Regulation of Necroptosis by Phospholipids and Sphingolipids



{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Necroptosis
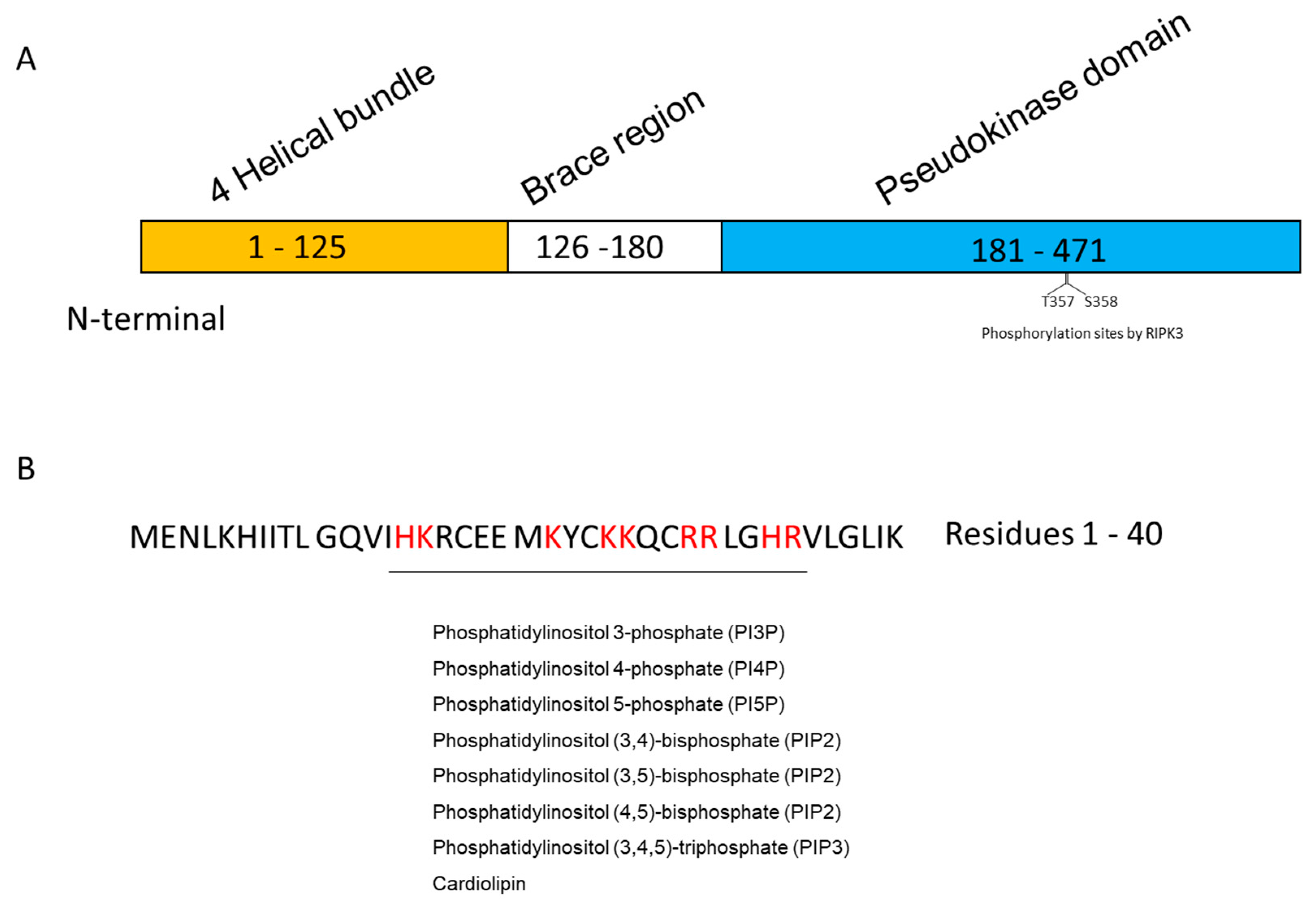
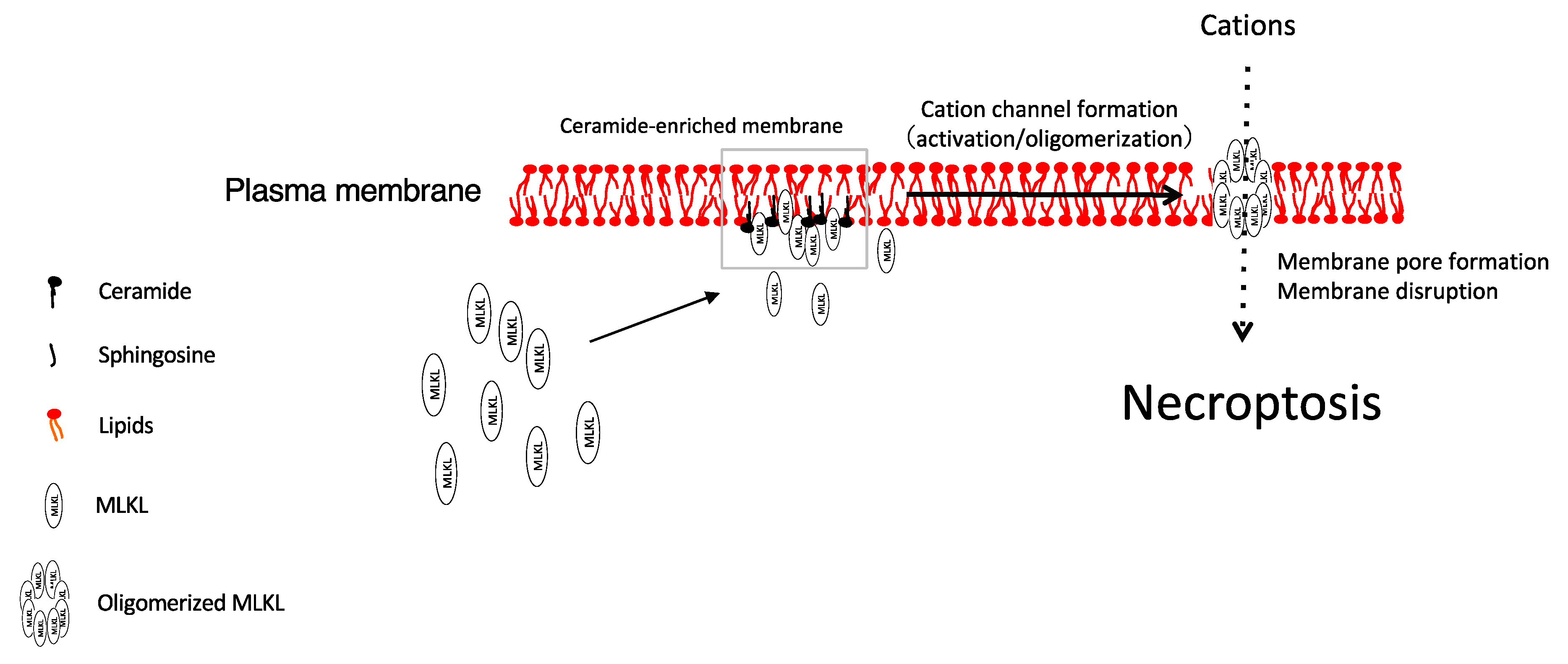
1.2. MLKL
1.3. Lipids
2. Phospholipids
2.1. Inositol Phospholipids
2.2. Cardiolipin
3. Sphingolipids
3.1. Ceramide
3.2. Sphingosine Analogue FTY720 (Fingolimod)
4. Very Long Chain Fatty Acids
5. Glycolipid Transfer Protein (GLTP)
6. Docosahexaenoic Acid (DHA)
7. Future Outlook and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S.; Tanaka, M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.I.; Harmon, B.V.; Gobe, G.C.; Kerr, J.F. Patterns of cell death. Methods Achiev. Exp. Pathol. 1988, 13, 18–54. [Google Scholar] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; Dixon, S.J. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016, 23, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.R.; Morrow, L.M.; Visser, M.B.; Atilla-Gokcumen, G.E. Turning the Spotlight on Lipids in Non-Apoptotic Cell Death. ACS Chem. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef]
- Diao, Y.; Ma, X.; Min, W.; Lin, S.; Kang, H.; Dai, Z.; Wang, X.; Zhao, Y. Dasatinib promotes paclitaxel-induced necroptosis in lung adenocarcinoma with phosphorylated caspase-8 by c-Src. Cancer Lett. 2016, 379, 12–23. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.B.; Fang, Y.L.; Zhang, Z.R.; Shao, J.; Hong, M.; Huang, C.J.; Liu, J.; Chen, R.Q. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal. 2017, 31, 112–123. [Google Scholar] [CrossRef]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Petrie, E.J.; Sandow, J.J.; Jacobsen, A.V.; Smith, B.J.; Griffin, M.D.W.; Lucet, I.S.; Dai, W.; Young, S.N.; Tanzer, M.C.; Wardak, A.; et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 2018, 9, 2422. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, Y.; Choi, H.M.C.; Sarkar, C.; Koh, E.Y.; Wu, J.; Lipinski, M.M. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018, 9, 476. [Google Scholar] [CrossRef]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Bell, R.M. Functions of sphingolipids and sphingolipid breakdown products in cellular regulation. Science 1989, 243, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Liscovitch, M.; Cantley, L.C. Lipid second messengers. Cell 1994, 77, 329–334. [Google Scholar] [CrossRef]
- Kolesnick, R.N.; Kronke, M. Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 1998, 60, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Nishizuka, Y. Protein kinase C isotypes and their specific functions: Prologue. J. Biochem. 2002, 132, 509–511. [Google Scholar] [CrossRef]
- Murakami, M.; Kudo, I. Phospholipase A2. J. Biochem. 2002, 131, 285–292. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.R.; Li, N.; Atilla-Gokcumen, G.E. Very Long Chain Fatty Acids Are Functionally Involved in Necroptosis. Cell Chem. Biol. 2017, 24, 1445–1454 e1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, K.; Mather, A.R.; Marimuthu, S.; Shah, P.P.; Snider, A.J.; Obeid, L.M.; Hannun, Y.A.; Beverly, L.J.; Siskind, L.J. Loss of neutral ceramidase protects cells from nutrient- and energy -deprivation-induced cell death. Biochem. J. 2016, 473, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nganga, R.; Oleinik, N.; Kim, J.; Selvam, S.P.; De Palma, R.; Johnson, K.A.; Parikh, R.Y.; Gangaraju, V.; Peterson, Y.; Dany, M.; et al. Receptor-interacting Ser/Thr kinase 1 (RIPK1) and myosin IIA-dependent ceramidosomes form membrane pores that mediate blebbing and necroptosis. J. Biol. Chem. 2019, 294, 502–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, W.; Stahelin, R.V. Membrane-protein interactions in cell signaling and membrane trafficking. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 119–151. [Google Scholar] [CrossRef]
- Dovey, C.M.; Diep, J.; Clarke, B.P.; Hale, A.T.; McNamara, D.E.; Guo, H.; Brown, N.W., Jr.; Cao, J.Y.; Grace, C.R.; Gough, P.J.; et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 2018, 70, 936–948 e937. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [Green Version]
- Kester, M.; Bassler, J.; Fox, T.E.; Carter, C.J.; Davidson, J.A.; Parette, M.R. Preclinical development of a C6-ceramide NanoLiposome, a novel sphingolipid therapeutic. Biol. Chem. 2015, 396, 737–747. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, X.; Zhou, Y.; Wang, H. C2-ceramide induces cell death and protective autophagy in head and neck squamous cell carcinoma cells. Int. J. Mol. Sci. 2014, 15, 3336–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitatani, K.; Usui, T.; Sriraman, S.K.; Toyoshima, M.; Ishibashi, M.; Shigeta, S.; Nagase, S.; Sakamoto, M.; Ogiso, H.; Okazaki, T.; et al. Ceramide limits phosphatidylinositol-3-kinase C2beta-controlled cell motility in ovarian cancer: Potential of ceramide as a metastasis-suppressor lipid. Oncogene 2016, 35, 2801–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnjuk, K.; Stockl, J.B.; Frohlich, T.; Arnold, G.J.; Behr, R.; Berg, U.; Berg, D.; Kunz, L.; Bishop, C.; Xu, J.; et al. Necroptosis in primate luteolysis: A role for ceramide. Cell Death Discov. 2019, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumura, K.; Justice, M.J.; Schweitzer, K.S.; Krishnan, S.; Bronova, I.; Berdyshev, E.V.; Hubbard, W.C.; Pewzner-Jung, Y.; Futerman, A.H.; Choi, A.M.K.; et al. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J. 2018, 32, 1880–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, L.J.; Alahari, S.; Tagliaferro, A.; Post, M.; Caniggia, I. Augmented trophoblast cell death in preeclampsia can proceed via ceramide-mediated necroptosis. Cell Death Dis. 2017, 8, e2590. [Google Scholar] [CrossRef] [PubMed]
- Malinina, L.; Malakhova, M.L.; Teplov, A.; Brown, R.E.; Patel, D.J. Structural basis for glycosphingolipid transfer specificity. Nature 2004, 430, 1048–1053. [Google Scholar] [CrossRef] [Green Version]
- Yamaji, T.; Hanada, K. Sphingolipid metabolism and interorganellar transport: Localization of sphingolipid enzymes and lipid transfer proteins. Traffic 2015, 16, 101–122. [Google Scholar] [CrossRef]
- Mishra, S.K.; Stephenson, D.J.; Chalfant, C.E.; Brown, R.E. Upregulation of human glycolipid transfer protein (GLTP) induces necroptosis in colon carcinoma cells. BBA-Mol. Cell Biol. Lipids 2019, 1864, 158–167. [Google Scholar] [CrossRef]
- Pacheco, F.J.; Almaguel, F.G.; Evans, W.; Rios-Colon, L.; Filippov, V.; Leoh, L.S.; Rook-Arena, E.; Mediavilla-Varela, M.; De Leon, M.; Casiano, C.A. Docosahexanoic acid antagonizes TNF-alpha-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflamm. Res. 2014, 63, 859–871. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Matsuda, M.; Yaegashi, N.; Nabe, T.; Kitatani, K. Regulation of Necroptosis by Phospholipids and Sphingolipids. Cells 2020, 9, 627. https://doi.org/10.3390/cells9030627
Zhang X, Matsuda M, Yaegashi N, Nabe T, Kitatani K. Regulation of Necroptosis by Phospholipids and Sphingolipids. Cells. 2020; 9(3):627. https://doi.org/10.3390/cells9030627
Chicago/Turabian StyleZhang, Xuewei, Masaya Matsuda, Nobuo Yaegashi, Takeshi Nabe, and Kazuyuki Kitatani. 2020. "Regulation of Necroptosis by Phospholipids and Sphingolipids" Cells 9, no. 3: 627. https://doi.org/10.3390/cells9030627
APA StyleZhang, X., Matsuda, M., Yaegashi, N., Nabe, T., & Kitatani, K. (2020). Regulation of Necroptosis by Phospholipids and Sphingolipids. Cells, 9(3), 627. https://doi.org/10.3390/cells9030627