Conducting Polymer Mediated Electrical Stimulation Induces Multilineage Differentiation with Robust Neuronal Fate Determination of Human Induced Pluripotent Stem Cells

 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
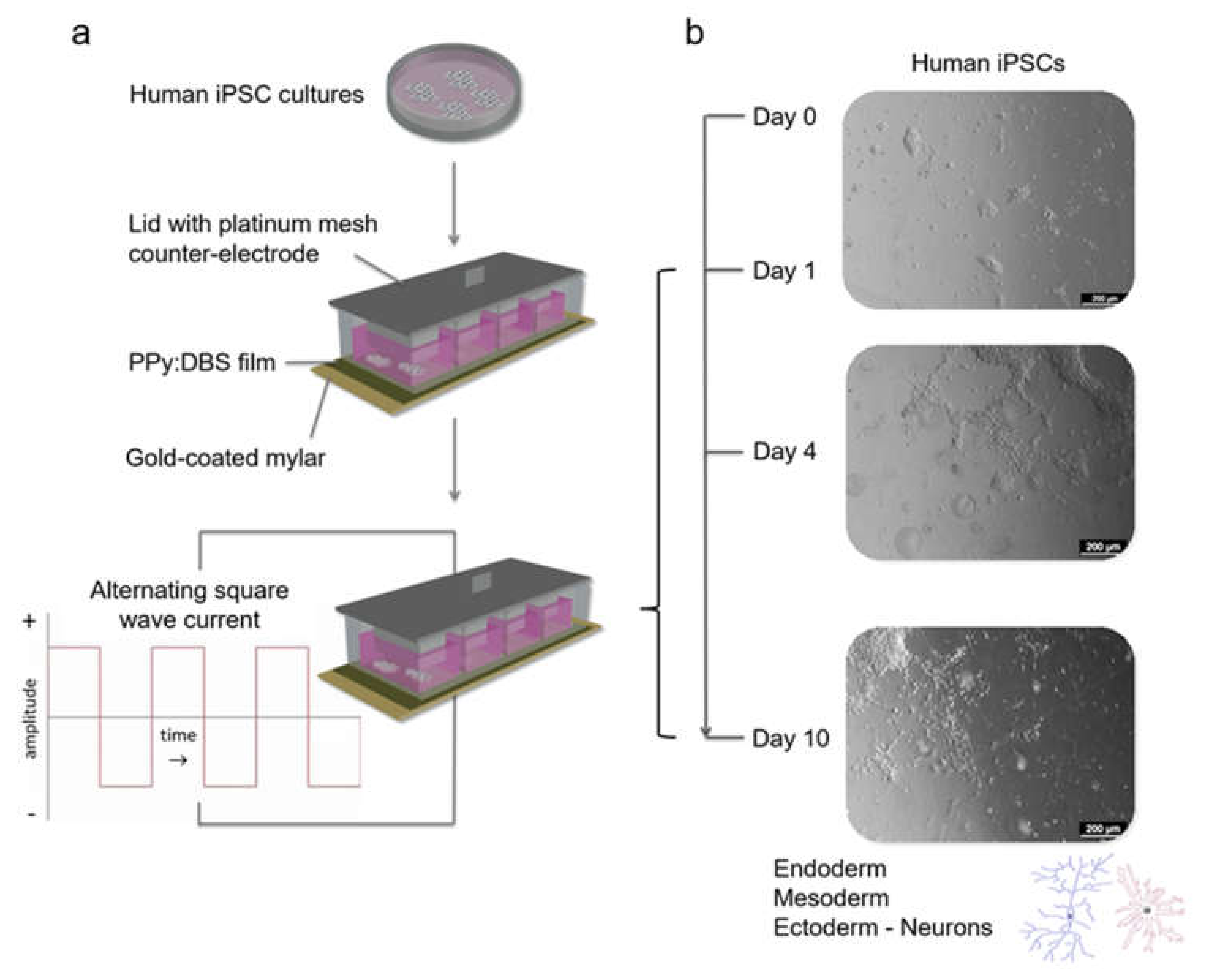
2.1. Preparation of Polymer Films and Stimulation Modules
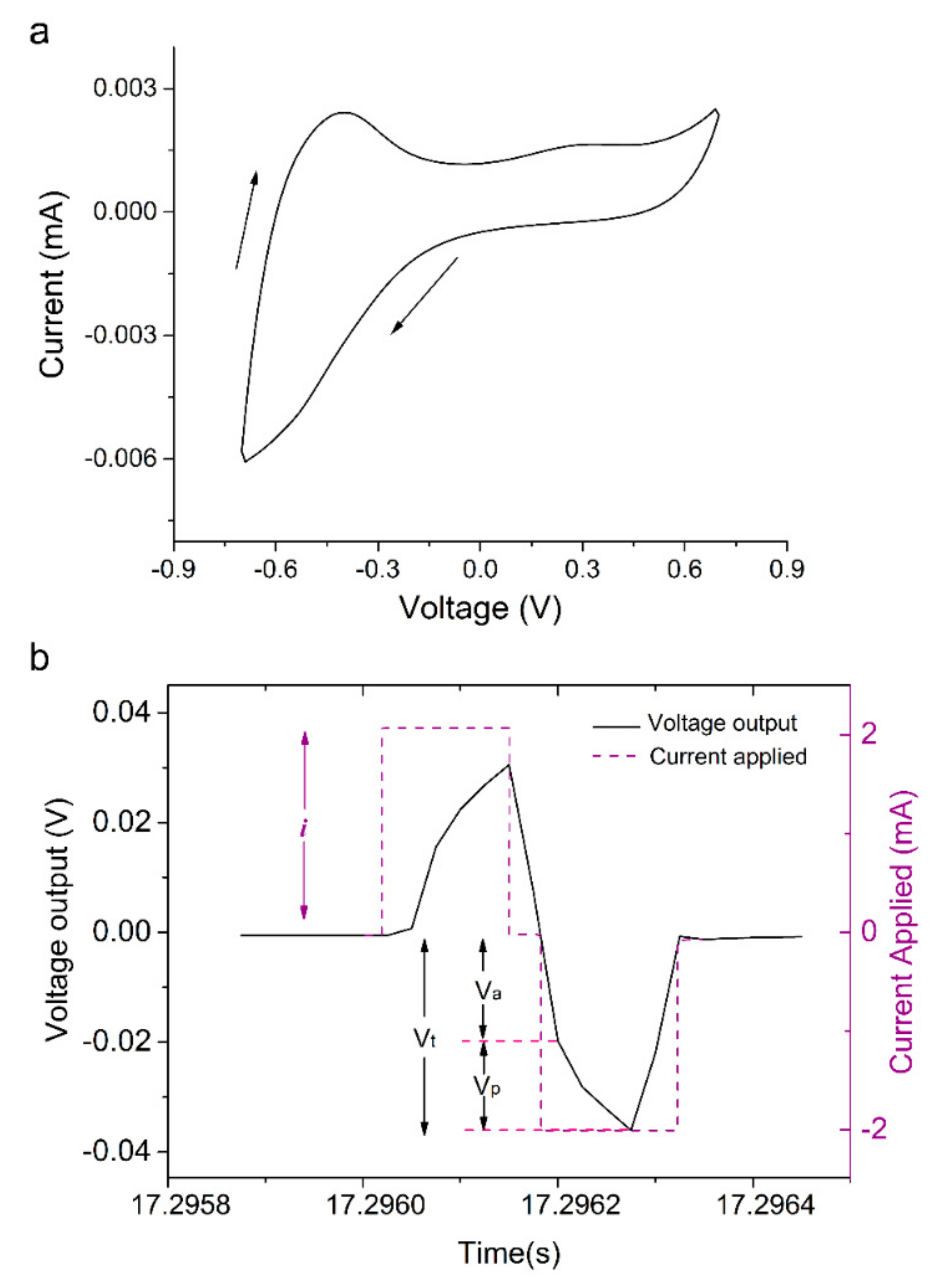
2.2. Cyclic Voltammetry
2.3. Human iPSC Culture
2.4. Conventional Human iPSC Differentiation
2.5. Electrical Stimulation
2.6. Immunocytochemistry and Analysis
2.7. Flow Cytometry
2.8. Real-Time Quantitative PCR (RT-qPCR)
2.9. Statistical Analysis
3. Results
3.1. Polymerisation, Electrochemical Activity and Stability of PPy:DBS Films
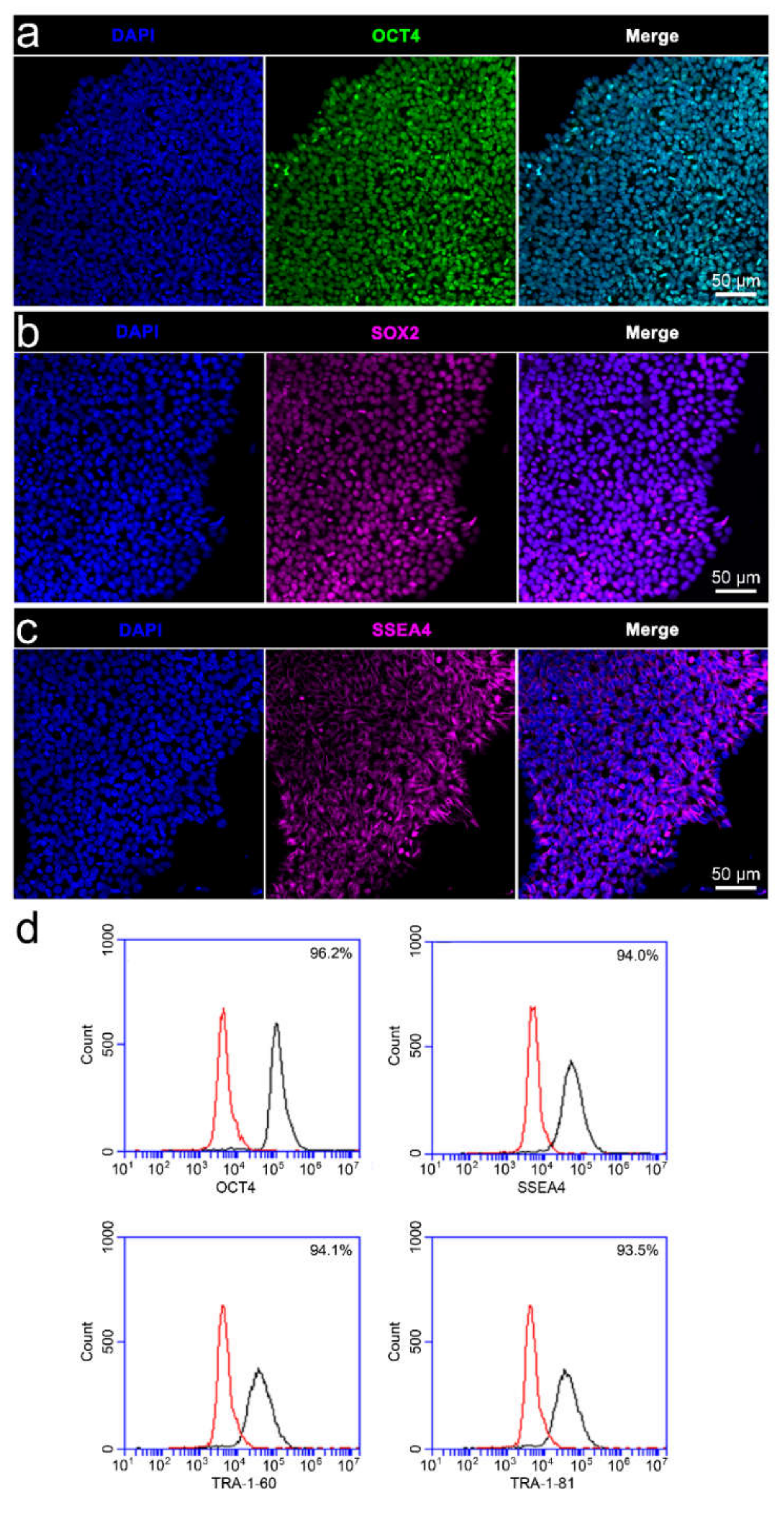
3.2. Characterisation of iPSC Pluripotency and Differentiability
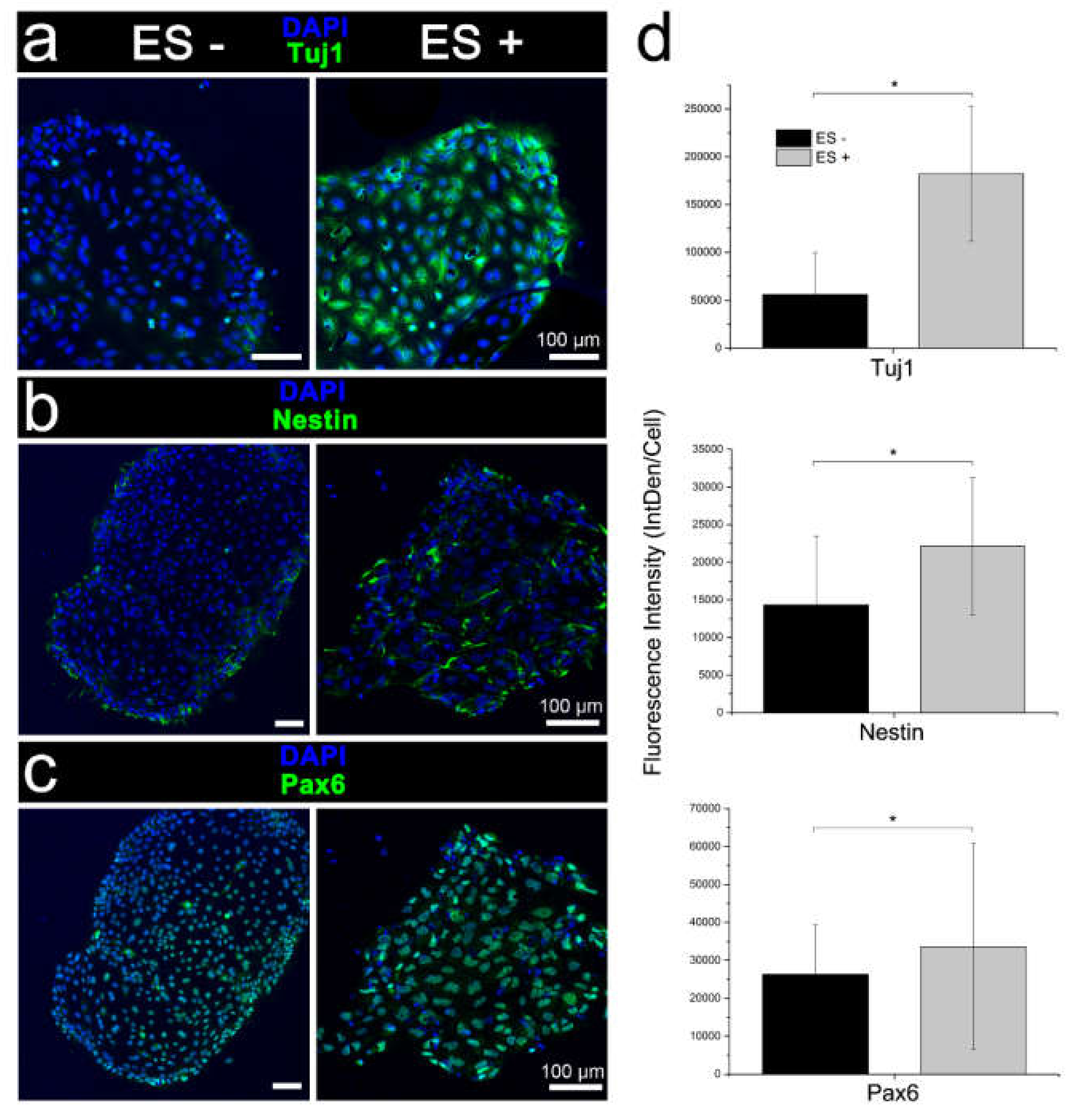
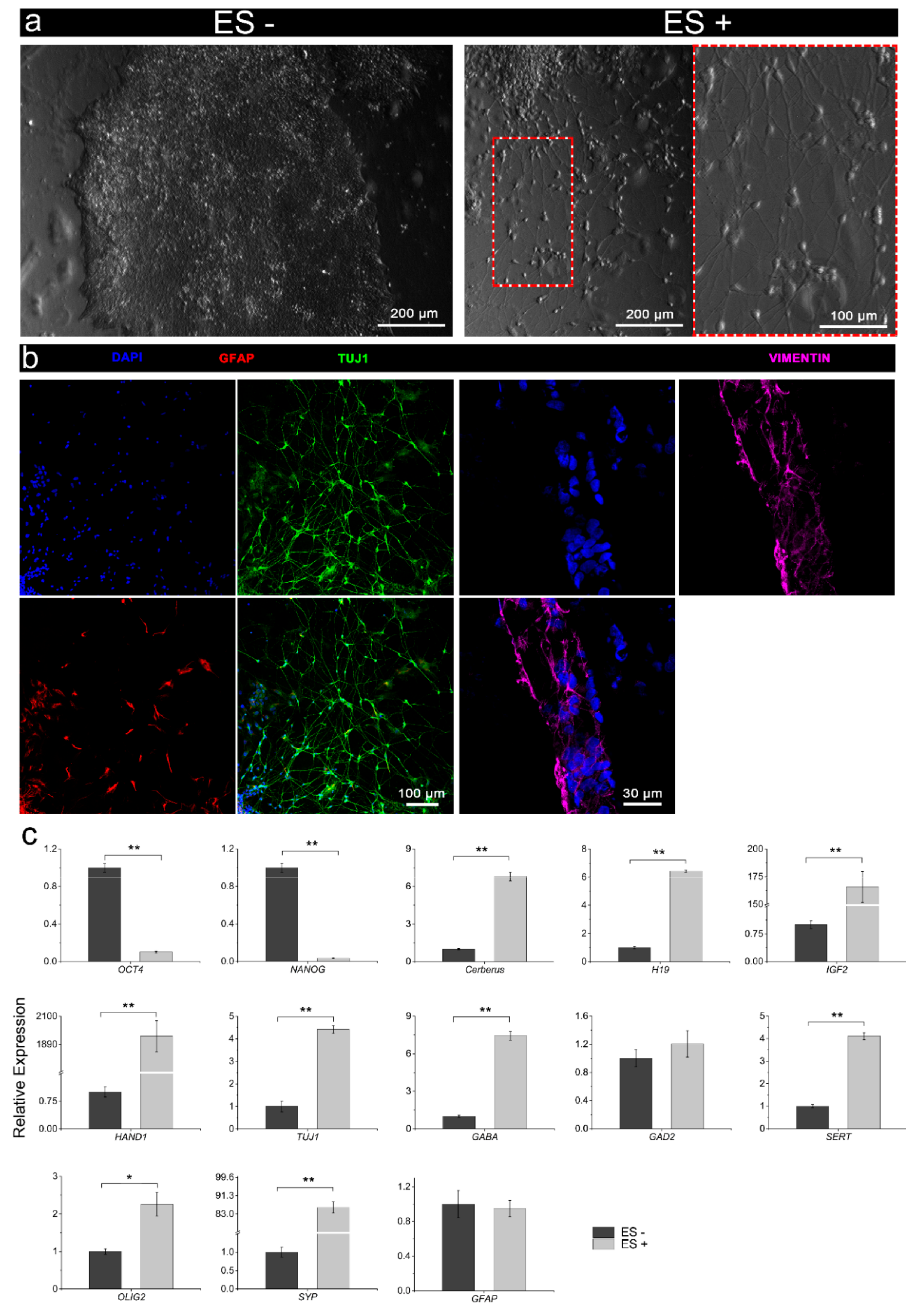
3.3. Characterisation of Stimulated iPSCs
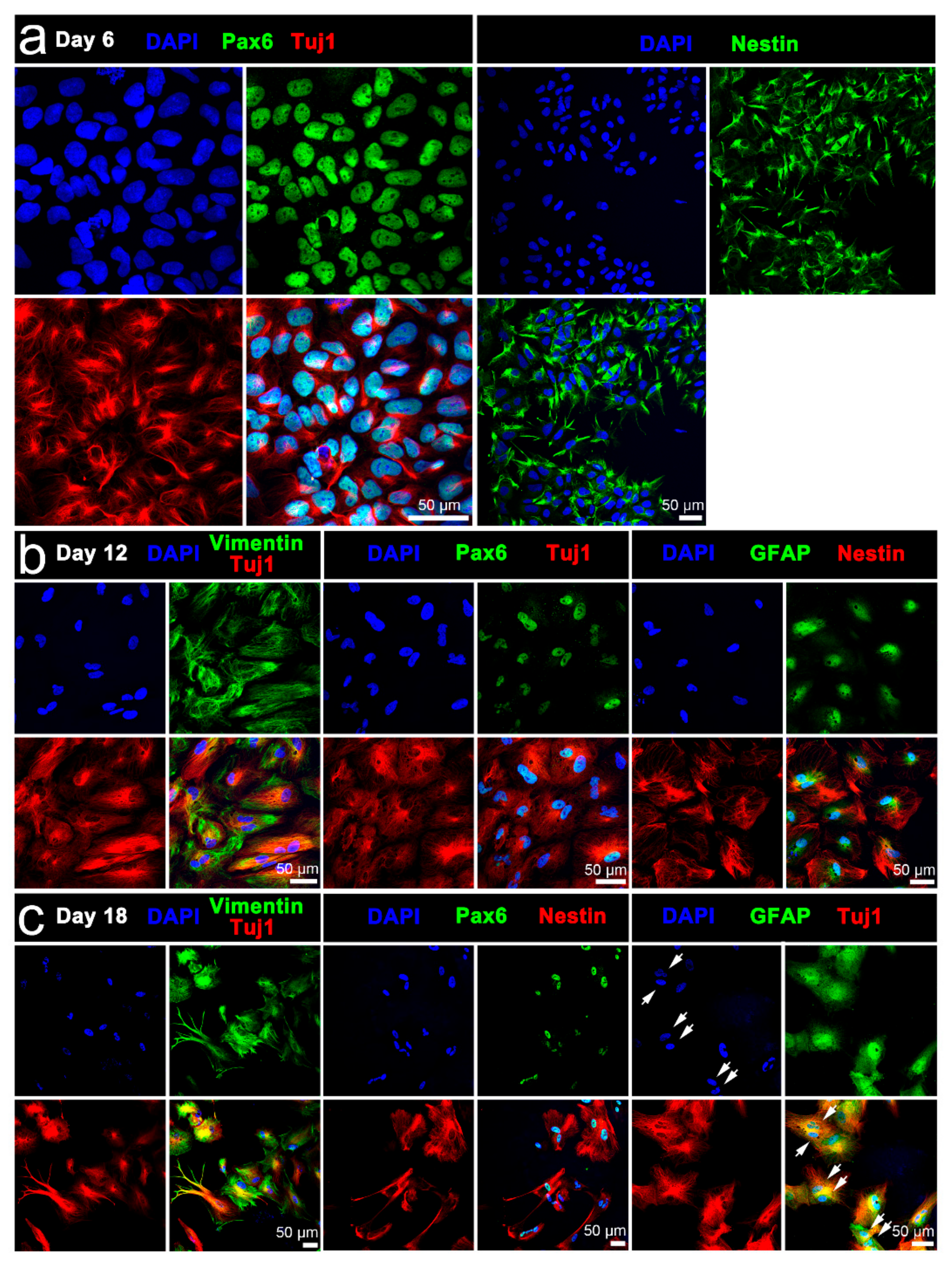
3.3.1. Conventional Electrical Stimulation with Chemical Induction
3.3.2. Electrical Stimulation without Chemical Induction
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balint, R.; Cassidy, N.J.; Cartmell, S.H. Electrical stimulation: A novel tool for tissue engineering. Tissue Eng. Part. B. Rev. 2013, 19, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kringelbach, M.L.; Jenkinson, N.; Owen, S.L.; Aziz, T.Z. Translational principles of deep brain stimulation. Nat. Rev. Neurosci 2007, 8, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Moffa, A.H.; Martin, D.; Alonzo, A.; Bennabi, D.; Blumberger, D.M.; Bensenor, I.M.; Daskalakis, Z.; Fregni, F.; Haffen, E.; Lisanby, S.H.; et al. Efficacy and acceptability of transcranial direct current stimulation (tDCS) for major depressive disorder: An individual patient data meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Deyo, R.A.; Walsh, N.E.; Martin, D.C.; Schoenfeld, L.S.; Ramamurthy, S. A controlled trial of transcutaneous electrical nerve stimulation (TENS) and exercise for chronic low back pain. N. Engl. J. Med. 1990, 322, 1627–1634. [Google Scholar] [CrossRef]
- Tomaskovic-Crook, E.; Zhang, P.; Ahtiainen, A.; Kaisvuo, H.; Lee, C.Y.; Beirne, S.; Aqrawe, Z.; Svirskis, D.; Hyttinen, J.; Wallace, G.G.; et al. Human Neural Tissues from Neural Stem Cells Using Conductive Biogel and Printed Polymer Microelectrode Arrays for 3D Electrical Stimulation. Adv. Healthc. Mater. 2019, 8, e1900425. [Google Scholar] [CrossRef]
- Stewart, E.; Kobayashi, N.R.; Higgins, M.J.; Quigley, A.F.; Jamali, S.; Moulton, S.E.; Kapsa, R.M.; Wallace, G.G.; Crook, J.M. Electrical stimulation using conductive polymer polypyrrole promotes differentiation of human neural stem cells: A biocompatible platform for translational neural tissue engineering. Tissue Eng. Part. C. Methods 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Yamada, M.; Tanemura, K.; Okada, S.; Iwanami, A.; Nakamura, M.; Mizuno, H.; Ozawa, M.; Ohyama-Goto, R.; Kitamura, N.; Kawano, M.; et al. Electrical stimulation modulates fate determination of differentiating embryonic stem cells. Stem Cells 2007, 25, 562–570. [Google Scholar] [CrossRef]
- Hernandez, D.; Millard, R.; Sivakumaran, P.; Wong, R.C.; Crombie, D.E.; Hewitt, A.W.; Liang, H.; Hung, S.S.; Pebay, A.; Shepherd, R.K.; et al. Electrical Stimulation Promotes Cardiac Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells Int. 2016, 2016, 1718041. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Mattis, V.B.; Svendsen, C.N. Induced pluripotent stem cells: A new revolution for clinical neurology? Lancet Neurol. 2011, 10, 383–394. [Google Scholar] [CrossRef]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Richardson, R.T.; Thompson, B.; Moulton, S.; Newbold, C.; Lum, M.G.; Cameron, A.; Wallace, G.; Kapsa, R.; Clark, G.; O’Leary, S. The effect of polypyrrole with incorporated neurotrophin-3 on the promotion of neurite outgrowth from auditory neurons. Biomaterials 2007, 28, 513–523. [Google Scholar] [CrossRef]
- von Bohlen Und Halbach, O. Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res. 2007, 329, 409–420. [Google Scholar] [CrossRef]
- Gellner, A.K.; Reis, J.; Fritsch, B. Glia: A Neglected Player in Non-invasive Direct Current Brain Stimulation. Front. Cell Neurosci. 2016, 10, 188. [Google Scholar] [CrossRef]
- Orkand, R.K.; Nicholls, J.G.; Kuffler, S.W. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef]
- Pannasch, U.; Rouach, N. Emerging role for astroglial networks in information processing: From synapse to behavior. Trends Neurosci. 2013, 36, 405–417. [Google Scholar] [CrossRef]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Nguyen, H.N.; Guo, Z.; Lalli, M.A.; Wang, X.; Su, Y.; Kim, N.S.; Yoon, K.J.; Shin, J.; Zhang, C.; et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 2014, 515, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Vermehren-Schmaedick, A.; Khanjian, R.A.; Balkowiec, A. Cellular mechanisms of activity-dependent BDNF expression in primary sensory neurons. Neuroscience 2015, 310, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Feng, L.; Yang, F.; Lu, B. Activity- and Ca(2+)-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J. Cell Biol. 2000, 150, 1423–1434. [Google Scholar] [CrossRef]
- Meyer-Franke, A.; Wilkinson, G.A.; Kruttgen, A.; Hu, M.; Munro, E.; Hanson, M.G., Jr.; Reichardt, L.F.; Barres, B.A. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998, 21, 681–693. [Google Scholar] [CrossRef]
- Shepherd, R.K.; Coco, A.; Epp, S.B.; Crook, J.M. Chronic depolarization enhances the trophic effects of brain-derived neurotrophic factor in rescuing auditory neurons following a sensorineural hearing loss. J. Comp. Neurol. 2005, 486, 145–158. [Google Scholar] [CrossRef]
- Zhao, R.; Liu, L.; Rittenhouse, A.R. Ca2+ influx through both L- and N-type Ca2+ channels increases c-fos expression by electrical stimulation of sympathetic neurons. Eur. J. Neurosci. 2007, 25, 1127–1135. [Google Scholar] [CrossRef]
- Yan, X.; Liu, J.; Ye, Z.; Huang, J.; He, F.; Xiao, W.; Hu, X.; Luo, Z. CaMKII-Mediated CREB Phosphorylation Is Involved in Ca2+-Induced BDNF mRNA Transcription and Neurite Outgrowth Promoted by Electrical Stimulation. PLoS ONE 2016, 11, e0162784. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward | Reverse | Length (bp) | |
|---|---|---|---|---|
| OCT4 | CAATTTGCCAAGCTCCTGA | CGTTTGGCTGAATACCTTCC | 105 | |
| NANOG | TACCTCAGCCTCCAGCAGAT | TGCGTCACACCATTGCTATT | 146 | |
| Cerberus | GCCATGAAGTACATTGGGAGA | CACAGCCTTCGTGGGTTATAG | 69 | |
| H19 | GCAAGAAGCGGGTCTGTTT | GCTGGGTAGCACCATTTCTT | 105 | |
| HAND1 | AAGCGGAAAAGGGAGCTG | ACTCCAGCGCCCAGACTT | 112 | |
| IGF2 | CTGTTTCCGCAGCTGTGAC | GGGGTATCTGGGGAAGTTGT | 118 | |
| SERT | CTCCGAGGACAACATCAC | CTTGCCAGAGGTCTTGAC | 199 | |
| GABA | GTCCAGGTCTGTCTGACTGTCTT | CTTCACTTCGGTTACACGCTCTC | 197 | |
| SYP | TTGCCTTCCTCTACTCCAT | GCCATCTTCACATCTGACA | 172 | |
| TUJ1 | ACACAGGCGTCCACAGTT | GTTCCAGGTCCACCAGAATG | 167 | |
| OLIG2 | TTGCTCCTCTTCCTCCTT | GGCTTCCAACTAACTTGTG | 129 | |
| GFAP | ATCAACTCACCGCCAACA | CTTCATCTGCTTCCTGTCTATA | 153 | |
| GAD2 | CGACCTGCTCCAGTCTCCAA | ATGCCGCCCGTGAACTTCT | 144 | |
| β-Actin | AGGCATCCTCACCCTGAAGTA | CACACGCAGCTCATTGTAGA | 103 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaskovic-Crook, E.; Gu, Q.; Rahim, S.N.A.; Wallace, G.G.; Crook, J.M. Conducting Polymer Mediated Electrical Stimulation Induces Multilineage Differentiation with Robust Neuronal Fate Determination of Human Induced Pluripotent Stem Cells. Cells 2020, 9, 658. https://doi.org/10.3390/cells9030658
Tomaskovic-Crook E, Gu Q, Rahim SNA, Wallace GG, Crook JM. Conducting Polymer Mediated Electrical Stimulation Induces Multilineage Differentiation with Robust Neuronal Fate Determination of Human Induced Pluripotent Stem Cells. Cells. 2020; 9(3):658. https://doi.org/10.3390/cells9030658
Chicago/Turabian StyleTomaskovic-Crook, Eva, Qi Gu, Siti N Abdul Rahim, Gordon G Wallace, and Jeremy M Crook. 2020. "Conducting Polymer Mediated Electrical Stimulation Induces Multilineage Differentiation with Robust Neuronal Fate Determination of Human Induced Pluripotent Stem Cells" Cells 9, no. 3: 658. https://doi.org/10.3390/cells9030658
APA StyleTomaskovic-Crook, E., Gu, Q., Rahim, S. N. A., Wallace, G. G., & Crook, J. M. (2020). Conducting Polymer Mediated Electrical Stimulation Induces Multilineage Differentiation with Robust Neuronal Fate Determination of Human Induced Pluripotent Stem Cells. Cells, 9(3), 658. https://doi.org/10.3390/cells9030658