The Peroxisome Proliferator-Activated Receptor α- Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells
2.3. Mycobacterial Strains and Culture
2.4. Mycobacterial Infection
2.5. Antibodies and Reagents
2.6. GEM Treatment
2.7. Immunopathology of Mycobacterium-Infected Mice
2.8. RNA Extraction, Semiquantitative Real-Time PCR, and Quantitative Real-Time PCR
2.9. Enzyme-Linked Immunosorbent Assay
2.10. Immunoblotting
2.11. Lentiviral shRNA Transduction
2.12. Immunofluorescence
2.13. Minimum Inhibitory Concentration (MIC) Determination Using Resazurin Microtiter Assay (REMA)
2.14. Statistical Analysis
3. Results
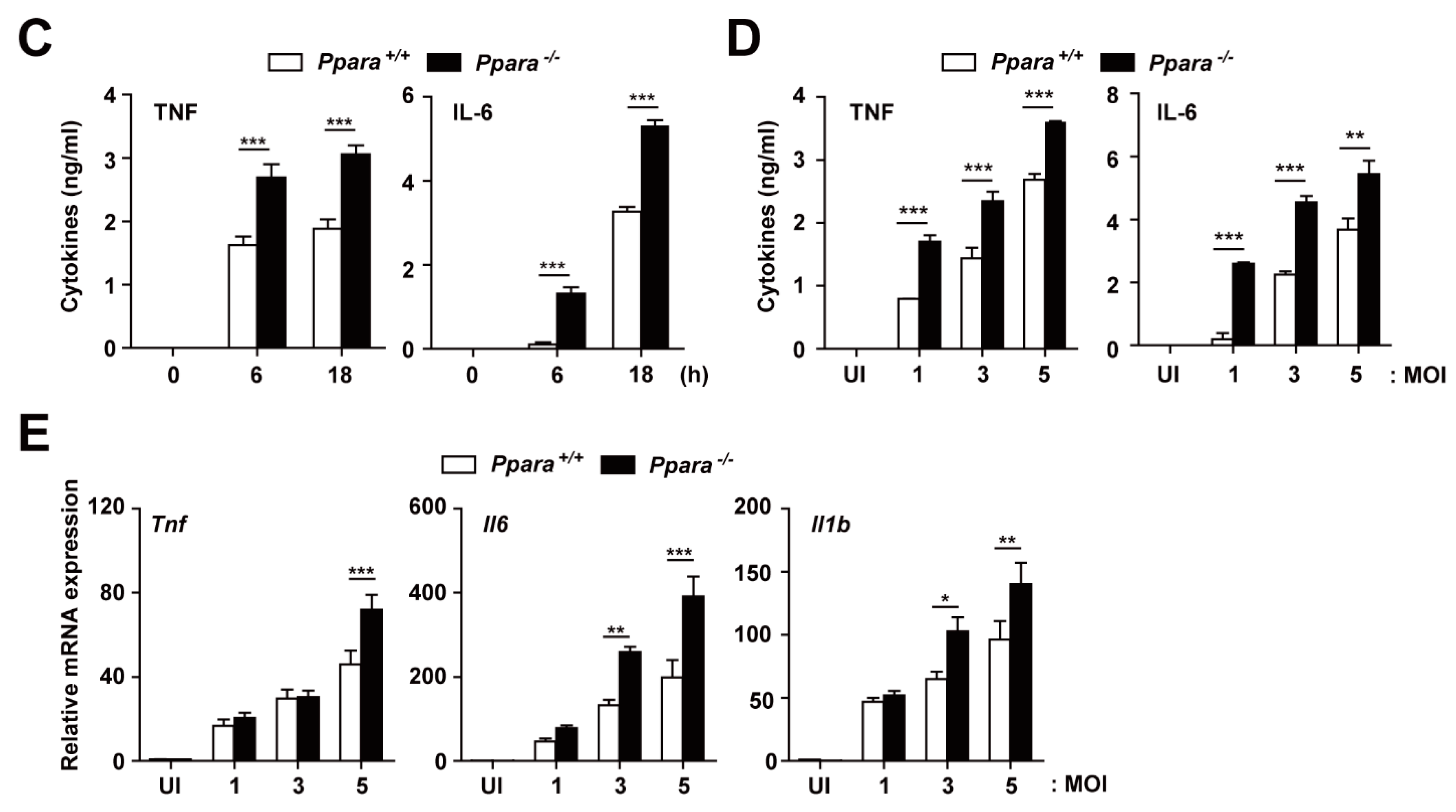
3.1. Ppara-/- Mice Exhibited an Upregulated Proinflammatory but Defective Antimicrobial Response During Mabc Infection Compared with Ppara+/+ Mice
3.2. PPARα Deficiency Increased the Inflammatory Response During Mabc Infection
3.3. PPARα Activation Enhances Macrophage Antimicrobial Responses Againt Mabc Infection
3.4. PPARα Activation Regulates the Inflammatory Response to Mabc Infection
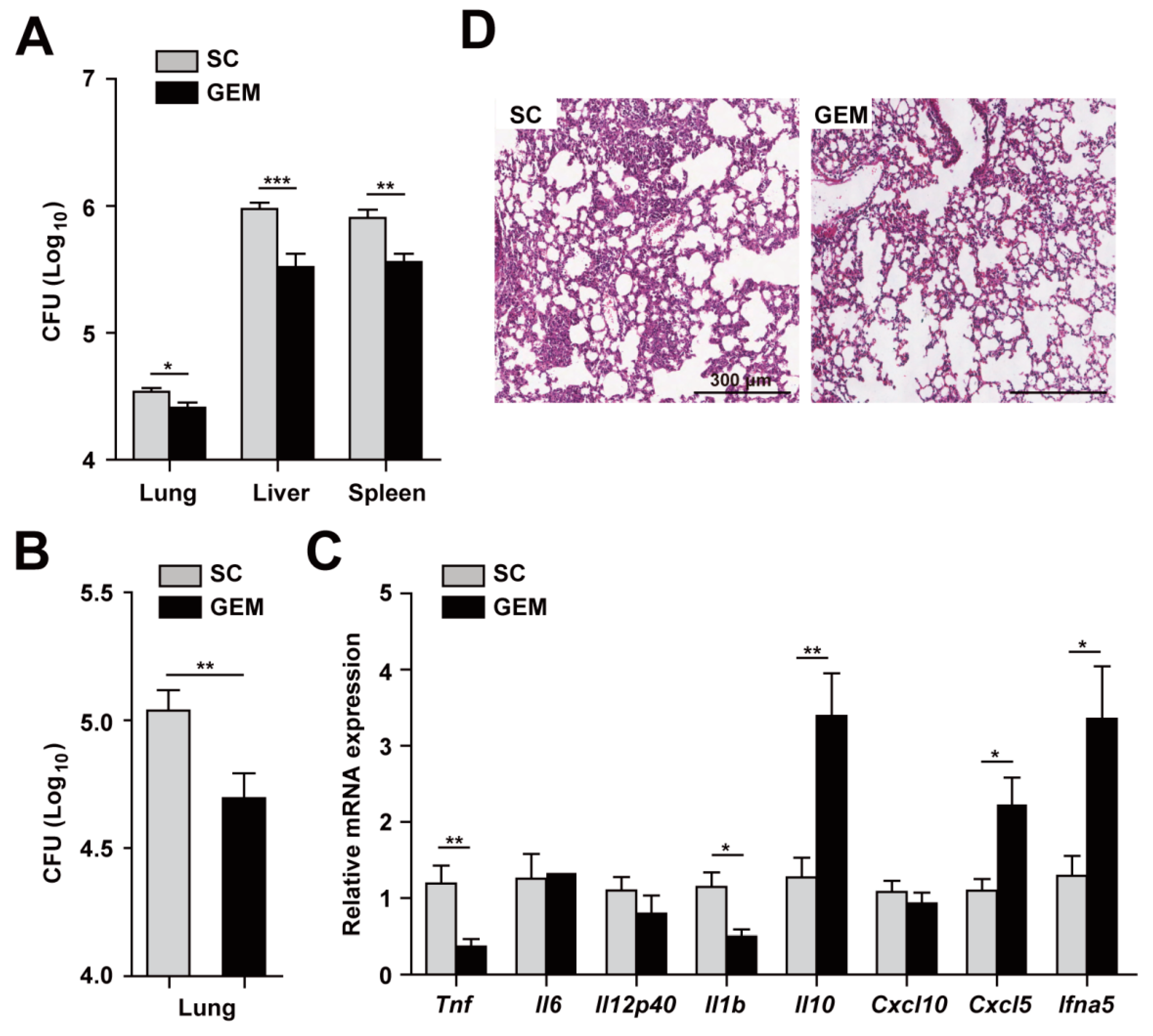
3.5. PPARα Activation Upregulates Antimicrobial Responses and Downregulates Pathological Inflammation during Mabc Infection
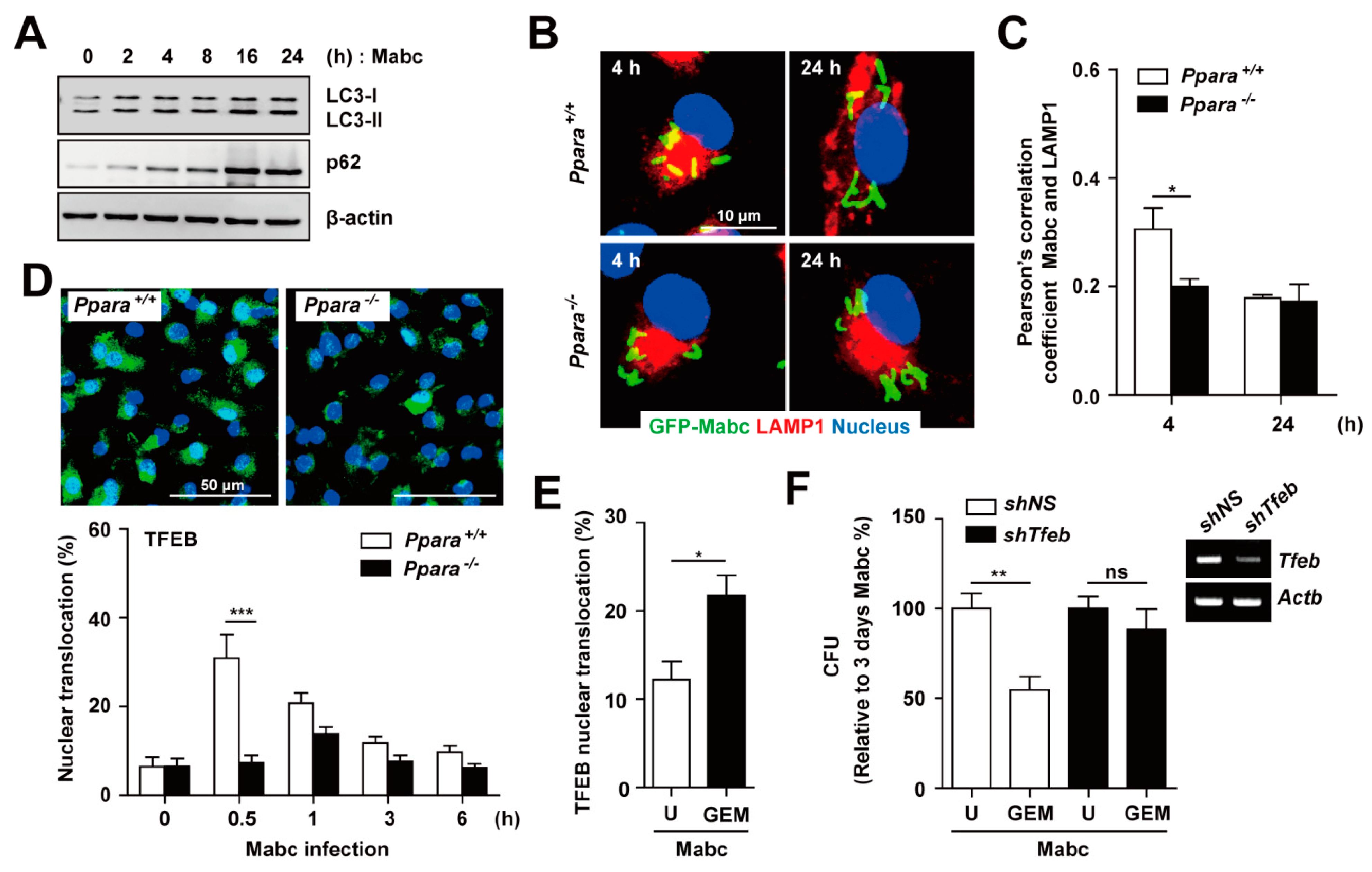
3.6. PPARα is Required for Nuclear Translocation of TFEB in Macrophages during Mabc Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cowman, S.; van Ingen, J.; Griffith, D.E.; Loebinger, M.R. Non-tuberculous mycobacterial pulmonary disease. Eur. Respir. J. 2019, 54, 1900250. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.O.; Lee, K.; Choi, H.K.; Ha, S.; Lee, S.M.; Seo, G.H. Incidence, comorbidities, and treatment patterns of nontuberculous mycobacterial infection in South Korea. Medicine 2019, 98, e17869. [Google Scholar] [CrossRef] [PubMed]
- Swenson, C.; Zerbe, C.S.; Fennelly, K. Host variability in NTM disease: Implications for research needs. Front. Microbiol. 2018, 9, 2901. [Google Scholar] [CrossRef]
- Rivero-Lezcano, O.M.; Gonzalez-Cortes, C.; Mirsaeidi, M. The unexplained increase of nontuberculous mycobacteriosis. Int. J. Mycobacteriol. 2019, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lopeman, R.C.; Harrison, J.; Desai, M.; Cox, J.A.G. Mycobacterium abscessus: Environmental bacterium turned clinical nightmare. Microorganisms 2019, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Pontis, S.; Ribeiro, A.; Sasso, O.; Piomelli, D. Macrophage-derived lipid agonists of PPAR-alpha as intrinsic controllers of inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 7–14. [Google Scholar] [CrossRef]
- Moran, E.P.; Ma, J.X. Therapeutic effects of PPAR alpha on neuronal death and microvascular impairment. PPAR Res. 2015, 2015, 595426. [Google Scholar] [CrossRef]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef]
- Hamblin, M.; Chang, L.; Fan, Y.; Zhang, J.; Chen, Y.E. PPARs and the cardiovascular system. Antioxid. Redox Signal. 2009, 11, 1415–1452. [Google Scholar] [CrossRef]
- Yu, X.H.; Zheng, X.L.; Tang, C.K. Peroxisome proliferator-activated receptor alpha in lipid metabolism and atherosclerosis. Adv. Clin. Chem. 2015, 71, 171–203. [Google Scholar]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Guo, S.; Liu, G. Impairment of fatty acid oxidation in alveolar epithelial cells mediates acute lung injury. Am. J. Respir. Cell Mol. Biol. 2019, 60, 167–178. [Google Scholar] [CrossRef]
- Iwaki, T.; Bennion, B.G.; Stenson, E.K.; Lynn, J.C.; Otinga, C.; Djukovic, D.; Raftery, D.; Fei, L.; Wong, H.R.; Liles, W.C.; et al. PPARalpha contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 2019, 7, e14078. [Google Scholar] [CrossRef]
- Liang, N.; Damdimopoulos, A.; Goni, S.; Huang, Z.; Vedin, L.L.; Jakobsson, T.; Giudici, M.; Ahmed, O.; Pedrelli, M.; Barilla, S.; et al. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARalpha. Nat. Commun. 2019, 10, 1684. [Google Scholar] [CrossRef]
- Gopal, R.; Mendy, A.; Marinelli, M.A.; Richwalls, L.J.; Seger, P.J.; Patel, S.; McHugh, K.J.; Rich, H.E.; Grousd, J.A.; Forno, E.; et al. Peroxisome proliferator-activated Receptor gamma (PPAR) suppresses inflammation and bacterial clearance during influenza-bacterial super-infection. Viruses 2019, 11, 505. [Google Scholar] [CrossRef] [PubMed]
- Leopold Wager, C.M.; Arnett, E.; Schlesinger, L.S. Mycobacterium tuberculosis and macrophage nuclear receptors: What we do and don’t know. Tuberculosis 2019, 116S, S98–S106. [Google Scholar] [CrossRef]
- Leopold Wager, C.M.; Arnett, E.; Schlesinger, L.S. Macrophage nuclear receptors: Emerging key players in infectious diseases. PLoS Pathog. 2019, 15, e1007585. [Google Scholar] [CrossRef] [PubMed]
- Paumelle, R.; Haas, J.T.; Hennuyer, N.; Bauge, E.; Deleye, Y.; Mesotten, D.; Langouche, L.; Vanhoutte, J.; Cudejko, C.; Wouters, K.; et al. Hepatic PPARalpha is critical in the metabolic adaptation to sepsis. J. Hepatol. 2019, 70, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Gugliandolo, E.; Fusco, R.; Ginestra, G.; D’Amico, R.; Bisignano, C.; Mandalari, G.; Cuzzocrea, S.; Di Paola, R. Involvement of TLR4 and PPAR-alpha receptors in host response and NLRP3 inflammasome activation, against pulmonary infection with Pseudomonas Aeruginosa. Shock 2019, 51, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.; Habib, N.; Guzzardi, M.A.; Kitsberg, D.; Bomze, D.; Ezra, E.; Uygun, B.E.; Uygun, K.; Trippler, M.; Schlaak, J.F.; et al. Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection. Nat. Chem. Biol. 2016, 12, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.J.; Jeon, K.; Lee, N.Y.; Kim, B.J.; Kook, Y.H.; Lee, S.H.; Park, Y.K.; Kim, C.K.; Shin, S.J.; Huitt, G.A.; et al. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am. J. Respir. Crit. Care Med. 2011, 183, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Shin, D.M.; Kim, K.H.; Lee, Z.W.; Lee, C.H.; Park, S.G.; Bae, Y.S.; Jo, E.K. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J. Immunol. 2009, 182, 3696–3705. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Yang, C.S.; Jin, H.S.; Kim, K.K.; Lee, Z.W.; Lee, S.H.; Kim, J.M.; Jo, E.K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Andreu, N.; Fletcher, T.; Krishnan, N.; Wiles, S.; Robertson, B.D. Rapid measurement of antituberculosis drug activity in vitro and in macrophages using bioluminescence. J. Antimicrob. Chemother. 2012, 67, 404–414. [Google Scholar] [CrossRef]
- Kim, T.S.; Choe, J.H.; Kim, Y.J.; Yang, C.S.; Kwon, H.J.; Jeong, J.; Kim, G.; Park, D.E.; Jo, E.K.; Cho, Y.L.; et al. Activity of LCB01-0371, a novel oxazolidinone, against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2017, 61, e02752-16. [Google Scholar] [CrossRef] [PubMed]
- Caverly, L.J.; Caceres, S.M.; Fratelli, C.; Happoldt, C.; Kidwell, K.M.; Malcolm, K.C.; Nick, J.A.; Nichols, D.P. Mycobacterium abscessus morphotype comparison in a murine model. PLoS ONE 2015, 10, e0117657. [Google Scholar] [CrossRef]
- Bernut, A.; Dupont, C.; Ogryzko, N.V.; Neyret, A.; Herrmann, J.L.; Floto, R.A.; Renshaw, S.A.; Kremer, L. CFTR protects against Mycobacterium abscessus infection by fine-tuning host oxidative defenses. Cell Rep. 2019, 26, 1828–1840. [Google Scholar] [CrossRef]
- Pahan, K.; Jana, M.; Liu, X.; Taylor, B.S.; Wood, C.; Fischer, S.M. Gemfibrozil, a lipid-lowering drug, inhibits the induction of nitric-oxide synthase in human astrocytes. J. Biol. Chem. 2002, 277, 45984–45991. [Google Scholar] [CrossRef]
- Kiesewetter, B.; Dolak, W.; Mayerhoefer, M.E.; Simonitsch-Klupp, I.; Raderer, M. Successful clarithromycin monotherapy in a patient with primary follicular lymphoma of the duodenum. Case Rep. Oncol. 2018, 11, 239–245. [Google Scholar] [CrossRef]
- Xu, H.E.; Stanley, T.B.; Montana, V.G.; Lambert, M.H.; Shearer, B.G.; Cobb, J.E.; McKee, D.D.; Galardi, C.M.; Plunket, K.D.; Nolte, R.T.; et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002, 415, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Kim, B.J.; Kook, Y.H.; Kim, B.J. Phagosome escape of rough Mycobacterium abscessus strains in murine macrophage via phagosomal rupture can lead to type I interferon production and their cell-to-cell spread. Front. Immunol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.L.; Viljoen, A.; Bah, A.; Simeone, R.; Bernut, A.; Laencina, L.; Deramaudt, T.; Rottman, M.; Gaillard, J.L.; Majlessi, L.; et al. The distinct fate of smooth and rough Mycobacterium abscessus variants inside macrophages. Open Biol. 2016, 6, 160185. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Singh, N.; Kansal, P.; Ahmad, Z.; Baid, N.; Kushwaha, H.; Khatri, N.; Kumar, A. Antimycobacterial effect of IFNG (interferon gamma)-induced autophagy depends on HMOX1 (heme oxygenase 1)-mediated increase in intracellular calcium levels and modulation of PPP3/calcineurin-TFEB (transcription factor EB) axis. Autophagy 2018, 14, 972–991. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.D.; Bai, X.; Kartalija, M.; Orme, I.M.; Ordway, D.J. Host immune response to rapidly growing mycobacteria, an emerging cause of chronic lung disease. Am. J. Respir. Cell Mol. Biol. 2010, 43, 387–393. [Google Scholar] [CrossRef]
- Koh, W.J.; Stout, J.E.; Yew, W.W. Advances in the management of pulmonary disease due to Mycobacterium abscessus complex. Int. J. Tuberc. Lung Dis. 2014, 18, 1141–1148. [Google Scholar] [CrossRef]
- Kasperbauer, S.H.; De Groote, M.A. The treatment of rapidly growing mycobacterial infections. Clin. Chest Med. 2015, 36, 67–78. [Google Scholar] [CrossRef]
- Holland, S.M. Host defense against nontuberculous mycobacterial infections. Semin. Respir. Infect. 1996, 11, 217–230. [Google Scholar]
- Honda, J.R.; Alper, S.; Bai, X.; Chan, E.D. Acquired and genetic host susceptibility factors and microbial pathogenic factors that predispose to nontuberculous mycobacterial infections. Curr. Opin. Immunol. 2018, 54, 66–73. [Google Scholar] [CrossRef]
- Malcolm, K.C.; Caceres, S.M.; Pohl, K.; Poch, K.R.; Bernut, A.; Kremer, L.; Bratton, D.L.; Herrmann, J.L.; Nick, J.A. Neutrophil killing of Mycobacterium abscessus by intra- and extracellular mechanisms. PLoS ONE 2018, 13, e0196120. [Google Scholar] [CrossRef] [PubMed]
- Bernut, A.; Nguyen-Chi, M.; Halloum, I.; Herrmann, J.L.; Lutfalla, G.; Kremer, L. Mycobacterium abscessus-induced granuloma formation is strictly dependent on TNF signaling and neutrophil trafficking. PLoS Pathog. 2016, 12, e1005986. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L.; Baxter, R.; Liu, L.; Varley, C.D.; Curtis, J.R.; Baddley, J.W.; McFarland, B.; Austin, D.; Radcliffe, L.; Suhler, E.; et al. Mycobacterial diseases and antitumour necrosis factor therapy in USA. Ann. Rheum. Dis. 2013, 72, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L.; Chang, E.; Yamashita, S.; Iademarco, M.F.; LoBue, P.A. Nontuberculous mycobacteria infections and anti-tumor necrosis factor-alpha therapy. Emerg. Infect. Dis. 2009, 15, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, K.C.; Nichols, E.M.; Caceres, S.M.; Kret, J.E.; Martiniano, S.L.; Sagel, S.D.; Chan, E.D.; Caverly, L.; Solomon, G.M.; Reynolds, P.; et al. Mycobacterium abscessus induces a limited pattern of neutrophil activation that promotes pathogen survival. PLoS ONE 2013, 8, e57402. [Google Scholar] [CrossRef]
- Gopalakrishnan, A.; Dietzold, J.; Verma, S.; Bhagavathula, M.; Salgame, P. Toll-like receptor 2 prevents neutrophil-driven immunopathology during infection with Mycobacterium tuberculosis by curtailing CXCL5 production. Infect. Immun. 2019, 87, e00760-18. [Google Scholar] [CrossRef]
- Kim, T.S.; Jin, Y.B.; Kim, Y.S.; Kim, S.; Kim, J.K.; Lee, H.M.; Suh, H.W.; Choe, J.H.; Kim, Y.J.; Koo, B.S.; et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy 2019, 15, 1356–1375. [Google Scholar] [CrossRef]
- Nouailles, G.; Dorhoi, A.; Koch, M.; Zerrahn, J.; Weiner, J., 3rd; Fae, K.C.; Arrey, F.; Kuhlmann, S.; Bandermann, S.; Loewe, D.; et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Investig. 2014, 124, 1268–1282. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Iqbal, A.J. Epigenetic control of macrophage polarisation and soluble mediator gene expression during inflammation. Mediat. Inflamm. 2016, 2016, 6591703. [Google Scholar] [CrossRef]
- Lim, A.; Allison, C.; Tan, D.B.; Oliver, B.; Price, P.; Waterer, G. Immunological markers of lung disease due to non-tuberculous mycobacteria. Dis. Markers 2010, 29, 103–109. [Google Scholar] [CrossRef][Green Version]
- Bamba, Y.; Moro, H.; Aoki, N.; Koizumi, T.; Ohshima, Y.; Watanabe, S.; Sakagami, T.; Koya, T.; Takada, T.; Kikuchi, T. Multiplex cytokine analysis in Mycobacterium avium complex lung disease: Relationship between CXCL10 and poor prognostic factors. BMC Infect. Dis. 2019, 19, 263. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chauhan, S.; Jain, A.; Ponpuak, M.; Choi, S.W.; Mudd, M.; Peters, R.; Mandell, M.A.; Johansen, T.; Deretic, V. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy 2017, 13, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Bhagyaraj, E.; Nanduri, R.; Ahuja, N.; Gupta, P. NR1D1 ameliorates Mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy 2015, 11, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Reich-Slotky, R.; Kabbash, C.A.; Della-Latta, P.; Blanchard, J.S.; Feinmark, S.J.; Freeman, S.; Kaplan, G.; Shuman, H.A.; Silverstein, S.C. Gemfibrozil inhibits Legionella pneumophila and Mycobacterium tuberculosis enoyl coenzyme A reductases and blocks intracellular growth of these bacteria in macrophages. J. Bacteriol. 2009, 191, 5262–5271. [Google Scholar] [CrossRef]
- Jana, M.; Mondal, S.; Gonzalez, F.J.; Pahan, K. Gemfibrozil, a lipid-lowering drug, increases myelin genes in human oligodendrocytes via peroxisome proliferator-activated receptor-beta. J. Biol. Chem. 2012, 287, 34134–34148. [Google Scholar] [CrossRef]
- Renna, M.; Schaffner, C.; Brown, K.; Shang, S.; Tamayo, M.H.; Hegyi, K.; Grimsey, N.J.; Cusens, D.; Coulter, S.; Cooper, J.; et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J. Clin. Investig. 2011, 121, 3554–3563. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.S.; Kim, J.K.; Hanh, B.T.B.; Kim, S.Y.; Kim, H.J.; Kim, Y.J.; Jeon, S.M.; Park, C.R.; Oh, G.T.; Park, J.-W.; et al. The Peroxisome Proliferator-Activated Receptor α- Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections. Cells 2020, 9, 648. https://doi.org/10.3390/cells9030648
Kim YS, Kim JK, Hanh BTB, Kim SY, Kim HJ, Kim YJ, Jeon SM, Park CR, Oh GT, Park J-W, et al. The Peroxisome Proliferator-Activated Receptor α- Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections. Cells. 2020; 9(3):648. https://doi.org/10.3390/cells9030648
Chicago/Turabian StyleKim, Yi Sak, Jin Kyung Kim, Bui Thi Bich Hanh, Soo Yeon Kim, Hyeon Ji Kim, Young Jae Kim, Sang Min Jeon, Cho Rong Park, Goo Taeg Oh, June-Woo Park, and et al. 2020. "The Peroxisome Proliferator-Activated Receptor α- Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections" Cells 9, no. 3: 648. https://doi.org/10.3390/cells9030648
APA StyleKim, Y. S., Kim, J. K., Hanh, B. T. B., Kim, S. Y., Kim, H. J., Kim, Y. J., Jeon, S. M., Park, C. R., Oh, G. T., Park, J.-W., Kim, J.-M., Jang, J., & Jo, E.-K. (2020). The Peroxisome Proliferator-Activated Receptor α- Agonist Gemfibrozil Promotes Defense Against Mycobacterium abscessus Infections. Cells, 9(3), 648. https://doi.org/10.3390/cells9030648