Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells


 and
and
Abstract
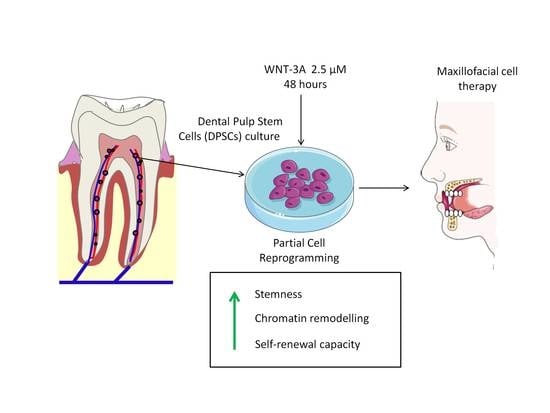
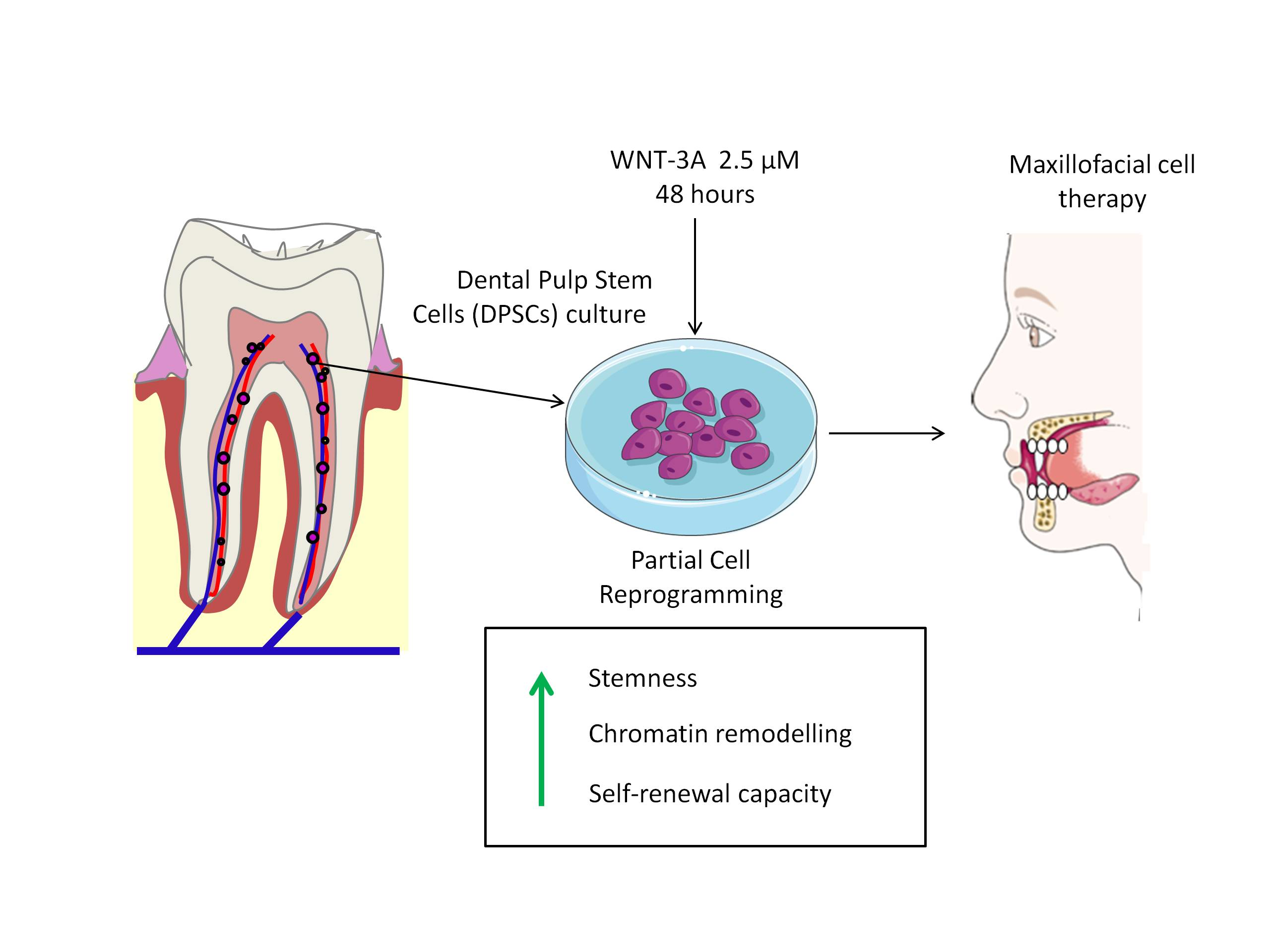
:
1. Introduction
2. Materials and Methods
2.1. DPSC Culture
2.2. ESC Culture
2.3. Notch and Wnt Pathway Pharmacological Modulation
2.4. Osteogenic Differentiation of DPSCs
2.5. Adipogenic Differentiation of DPSCs
2.6. RNA Extraction, Reverse Transcription and Quantitative Real-Time PCR (qPCR)
2.7. Protein Extraction
2.8. Western Blot (WB)
2.9. DNA Extraction
2.10. Quantification of DNA Methylation by Mass Spectroscopy (MS)
2.11. Cell Cycle Phase Determination
2.12. Statistical Analyses
3. Results
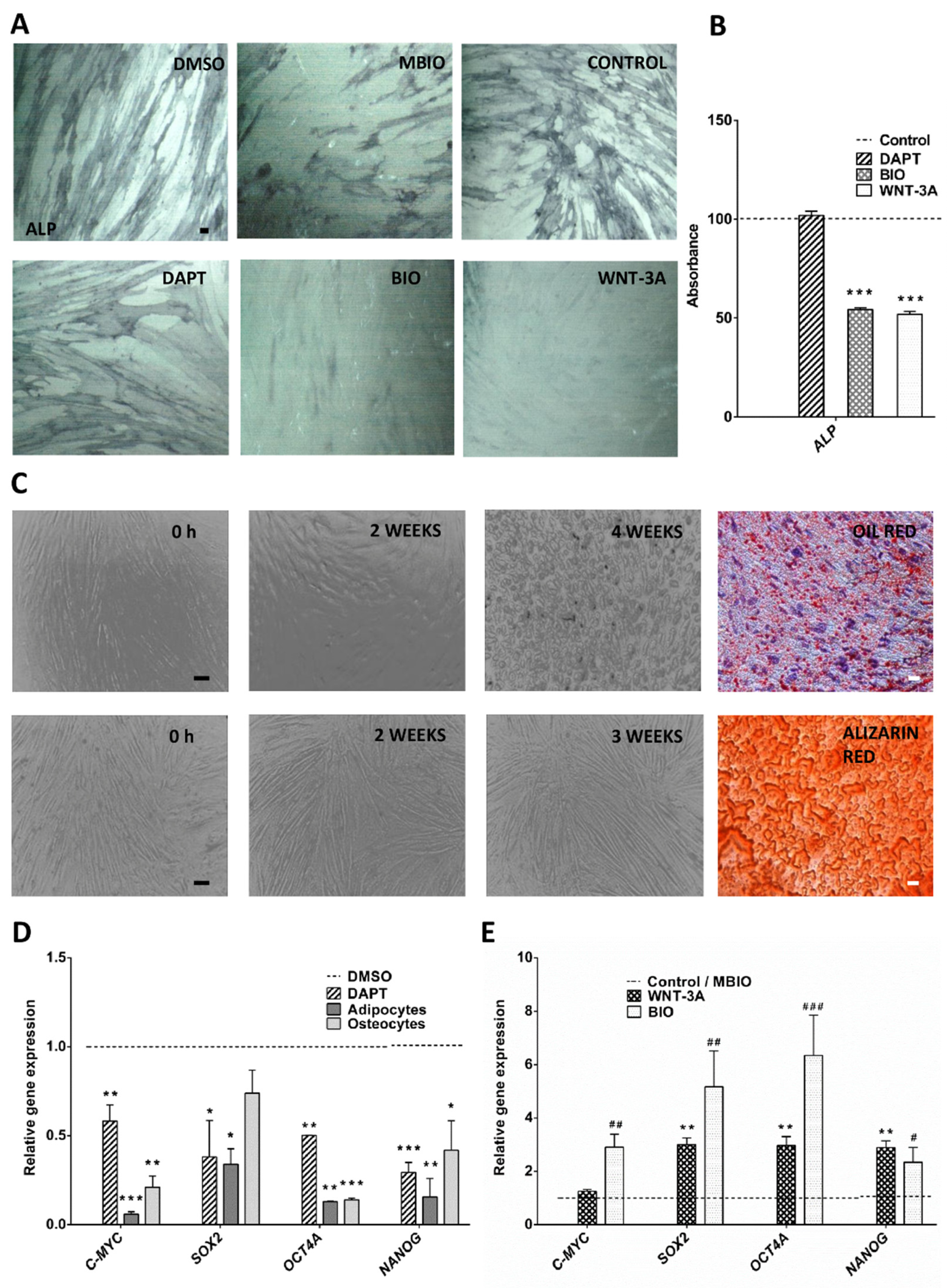
3.1. Wnt Activity Reverses Osteogenic Cell Differentiation and Increases the Expression of Core Pluripotency Factors in DPSCs
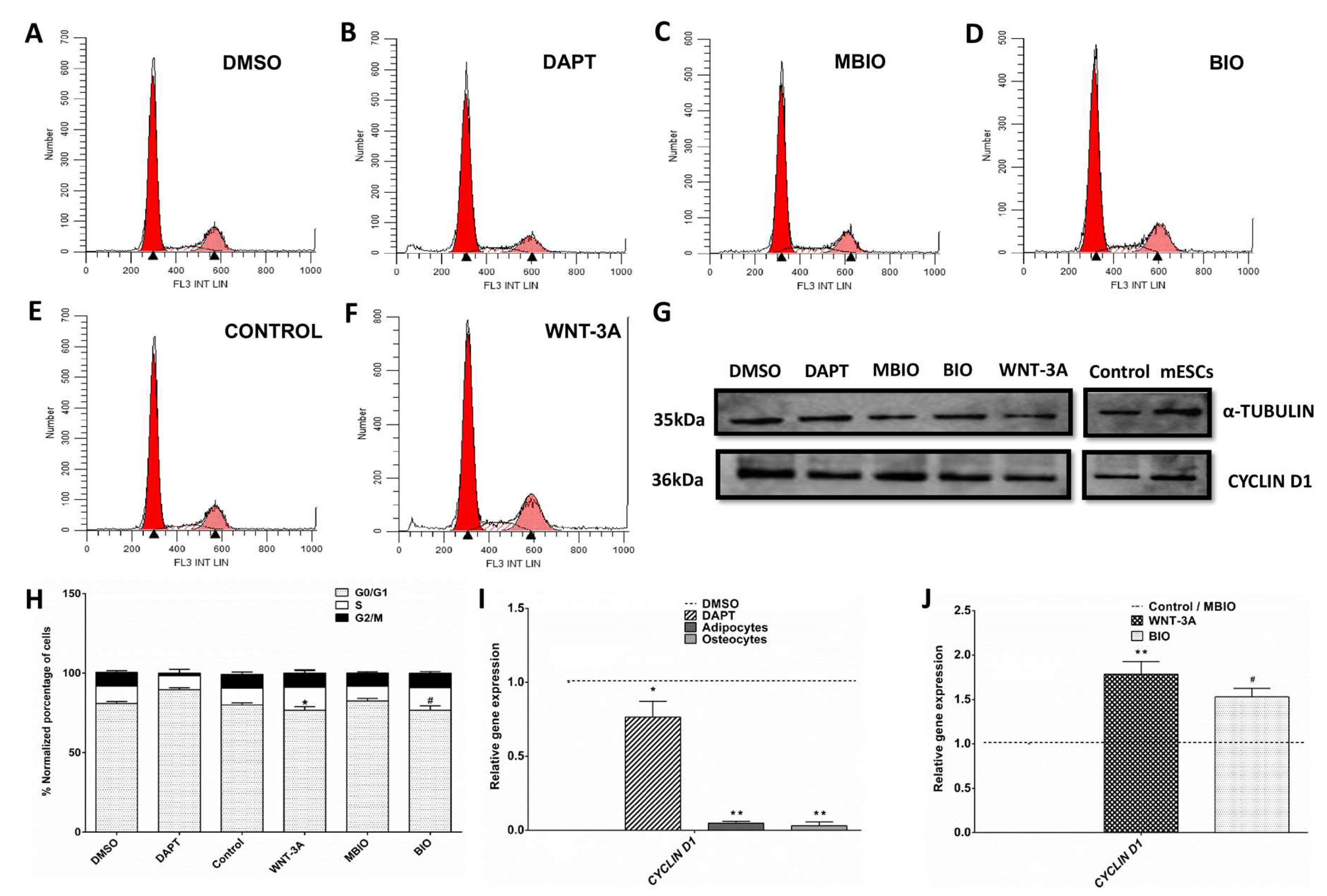
3.2. Notch and Wnt/β-Catenin Signaling Regulates the Cell Cycle in DPSCs
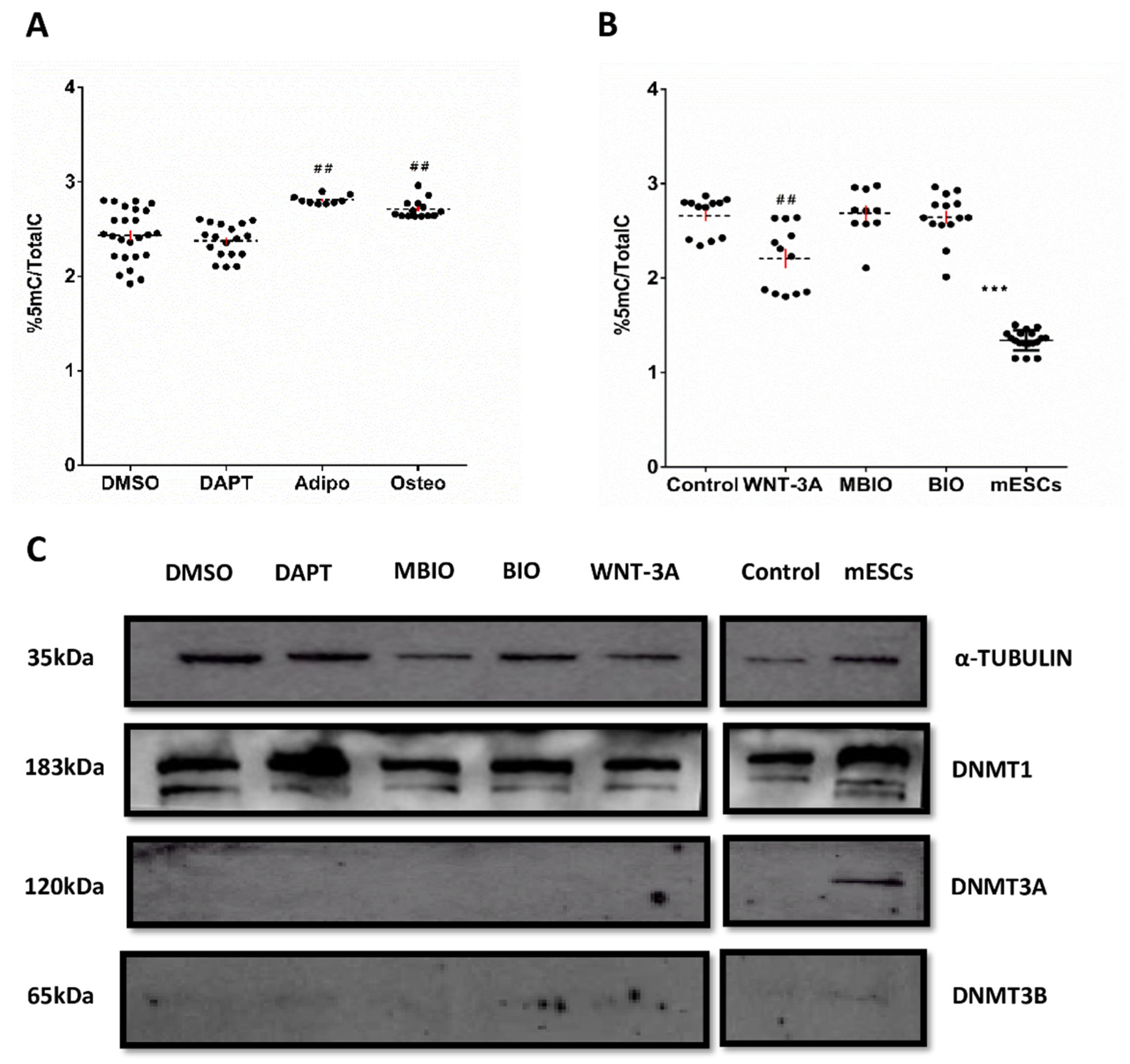
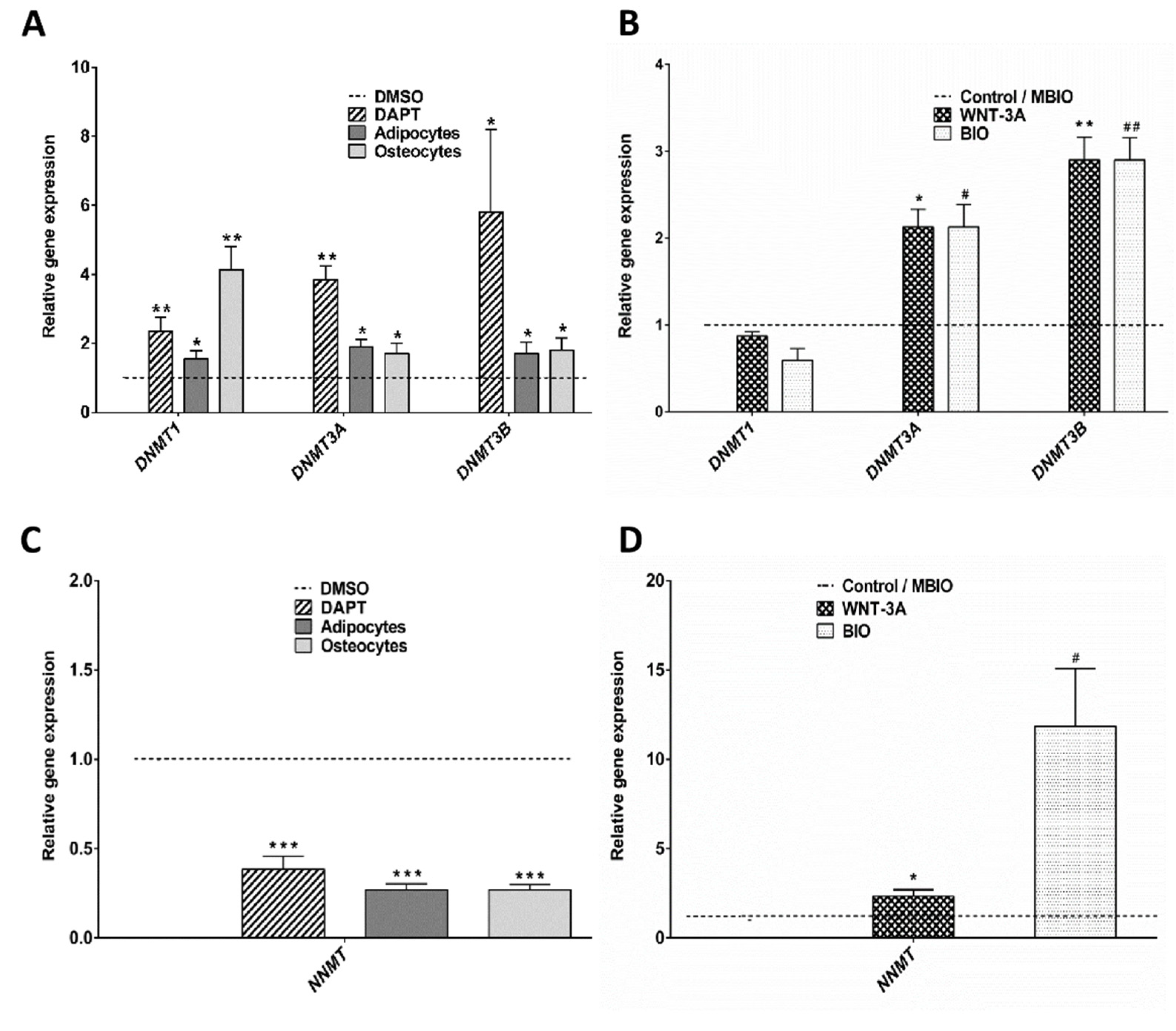
3.3. WNT-3A Leads to a DNA Hypo-Methylation State in DPSCs
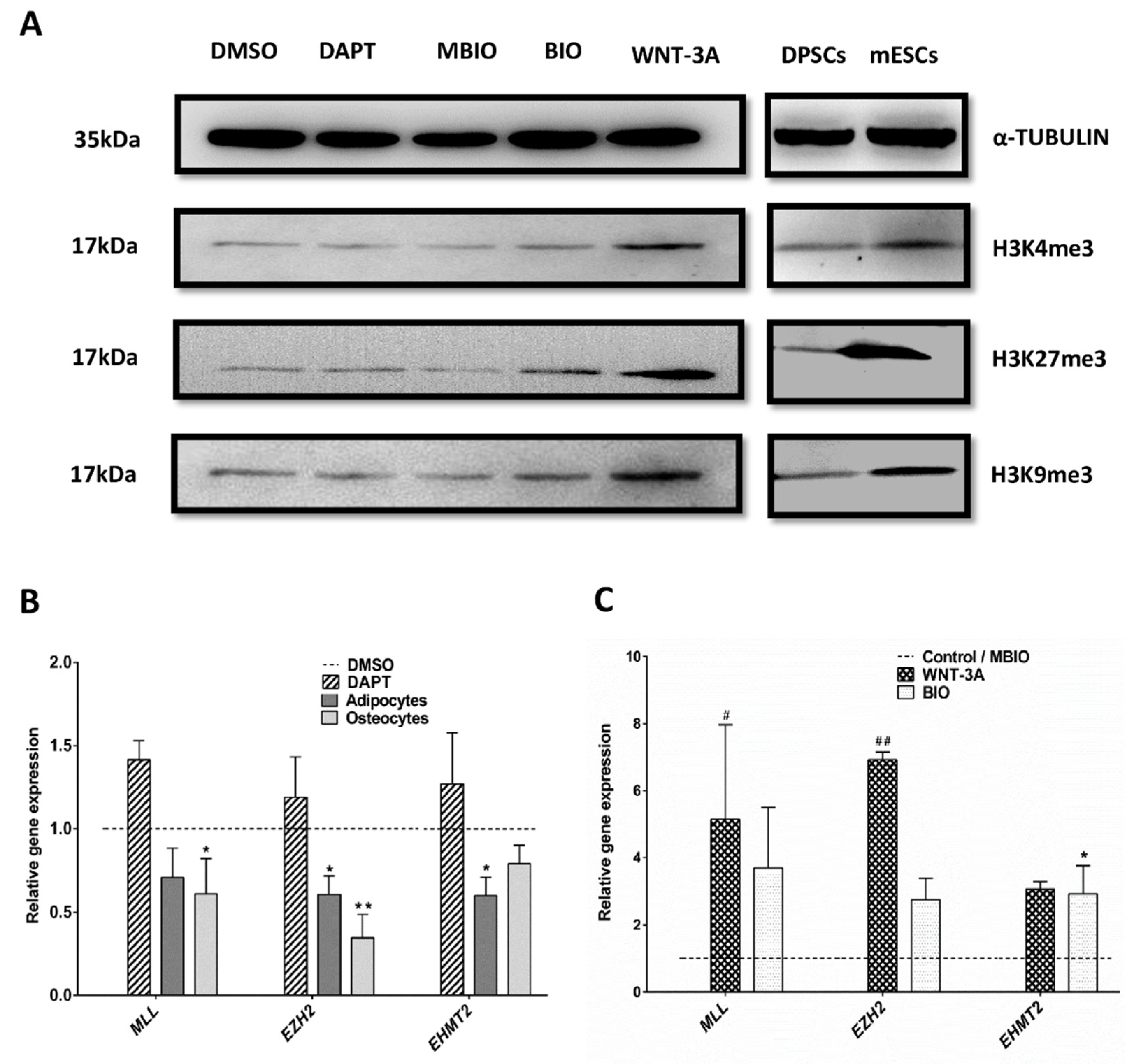
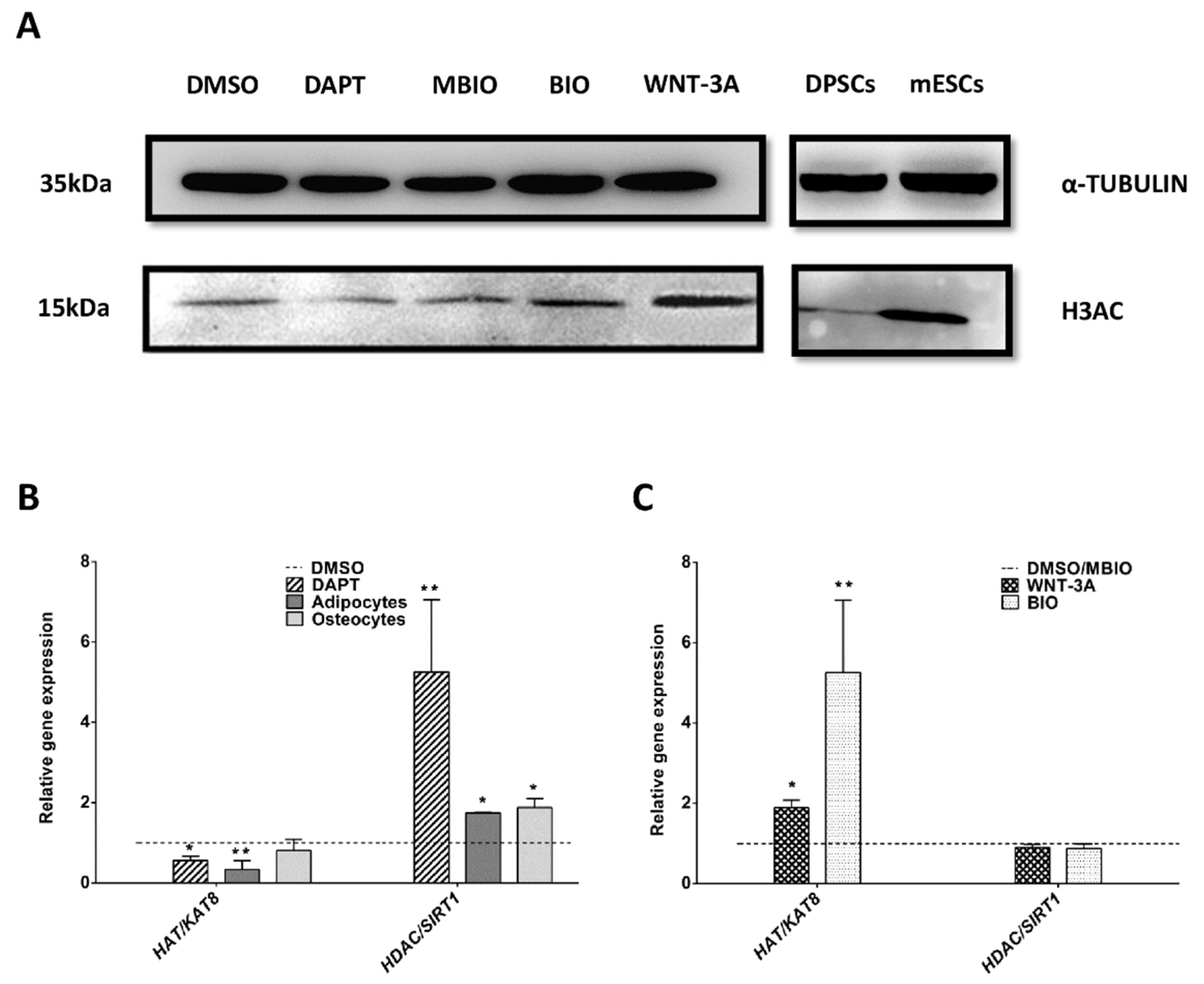
3.4. Wnt Activation Increases Histone Acetylation in DPSCs
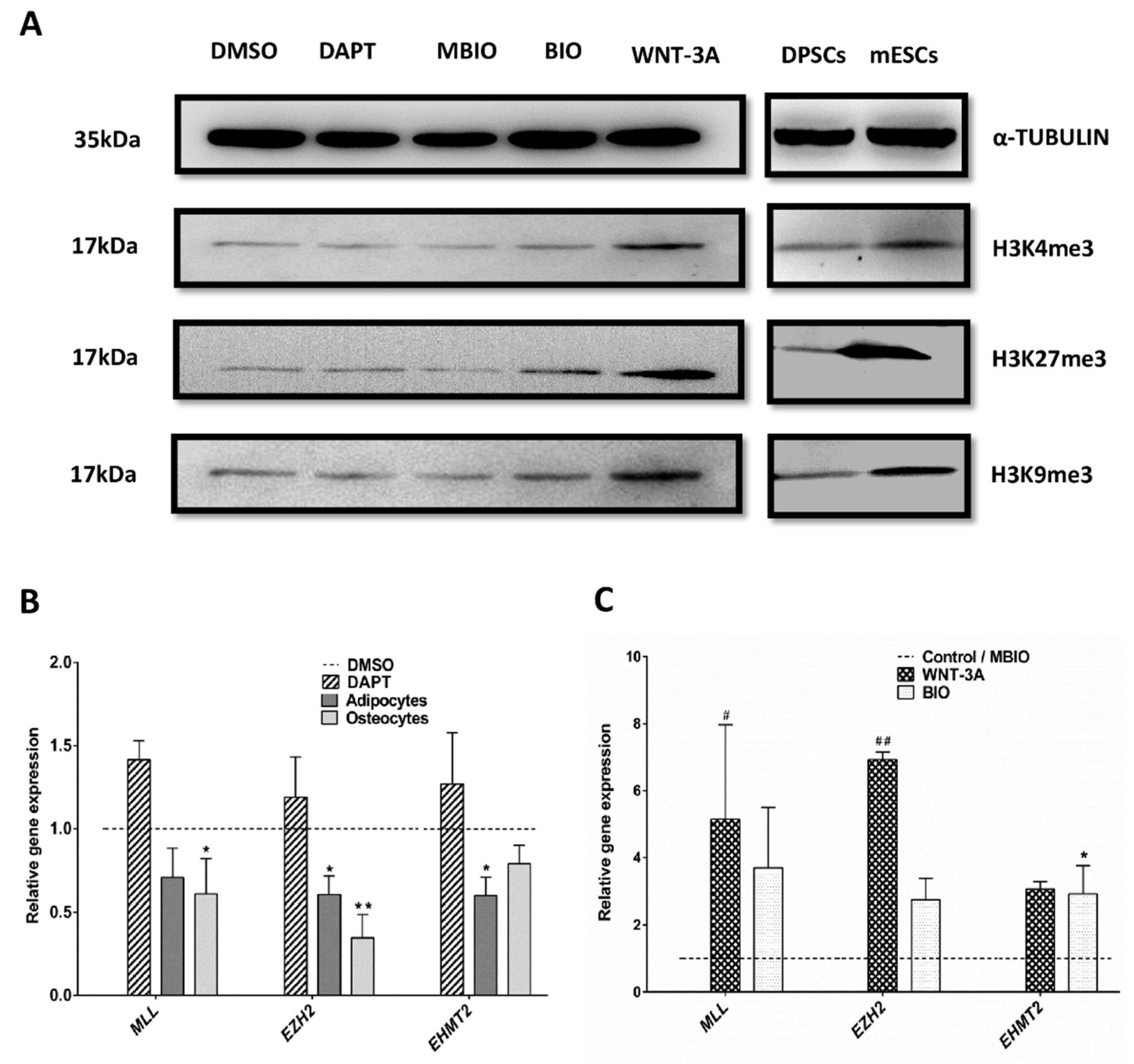
3.5. Wnt Activation Modifies the Histone H3 Methylation Pattern in DPSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaukua, N.; Shahidi, M.K.; Konstantinidou, C.; Dyachuk, V.; Kaucka, M.; Furlan, A.; An, Z.; Wang, L.; Hultman, I.; Ahrlund-Richter, L.; et al. Glial origin of mesenchymal stem cells in a tooth model system. Nature 2014, 513, 551–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janebodin, K.; Horst, O.V.; Ieronimakis, N.; Balasundaram, G.; Reesukumal, K.; Pratumvinit, B.; Reyes, M. Isolation and characterization of neural crest-derived stem cells from dental pulp of neonatal mice. PLoS ONE 2011, 6, e27526. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, F.; Sun, Y.; Jiang, B.; Zhang, W.; Yang, J.; Xu, G.T.; Liang, A.; Liu, S. Concise reviews: Characteristics and potential applications of human dental tissue-derived mesenchymal stem cells. Stem Cells (Dayton, Ohio) 2015, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Aurrekoetxea, M.; Garcia-Gallastegui, P.; Irastorza, I.; Luzuriaga, J.; Uribe-Etxebarria, V.; Unda, F.; Ibarretxe, G. Dental pulp stem cells as a multifaceted tool for bioengineering and the regeneration of craniomaxillofacial tissues. Front. Physiol. 2015, 6, 289. [Google Scholar] [CrossRef] [Green Version]
- Gronthos, S.; Brahim, J.; Li, W.; Fisher, L.W.; Cherman, N.; Boyde, A.; DenBesten, P.; Robey, P.G.; Shi, S. Stem cell properties of human dental pulp stem cells. J. Dent. Res. 2002, 81, 531–535. [Google Scholar] [CrossRef]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [Green Version]
- Ibarretxe, G.; Crende, O.; Aurrekoetxea, M.; Garcia-Murga, V.; Etxaniz, J.; Unda, F. Neural crest stem cells from dental tissues: A new hope for dental and neural regeneration. Stem Cells Int. 2012, 2012, 103503. [Google Scholar] [CrossRef] [Green Version]
- Atari, M.; Barajas, M.; Hernandez-Alfaro, F.; Gil, C.; Fabregat, M.; Ferres Padro, E.; Giner, L.; Casals, N. Isolation of pluripotent stem cells from human third molar dental pulp. Histol. Histopathol. 2011, 26, 1057–1070. [Google Scholar] [CrossRef]
- Atari, M.; Gil-Recio, C.; Fabregat, M.; Garcia-Fernandez, D.; Barajas, M.; Carrasco, M.A.; Jung, H.S.; Alfaro, F.H.; Casals, N.; Prosper, F.; et al. Dental pulp of the third molar: A new source of pluripotent-like stem cells. J. Cell Sci. 2012, 125, 3343–3356. [Google Scholar] [CrossRef] [Green Version]
- Rosa, V.; Dubey, N.; Islam, I.; Min, K.S.; Nor, J.E. Pluripotency of Stem Cells from Human Exfoliated Deciduous Teeth for Tissue Engineering. Stem Cells Int. 2016, 2016, 5957806. [Google Scholar] [CrossRef] [Green Version]
- Ferro, F.; Spelat, R.; D’Aurizio, F.; Puppato, E.; Pandolfi, M.; Beltrami, A.P.; Cesselli, D.; Falini, G.; Beltrami, C.A.; Curcio, F. Dental pulp stem cells differentiation reveals new insights in Oct4A dynamics. PLoS ONE 2012, 7, e41774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkis, I.; Kerkis, A.; Dozortsev, D.; Stukart-Parsons, G.C.; Gomes Massironi, S.M.; Pereira, L.V.; Caplan, A.I.; Cerruti, H.F. Isolation and characterization of a population of immature dental pulp stem cells expressing OCT-4 and other embryonic stem cell markers. Cells Tissues Organs 2006, 184, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Uribe-Etxebarria, V.; Luzuriaga, J.; Garcia-Gallastegui, P.; Agliano, A.; Unda, F.; Ibarretxe, G. Notch/Wnt cross-signalling regulates stemness of dental pulp stem cells through expression of neural crest and core pluripotency factors. Eur. Cell Mater. 2017, 34, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.; Tomlinson, S.R. The transcriptional foundation of pluripotency. Development 2009, 136, 2311–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Hackett, J.A.; Surani, M.A. Regulatory principles of pluripotency: From the ground state up. Cell Stem Cell 2014, 15, 416–430. [Google Scholar] [CrossRef] [Green Version]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef] [Green Version]
- Pierdomenico, L.; Bonsi, L.; Calvitti, M.; Rondelli, D.; Arpinati, M.; Chirumbolo, G.; Becchetti, E.; Marchionni, C.; Alviano, F.; Fossati, V.; et al. Multipotent mesenchymal stem cells with immunosuppressive activity can be easily isolated from dental pulp. Transplantation 2005, 80, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.; Urraca, N.; Skobowiat, C.; Hope, K.A.; Miravalle, L.; Chamberlin, R.; Donaldson, M.; Seagroves, T.N.; Reiter, L.T. Assessment of the Tumorigenic Potential of Spontaneously Immortalized and hTERT-Immortalized Cultured Dental Pulp Stem Cells. Stem Cells Transl. Med. 2015, 4, 905–912. [Google Scholar] [CrossRef]
- Kellner, M.; Steindorff, M.M.; Strempel, J.F.; Winkel, A.; Kuhnel, M.P.; Stiesch, M. Differences of isolated dental stem cells dependent on donor age and consequences for autologous tooth replacement. Arch. Oral. Biol. 2014, 59, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhou, J.; Xu, C.T.; Zhang, J.; Jin, Y.J.; Sun, G.L. Derivation and growth characteristics of dental pulp stem cells from patients of different ages. Mol. Med. Rep. 2015, 12, 5127–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thekkeparambil Chandrabose, S.; Sriram, S.; Subramanian, S.; Cheng, S.; Ong, W.K.; Rozen, S.; Kasim, N.H.A.; Sugii, S. Amenable epigenetic traits of dental pulp stem cells underlie high capability of xeno-free episomal reprogramming. Stem Cell Res. Ther. 2018, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Diomede, F.; Zini, N.; Pizzicannella, J.; Merciaro, I.; Pizzicannella, G.; D’Orazio, M.; Piattelli, A.; Trubiani, O. 5-Aza Exposure Improves Reprogramming Process Through Embryoid Body Formation in Human Gingival Stem Cells. Front. Genet. 2018, 9, 419. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- David, L.; Samavarchi-Tehrani, P.; Golipour, A.; Wrana, J.L. Looking into the black box: Insights into the mechanisms of somatic cell reprogramming. Genes 2011, 2, 81–106. [Google Scholar] [CrossRef]
- Wang, A.; Du, Y.; He, Q.; Zhou, C. A quantitative system for discriminating induced pluripotent stem cells, embryonic stem cells and somatic cells. PLoS ONE 2013, 8, e56095. [Google Scholar] [CrossRef] [Green Version]
- Nashun, B.; Hill, P.W.; Hajkova, P. Reprogramming of cell fate: Epigenetic memory and the erasure of memories past. EMBO J. 2015, 34, 1296–1308. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattout, A.; Aaronson, Y.; Sailaja, B.S.; Raghu Ram, E.V.; Harikumar, A.; Mallm, J.P.; Sim, K.H.; Nissim-Rafinia, M.; Supper, E.; Singh, P.B.; et al. Heterochromatin Protein 1beta (HP1beta) has distinct functions and distinct nuclear distribution in pluripotent versus differentiated cells. Genome Biol. 2015, 16, 213. [Google Scholar] [CrossRef] [Green Version]
- Vastenhouw, N.L.; Schier, A.F. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012, 24, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Harikumar, A.; Meshorer, E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep. 2015, 16, 1609–1619. [Google Scholar] [CrossRef] [Green Version]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Borghese, L.; Dolezalova, D.; Opitz, T.; Haupt, S.; Leinhaas, A.; Steinfarz, B.; Koch, P.; Edenhofer, F.; Hampl, A.; Brustle, O. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 2010, 28, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Bardin, A.J. Sending the right signal: Notch and stem cells. Biochim. et Biophys. Acta 2013, 1830, 2307–2322. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Chang, J.; Wang, C.Y. Wnt/beta-catenin inhibits dental pulp stem cell differentiation. J. Dent. Res. 2008, 87, 126–130. [Google Scholar] [CrossRef]
- Yiew, N.K.; Chatterjee, T.K.; Tang, Y.L.; Pellenberg, R.; Stansfield, B.K.; Bagi, Z.; Fulton, D.J.; Stepp, D.W.; Chen, W.; Patel, V.; et al. Novel role for Wnt inhibitor APCDD1 in adipocyte differentiation: Implications for diet-induced obesity. J. Biol. Chem. 2017, 292, 6312–6324. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Yoon, K.; Dang, L.; Tokunaga, A.; Gaiano, N. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 2007, 449, 351–355. [Google Scholar] [CrossRef]
- Huang, G.T.; Gronthos, S.; Shi, S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: Their biology and role in regenerative medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [CrossRef]
- Vasanthan, P.; Govindasamy, V.; Gnanasegaran, N.; Kunasekaran, W.; Musa, S.; Abu Kasim, N.H. Differential expression of basal microRNAs’ patterns in human dental pulp stem cells. J. Cell. Mol. Med. 2015, 19, 566–580. [Google Scholar] [CrossRef] [Green Version]
- Luzuriaga, J.; Pineda, J.R.; Irastorza, I.; Uribe-Etxebarria, V.; Garcia-Gallastegui, P.; Encinas, J.M.; Chamero, P.; Unda, F.; Ibarretxe, G. BDNF and NT3 Reprogram Human Ectomesenchymal Dental Pulp Stem Cells to Neurogenic and Gliogenic Neural Crest Progenitors Cultured in Serum-Free Medium. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 52, 1361–1380. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisciotta, A.; Riccio, M.; Carnevale, G.; Beretti, F.; Gibellini, L.; Maraldi, T.; Cavallini, G.M.; Ferrari, A.; Bruzzesi, G.; De Pol, A. Human serum promotes osteogenic differentiation of human dental pulp stem cells in vitro and in vivo. PLoS ONE 2012, 7, e50542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; He, H.; Tang, C.; Zhang, G.; Li, Y.; Wang, R.; Shi, J.; Jin, Y. Differentiation potential of STRO-1+ dental pulp stem cells changes during cell passaging. BMC Cell Biol. 2010, 11, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenbach, F.; Handschel, J. Effects of dexamethasone, ascorbic acid and beta-glycerophosphate on the osteogenic differentiation of stem cells in vitro. Stem Cell Res. Ther. 2013, 4, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, J.; Smith, A. Naive and primed pluripotent states. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat. Cell Biol. 2015, 17, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F. The DNMT3 family of mammalian de novo DNA methyltransferases. Prog. Mol. Biol. Transl. Sci. 2011, 101, 255–285. [Google Scholar] [CrossRef]
- Hanna, J.; Cheng, A.W.; Saha, K.; Kim, J.; Lengner, C.J.; Soldner, F.; Cassady, J.P.; Muffat, J.; Carey, B.W.; Jaenisch, R. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 2010, 107, 9222–9227. [Google Scholar] [CrossRef] [Green Version]
- Lin, H. S-Adenosylmethionine-dependent alkylation reactions: When are radical reactions used? Bioorg. Chem. 2011, 39, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. TEM 2017, 28, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Griscelli, F.; Feraud, O.; Oudrhiri, N.; Gobbo, E.; Casal, I.; Chomel, J.C.; Bieche, I.; Duvillard, P.; Opolon, P.; Turhan, A.G.; et al. Malignant germ cell-like tumors, expressing Ki-1 antigen (CD30), are revealed during in vivo differentiation of partially reprogrammed human-induced pluripotent stem cells. Am. J. Pathol. 2012, 180, 2084–2096. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Mostoslavsky, G. Experimental approaches for the generation of induced pluripotent stem cells. Stem Cell Res. Ther. 2010, 1, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, C.A.; Christodoulou, C.; Gianotti-Sommer, A.; Shen, S.S.; Sailaja, B.S.; Hezroni, H.; Spira, A.; Meshorer, E.; Kotton, D.N.; Mostoslavsky, G. Residual expression of reprogramming factors affects the transcriptional program and epigenetic signatures of induced pluripotent stem cells. PLoS ONE 2012, 7, e51711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Simandi, Z.; Horvath, A.; Wright, L.C.; Cuaranta-Monroy, I.; De Luca, I.; Karolyi, K.; Sauer, S.; Deleuze, J.F.; Gudas, L.J.; Cowley, S.M.; et al. OCT4 Acts as an Integrator of Pluripotency and Signal-Induced Differentiation. Mol. Cell 2016, 63, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Qin, H.; Qu, C.; Tuan, R.S.; Shi, S.; Huang, G.T. iPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev. 2010, 19, 469–480. [Google Scholar] [CrossRef]
- Li, J.; Chen, B. Oct4 was a novel target of Wnt signaling pathway. Mol. Cell. Biochem. 2012, 362, 233–240. [Google Scholar] [CrossRef]
- Lluis, F.; Pedone, E.; Pepe, S.; Cosma, M.P. Periodic activation of Wnt/beta-catenin signaling enhances somatic cell reprogramming mediated by cell fusion. Cell Stem Cell 2008, 3, 493–507. [Google Scholar] [CrossRef]
- Lowell, S.; Benchoua, A.; Heavey, B.; Smith, A.G. Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol. 2006, 4, e121. [Google Scholar] [CrossRef]
- Dravid, G.; Ye, Z.; Hammond, H.; Chen, G.; Pyle, A.; Donovan, P.; Yu, X.; Cheng, L. Defining the role of Wnt/beta-catenin signaling in the survival, proliferation, and self-renewal of human embryonic stem cells. Stem Cells 2005, 23, 1489–1501. [Google Scholar] [CrossRef]
- Fox, V.; Gokhale, P.J.; Walsh, J.R.; Matin, M.; Jones, M.; Andrews, P.W. Cell-cell signaling through NOTCH regulates human embryonic stem cell proliferation. Stem Cells 2008, 26, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, K.; Nakajima, M.; Kanokoda, M.; Kyba, M.; Dandapat, A.; Tolar, J.; Saito, M.K.; Toyoda, M.; Umezawa, A.; Hara, T. GSK3beta inhibition activates the CDX/HOX pathway and promotes hemogenic endothelial progenitor differentiation from human pluripotent stem cells. Exp. Hematol. 2016, 44, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichida, J.K.; Tcw, J.; Williams, L.A.; Carter, A.C.; Shi, Y.; Moura, M.T.; Ziller, M.; Singh, S.; Amabile, G.; Bock, C.; et al. Notch inhibition allows oncogene-independent generation of iPS cells. Nature Chem. Biol. 2014, 10, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Li, V.C.; Kirschner, M.W. Molecular ties between the cell cycle and differentiation in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9503–9508. [Google Scholar] [CrossRef] [Green Version]
- Ghule, P.N.; Medina, R.; Lengner, C.J.; Mandeville, M.; Qiao, M.; Dominski, Z.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Reprogramming the pluripotent cell cycle: Restoration of an abbreviated G1 phase in human induced pluripotent stem (iPS) cells. J. Cell Physiol. 2011, 226, 1149–1156. [Google Scholar] [CrossRef] [Green Version]
- Coronado, D.; Godet, M.; Bourillot, P.Y.; Tapponnier, Y.; Bernat, A.; Petit, M.; Afanassieff, M.; Markossian, S.; Malashicheva, A.; Iacone, R.; et al. A short G1 phase is an intrinsic determinant of naive embryonic stem cell pluripotency. Stem. Cell Res. 2013, 10, 118–131. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, T.; Sun, W.; Yuan, Z.; Yu, M.; Chen, G.; Guo, W.; Xiao, J.; Tian, W. DNA Demethylation Rescues the Impaired Osteogenic Differentiation Ability of Human Periodontal Ligament Stem Cells in High Glucose. Sci. Rep. 2016, 6, 27447. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Q.; Rao, L.; Yi, B.; Xu, Q. Effect of 5-Aza-2’-deoxycytidine on odontogenic differentiation of human dental pulp cells. J. Endod. 2015, 41, 640–645. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Ning, Y.; Xu, Q. The histone acetyltransferase p300 regulates the expression of pluripotency factors and odontogenic differentiation of human dental pulp cells. PLoS ONE 2014, 9, e102117. [Google Scholar] [CrossRef] [Green Version]
- Klinz, F.J.; Korkmaz, Y.; Bloch, W.; Raab, W.H.; Addicks, K. Histone deacetylases 2 and 9 are coexpressed and nuclear localized in human molar odontoblasts in vivo. Histochem. Cell Biol. 2012, 137, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Park, J.Y.; Choi, H.; Choung, P.H. HDAC inhibitor trichostatin A promotes proliferation and odontoblast differentiation of human dental pulp stem cells. Tissue Eng. Part A 2013, 19, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Paino, F.; Ricci, G.; De Rosa, A.; D’Aquino, R.; Laino, L.; Pirozzi, G.; Tirino, V.; Papaccio, G. Ecto-mesenchymal stem cells from dental pulp are committed to differentiate into active melanocytes. Eur. Cell Mater. 2010, 20, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.; Kim, J.J.; Kim, Y.; Tong, E.; Trammell, B.; Liu, Y.; Shi, S.; Lee, C.R.; Hong, C.; Wang, C.Y.; et al. Alcohol-induced suppression of KDM6B dysregulates the mineralization potential in dental pulp stem cells. Stem Cell Res. 2016, 17, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Shen, Y.; Xue, Z.; Bibikova, M.; April, C.; Liu, Z.; Cheng, L.; Nagy, A.; Pellegrini, M.; Fan, J.B.; et al. A panel of CpG methylation sites distinguishes human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterberger, A.; Andrews, S.D.; Weaver, I.C.; Szyf, M. DNA methyltransferase 1 knockdown activates a replication stress checkpoint. Mol. Cell. Biol. 2006, 26, 7575–7586. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, Y.; Szabo, P.; Szuts, D.; Bacquet, C.; Aranyi, T.; Paldi, A. Rapid turnover of DNA methylation in human cells. Epigenetics 2012, 7, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Ulanovskaya, O.A.; Zuhl, A.M.; Cravatt, B.F. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat. Chem. Biol. 2013, 9, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Vaissiere, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef]
- Uribe-Etxebarria, V.; Agliano, A. Wnt signaling reprograms metabolism in dental pulp stem cells. J. Cell Physiol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [Green Version]
- D’Alessio, A.C.; Weaver, I.C.; Szyf, M. Acetylation-induced transcription is required for active DNA demethylation in methylation-silenced genes. Mol. Cell. Biol. 2007, 27, 7462–7474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuara, V.; Perry, P.; Sauer, S.; Spivakov, M.; Jorgensen, H.F.; John, R.M.; Gouti, M.; Casanova, M.; Warnes, G.; Merkenschlager, M.; et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006, 8, 532–538. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence 5′–3′ | Annealing (ºC) | Amplicon (bp) | |
|---|---|---|---|---|
| β-ACTIN | Upstream | GTTGTCGACGACGAGCG | 58.5 | 93 |
| Downstream | GCACAGAGCCTCGCCTT | 59.7 | ||
| GAPDH | Upstream | CTTTTGCGTCGCCAG | 60.3 | 131 |
| Downstream | TTGATGGCAACAATATCCAC | 60.8 | ||
| DNMT1 | Upstream | CGTAAAGAAGAATTATCCGAGG | 60.5 | 123 |
| Downstream | GTTTTCTAGACGTCCATTCAC | 57.7 | ||
| DNMT3A | Upstream | GAAGAGAAGAATCCCTACAAAG | 57.6 | 136 |
| Downstream | CAATAATCTCCTTGACCTTGG | 60 | ||
| DNMT3B | Upstream | CTTACCTTACCATCGACCTC | 57.7 | 167 |
| Downstream | ATCCTGATACTCTGAACTGTC | 54.7 | ||
| NNMT | Upstream | CTGACTACTCAGACCAGAAC | 53.6 | 113 |
| Downstream | TCTGTTCCCTTCAAGATCAC | 59.3 | ||
| KAT8/HAC | Upstream | GAAATATGAGAAGAGCTACCG | 57.2 | 123 |
| Downstream | ATCTTATGGTCTTTGCCATC | 58 | ||
| SIRT1/HDAC | Upstream | AAGGAAAACTACTTCGCAAC | 57.6 | 89 |
| Downstream | GGAACCATGACACTGAATTATC | 59.7 | ||
| OCT4A | Upstream | CGTGAAGCTGGAGAAGGAGA | 60.7 | 137 |
| Downstream | CATCGGCCTGTGTATATCCC | 60.1 | ||
| CMYC | Upstream | GTCAAGAGGCGAACACACAAC | 59.8 | 162 |
| Downstream | TGGACGGACAGGATGTATGC | 59.8 | ||
| NANOG | Upstream | GTCAAGAAACAGAAGACCAG | 56.4 | 184 |
| Downstream | GCCACCTCTTAGATTTCATTC | 59.2 | ||
| SOX2 | Upstream | ATAATAACAATCATCGGCGG | 61.1 | 90 |
| Downstream | AAAAAGAGAGAGGCAAACTG | 57.8 | ||
| CYCLIN D1 | Upstream | TGAGGCGGTAGTAGGACAGG | 60.4 | 140 |
| Downstream | GACCTTCGTTGCCCTCTGT | 59.6 | ||
| EZH2 | Upstream | CCAACACAAGTCATCC | 60.4 | 91 |
| Downstream | CCATAAAATTCTGCTGTAGGG | 59.6 | ||
| MLL | Upstream | AAAGACTTCTAAGGAGGCAG | 61.1 | 183 |
| Downstream | AACATATAGCAACCAATGCC | 58.7 | ||
| EHMT2 | Upstream | CTGTCAGAGGAGTTAGGTTC | 60.4 | 135 |
| Downstream | ATCCACAGAGTAGGAATCATAG | 59.6 | ||
| PPARγ | Upstream | AAAGAAGCCAACACTAAACC | 57.4 | 78 |
| Downstream | TGGTCATTTCGTTAAAGGC | 60.1 | ||
| LPL | Upstream | ACACAGAGGTAGATATTGGAG | 54.8 | 143 |
| Downstream | CTTTTTCTGAGTCTCTCCTG | 55.7 | ||
| SPARC | Upstream | CTTCAGACTGCCCGGAGA | 61.1 | 90 |
| Downstream | GAAAGAAGATCCAGGCCCTC | 60.2 | ||
| OSTERIX | Upstream | TGAGGAGGAAGTTCACTATG | 53.8 | 200 |
| Downstream | CATTAGTGCTTGTAAAGGGG | 54.0 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uribe-Etxebarria, V.; García-Gallastegui, P.; Pérez-Garrastachu, M.; Casado-Andrés, M.; Irastorza, I.; Unda, F.; Ibarretxe, G.; Subirán, N. Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells. Cells 2020, 9, 652. https://doi.org/10.3390/cells9030652
Uribe-Etxebarria V, García-Gallastegui P, Pérez-Garrastachu M, Casado-Andrés M, Irastorza I, Unda F, Ibarretxe G, Subirán N. Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells. Cells. 2020; 9(3):652. https://doi.org/10.3390/cells9030652
Chicago/Turabian StyleUribe-Etxebarria, Verónica, Patricia García-Gallastegui, Miguel Pérez-Garrastachu, María Casado-Andrés, Igor Irastorza, Fernando Unda, Gaskon Ibarretxe, and Nerea Subirán. 2020. "Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells" Cells 9, no. 3: 652. https://doi.org/10.3390/cells9030652
APA StyleUribe-Etxebarria, V., García-Gallastegui, P., Pérez-Garrastachu, M., Casado-Andrés, M., Irastorza, I., Unda, F., Ibarretxe, G., & Subirán, N. (2020). Wnt-3a Induces Epigenetic Remodeling in Human Dental Pulp Stem Cells. Cells, 9(3), 652. https://doi.org/10.3390/cells9030652