on CTCs to overcome the complexity of isolating different phenotypes of CTCs (CellSearch® and Parsortix systems).
For CTCs analysis, we implemented the MET expression detection using the CellSearch
® and the Parsortix platforms routine. Due to the ultra-rare nature of CTCs, with numbers as low as 1 CTC per 10
6-10
7 leukocytes, most assays for CTCs analyses use a combination of enrichment and detection/characterization techniques [
18]. It is important to remark that the most widely used CTCs enrichment technique is the immunomagnetic-based selection of CTCs, being CellSearch
® system the only FDA approved device for CTCs enumeration, although there are others based on this strategy (e.g., IsoFlux™, AdnaTest, and MACS
®). The main limitation of this approach is the lack of a “universal marker” that can be used independently of both the tumor type and the stage of disease progression [
19]. To solve this problem, new CTCs isolation strategies have been developed such as size-based enrichment (e.g., Parsortix, ScreenCell
®, and ISET
®) and microfluidic-based enrichment (e.g., Parsortix, CTC-Chip/iChip, IsoFlux™, and GILUPI CellCollector™). The Parsortix system uses a combination of microfluidic and size/deformability-based approaches to separate CTCs from blood samples. Its main advantage over CellSearch
® is the ability to enumerate CTCs independently of their epithelial or mesenchymal status. However, cell size-based systems have difficulty to completely separate cancer cells and leukocyte by their size since patient’s CTCs exhibit a high degree of pleomorphism, where the size of the captured tumor cells can vary from 4-30 µm [
20]. Therefore, size overlapping between CTCs and leukocytes will result in the loss of small CTCs. Besides, although CTCs can be detected in non-metastatic patients using both approaches, their low levels of non-advanced disease limit the application of these technologies as diagnostic tools [
21].
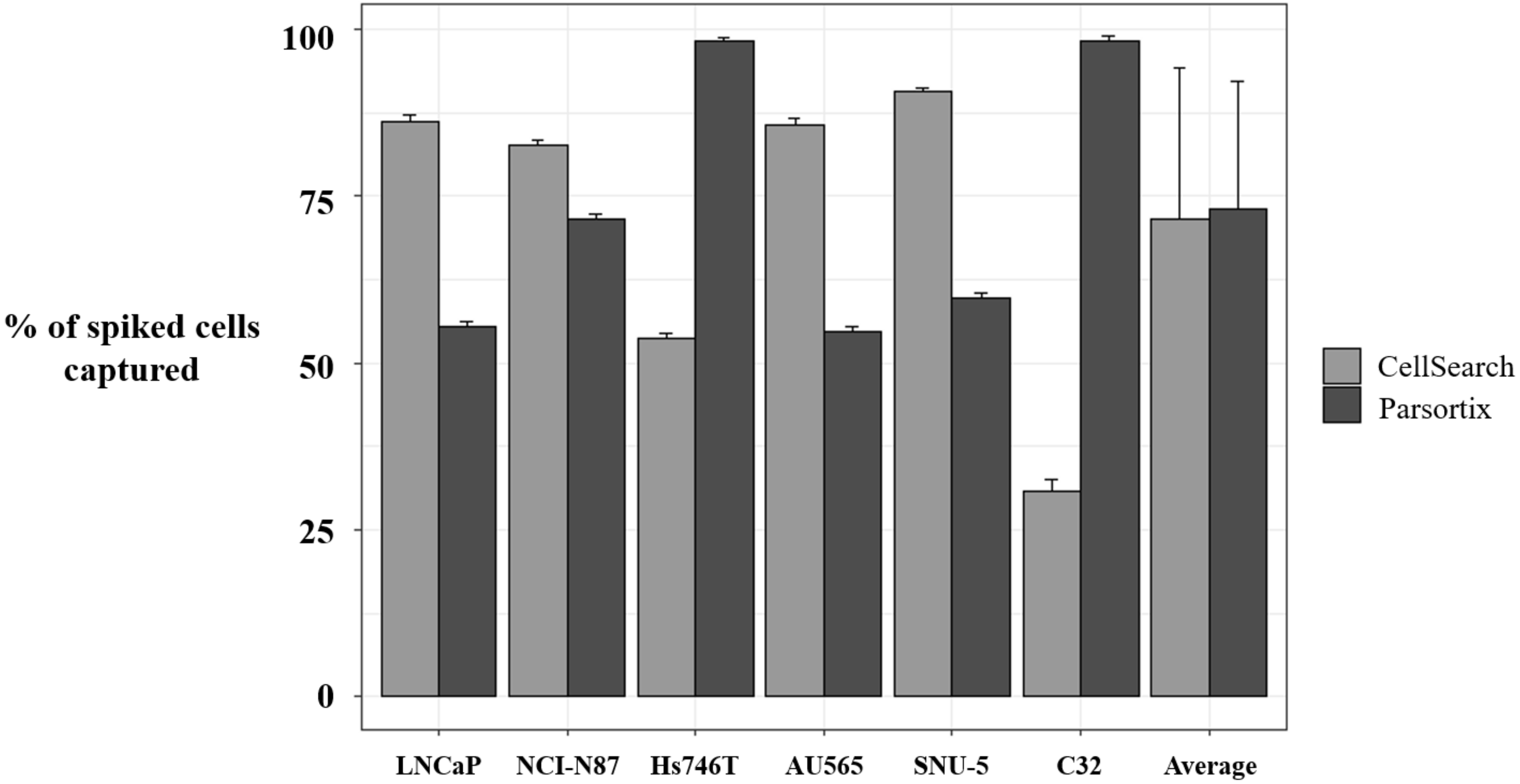
In our cohort of patients with head and neck and NSCLC, we found that the Parsortix approach identified a higher proportion of CTC-positive patients than the CellSearch® system, indicating that an antigen CTCs independent isolation approach can overcome the EpCAM expression heterogeneity that characterizes these tumors. Therefore, some non-epithelial or mesenchymal CTCs from cancer patients would be detected on the Parsortix platform but could escape the antigen-dependent enrichment method of the CellSearch® EpCAM assay, and vice versa with epithelial CTCs, such as reflected our spiking experiments with different cell lines expressing different levels of EpCAM. Therefore, although the good results obtained using Parsortix system, a larger cohort of patients would be necessary to clearly determine which technology should be used to characterize CTCs from patients with both tumors.
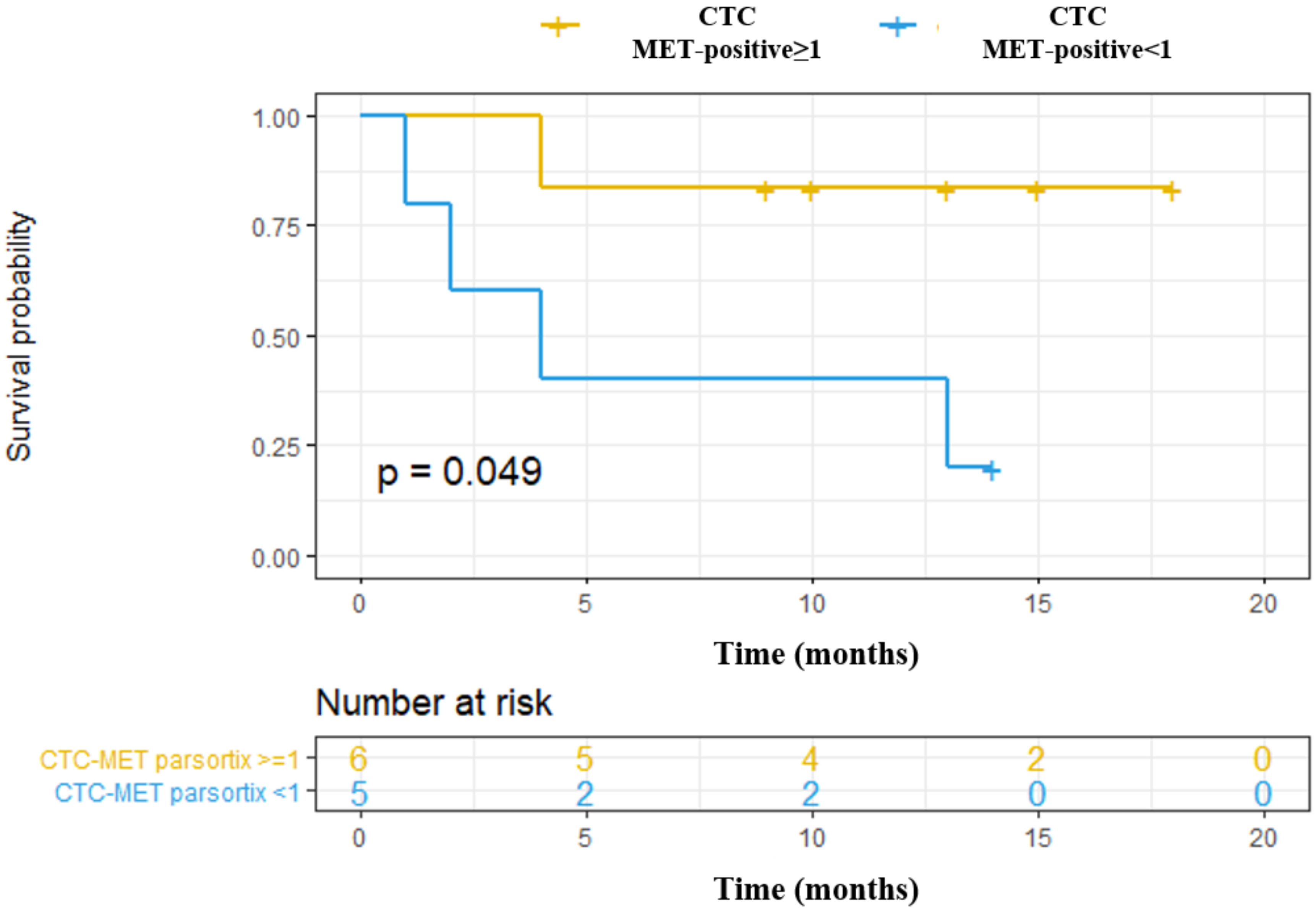
Independently to the CTCs enumeration rate showed by both technologies, our study provides a proof-of-concept for the analysis of MET expression in CTCs to determine MET status in patients that might be eligible in clinical trial testing MET inhibitors, and could be useful to monitor the appearance of therapy resistance. Previously, Zhang et al. developed a noninvasive assay for MET+ CTCs capture and characterization from multiple patients with metastatic carcinomas using the CellSearch
® system [
22]. Due to the strategy used, they identified a high prevalence of CD45-positive leukocytes expressing MET in patients with metastatic solid tumors, although the clinical relevance of this cell population is unknown. Ilie et al. also evaluated the prevalence of MET expression in CTCs from NSCLC patients [
23], using a combined strategy similar to the one employed in the present work. In their study, MET expression was assessed using a CellSearch
® system and by immunocytochemistry on ISET-enriched CTCs from advanced-stage NSCLC patients. They found that MET expression was lower in CTCs analyzed by CellSearch
® than using ISET. In fact, only 9 of 256 (3.52%) patients showed CTCs with high MET expression (scores of 2
+ or 3
+) in CellSearch
®, whereas MET-positive CTCs from advanced-stage NSCLC patients were successfully detected using the ISET platform, being 54 of the 75 (72%) patients’ blood samples defined as MET positive. Our data showed that the prevalence of MET expression in CTCs isolated by CellSearch
® is greater than previously reported data (20% MET-positive scores of 2+ or 3+), and even higher to the expected MET expression in patients with metastatic cancer. Importantly, using Parsortix system, we found 26.6% of patients with MET-positive scores of 2
+ or 3
+, being the concordance to detect the presence of CTCs overexpressing MET between both technologies, of 50%. These results showed that MET is expressed on CTCs with a considerable patient dependent and technology variability. Obviously, these results reflect the distinct CTCs enrichment methodologies of the two platforms, and the importance of tumor lineage for the MET-CTC-assay, highly diverse in our cohort of patient.
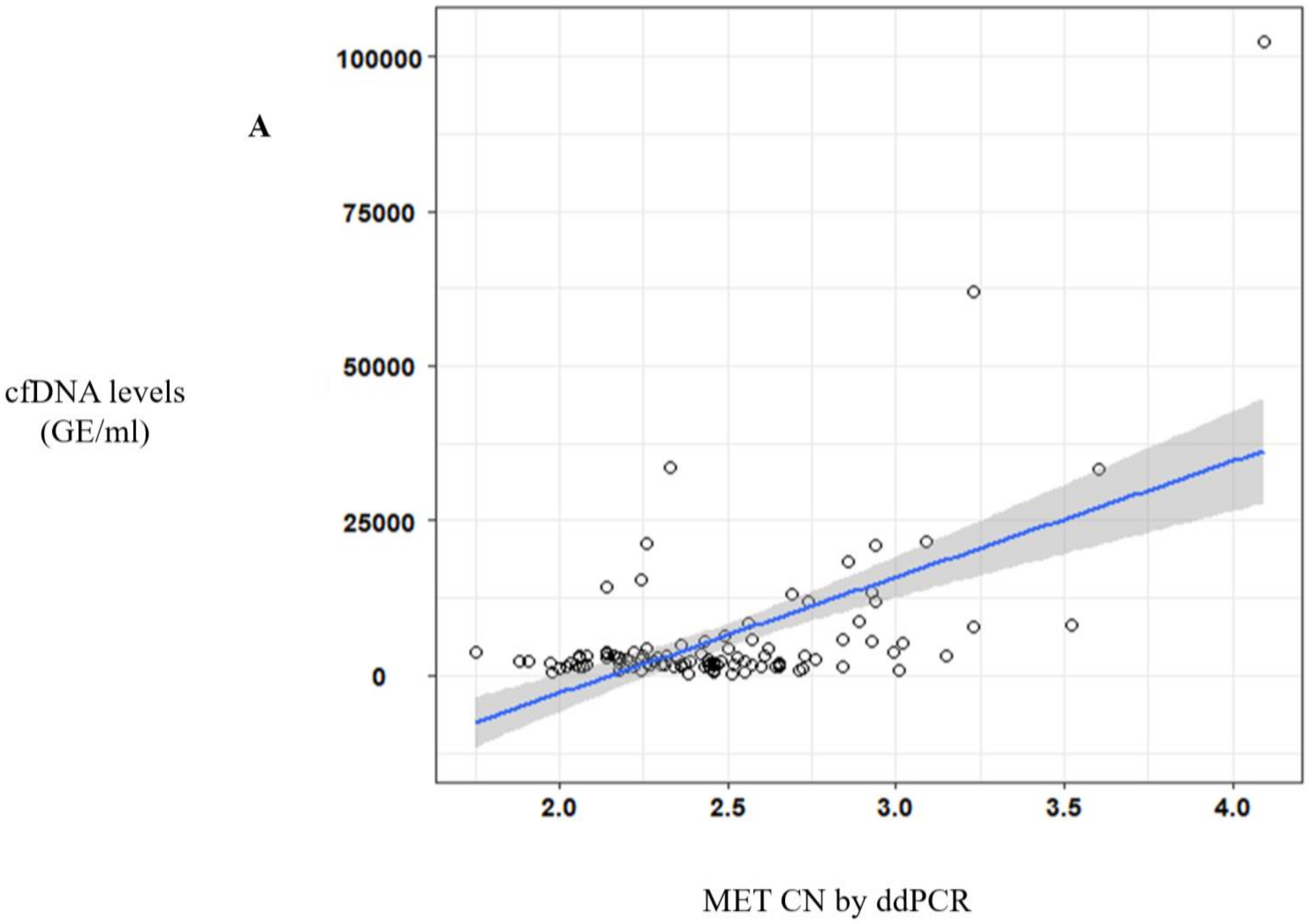
In addition to the CTCs analysis, we demonstrated the ddPCR approach to be a useful tool for detecting
MET amplification in plasma samples from cancer patients. Zhang et al. had already shown this capacity, detecting
MET CN in genomic DNA from cancer cell lines and tumor samples [
24]. Recently, a study in cfDNA obtained from plasma of different tumor types showed that detection of MET alterations (amplification and point mutations) by liquid biopsy was feasible using the Guardant360 NGS panel [
25]. In this study, they found 7% of patients with
MET amplification in plasma, most of them with metastatic disease. Although NGS panels can provide more complete information regarding the status of other driver genes, our goal was the validation of a more sensitive and easier to implement test to monitor
MET CN in plasma using ddPCR. For this aim, a cohort of healthy controls and cancer patients with different tumor types and metastatic stages were analyzed to detect
MET CN by ddPCR. We found 16.4% of the metastatic patients and 3% of the non-metastatic setting as positive for
MET amplification, being higher rates than the previously described using an NGS approach. Besides, we evaluated the concordance of the
MET CN detection between plasma and tumor samples obtained at the same time as the disease evolution. Importantly,
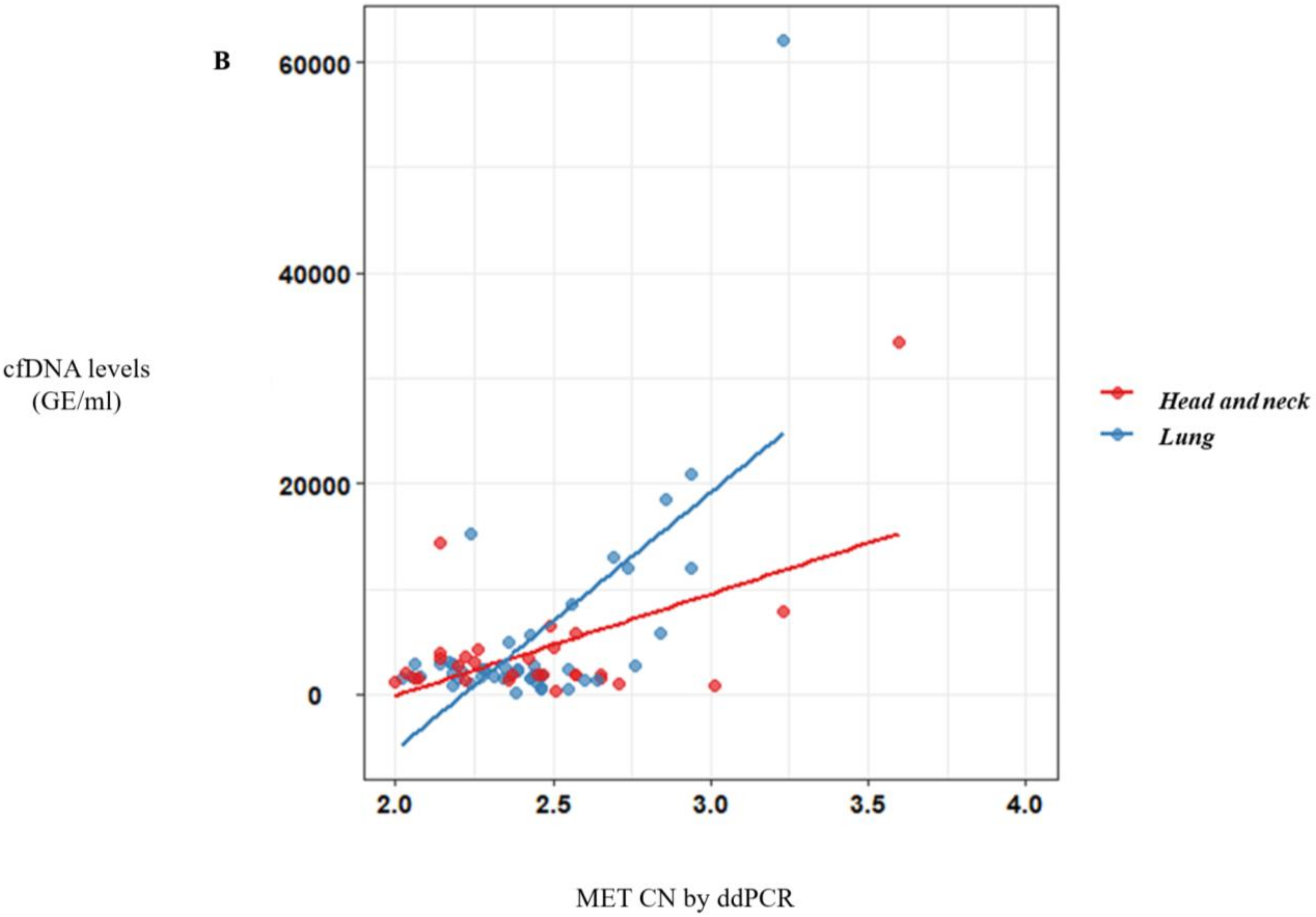
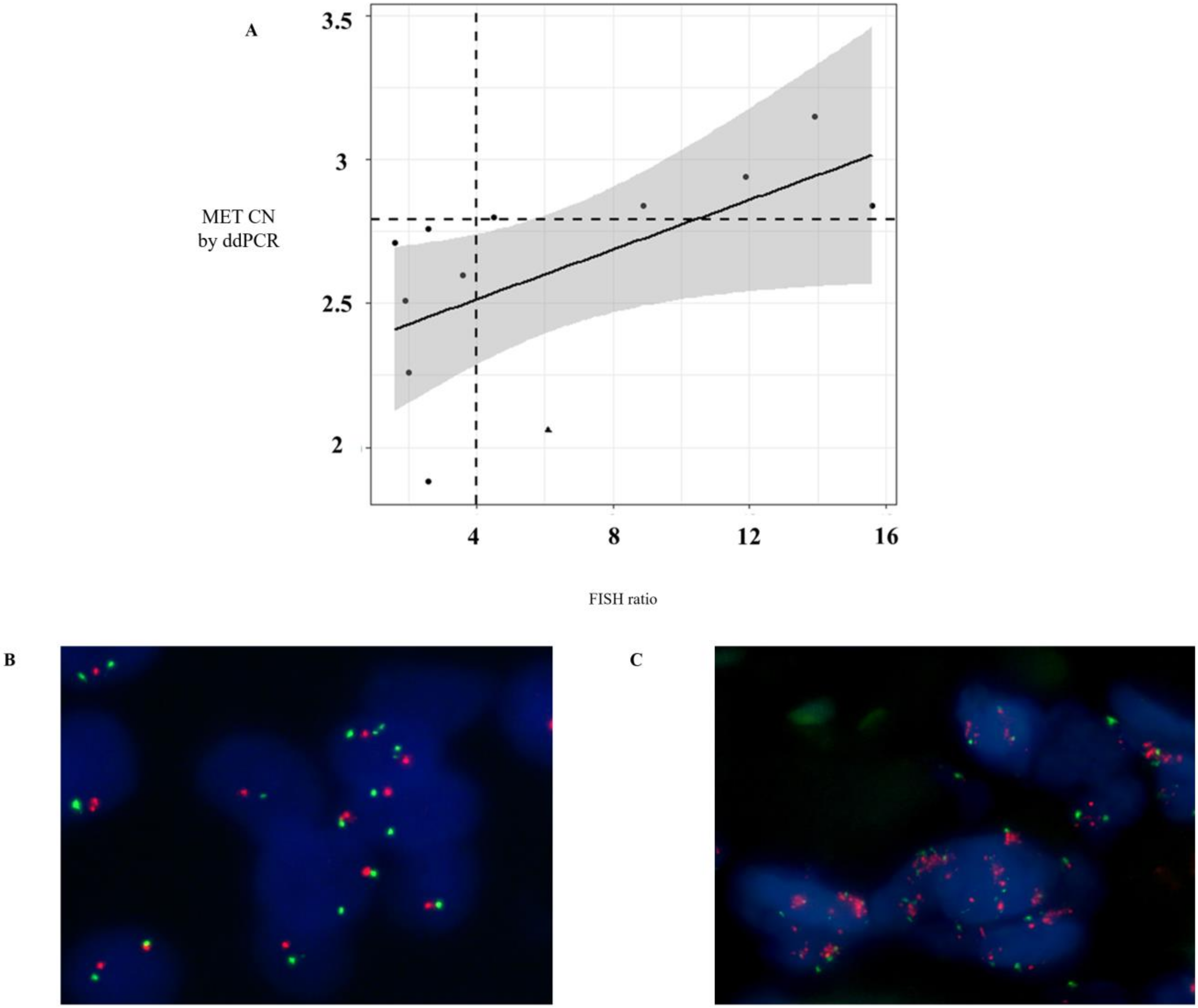
MET amplification positivity detected by ddPCR was comparable to that detected by FISH. Only one melanoma patient with
MET amplification determined by FISH (MET/CEN-7 ratio = 6.1) did not show
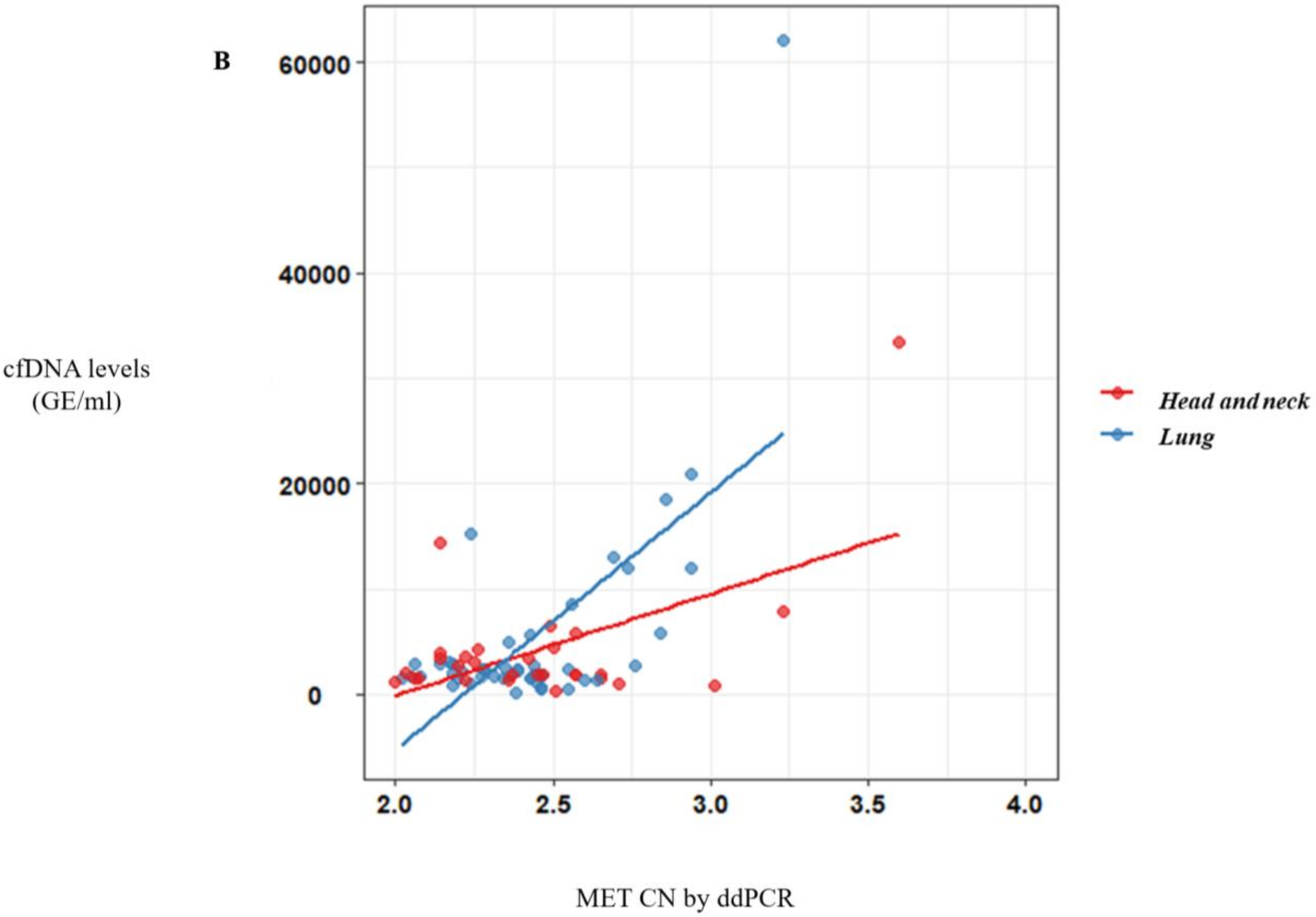
MET amplification in plasma using ddPCR (CN > 2.8). These results reinforced the value of the MET analysis in plasma samples as a more accessible monitoring tool to guide anti-MET therapies.
Actually,
MET amplification is a relevant resistance mechanism to first-generation EGFR-TKIs, being detectable in approximately 5%-22% of NSCLC patients with acquired resistance to these drugs [
26]. Osimertinib inhibits the EGFR variants Del19 and L858R, as well as the resistant T790M mutation. About 20% of such patients do not respond well to osimertinib, and almost all patients have eventually relapsed and developed resistance to the treatment. These resistance mechanisms are largely unknown, except for the C797S mutation and
MET amplification [
26]. There have been preclinical studies suggesting that monotherapy with osimertinib is not effective for the treatment of EGFR mutant NSCLC with acquired resistance to first-, second-, or third-generation EGFR-TKIs, due to
MET amplification, or even MET hyperactivation [
27]. Targeting EGFR and MET simultaneously may be required to overcome resistance to EGFR-TKIs by MET alteration, making it necessary to assess MET status before osimertinib treatment. Thus, some studies have reported the successful administration of dual EGFR and MET therapies [
28,
29,
30]. The combination of osimertinib and MET inhibitors can be safe and effective in NSCLC patients with
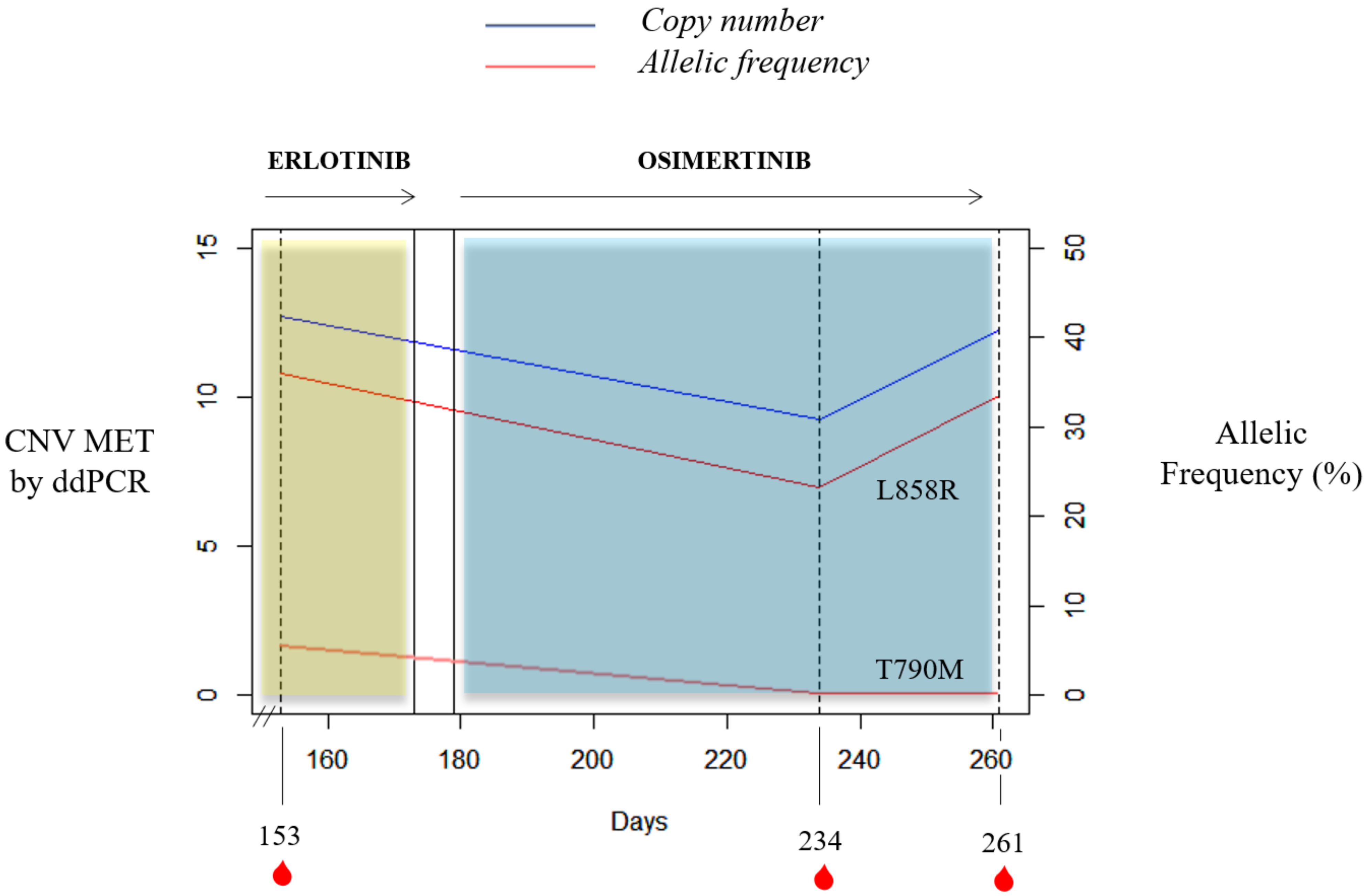
MET amplification detected by ddPCR as an acquired resistance mechanism. In this context, our results demonstrated the utility of plasma MET determination as a valuable biomarker for monitoring the appearance of resistance to anti-EGFR therapy. In fact, we reported a case of
MET amplification detected in plasma from a NSCLC patient harboring EGFR L858R mutation after disease progression to erlotinib.
Regardless of the value of our findings, some limitations to our study should be kept in mind. The small number of tissue samples (obtained close to plasma collection) that were available did not allow us to carry out a robust comparative study between MET CN obtained from plasma and tissue. Despite this limitation, we showed that plasma may be a good option for monitoring the molecular evolution of the disease, which is even more relevant in patients for which a tissue-rebiopsy is not feasible. In addition, we implemented the detection of MET expression in CTCs from cancer patients using two complementary technologies and obtaining promising results. Nevertheless, taking into account the limited follow-up of the patients and the heterogeneity of MET overexpression found within the different tumor types and the technologies, the real clinical value of the assessment of MET expression in CTCs must be evaluated also in a larger cohort of patients.
In conclusion, we developed specific and noninvasive assays to monitor MET expression on CTCs and MET CN on cfDNA using blood from cancer patients. Both molecular alterations were detected in patients with different tumors and represent a good opportunity to characterize these tumors in a noninvasive and dynamic way, as well as guiding the addition of MET inhibitors.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}