Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer


Abstract
:1. Introduction
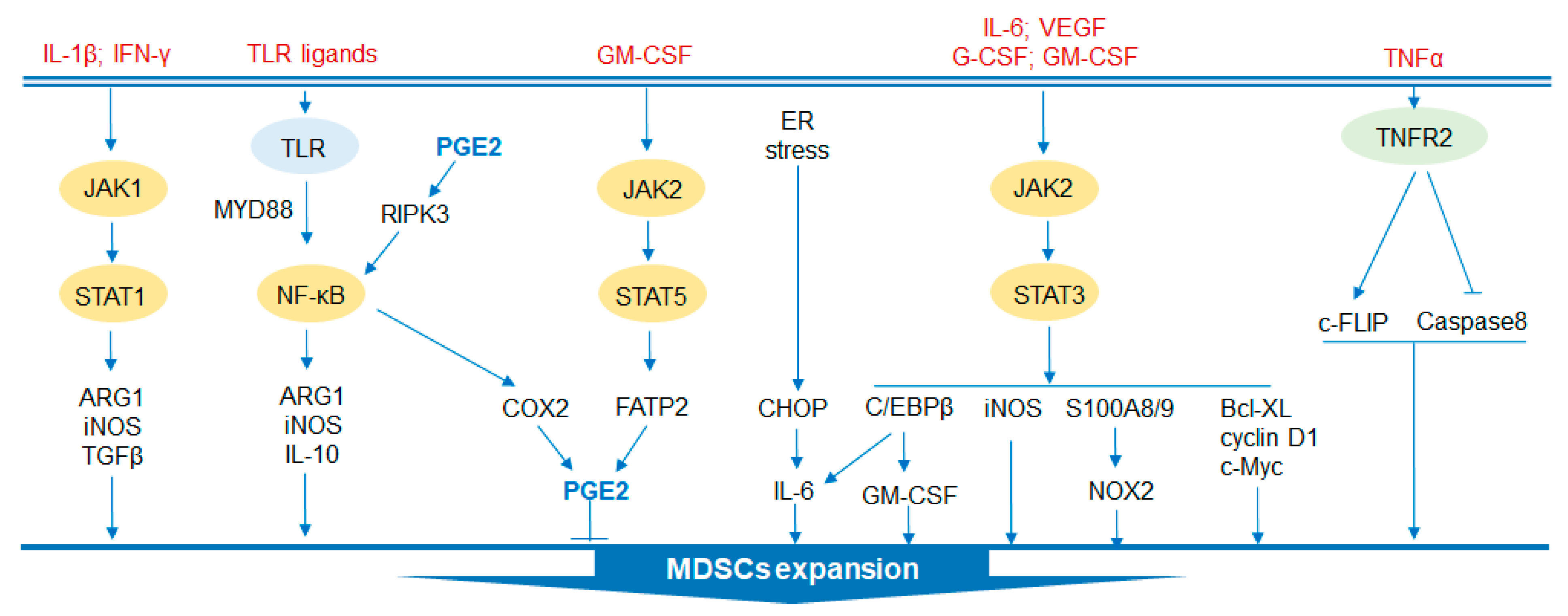
2. Signaling Pathways that Regulate MDSC Functions
2.1. STAT Signaling Pathway
2.2. C/EBPβ
2.3. TLR Signaling Pathway
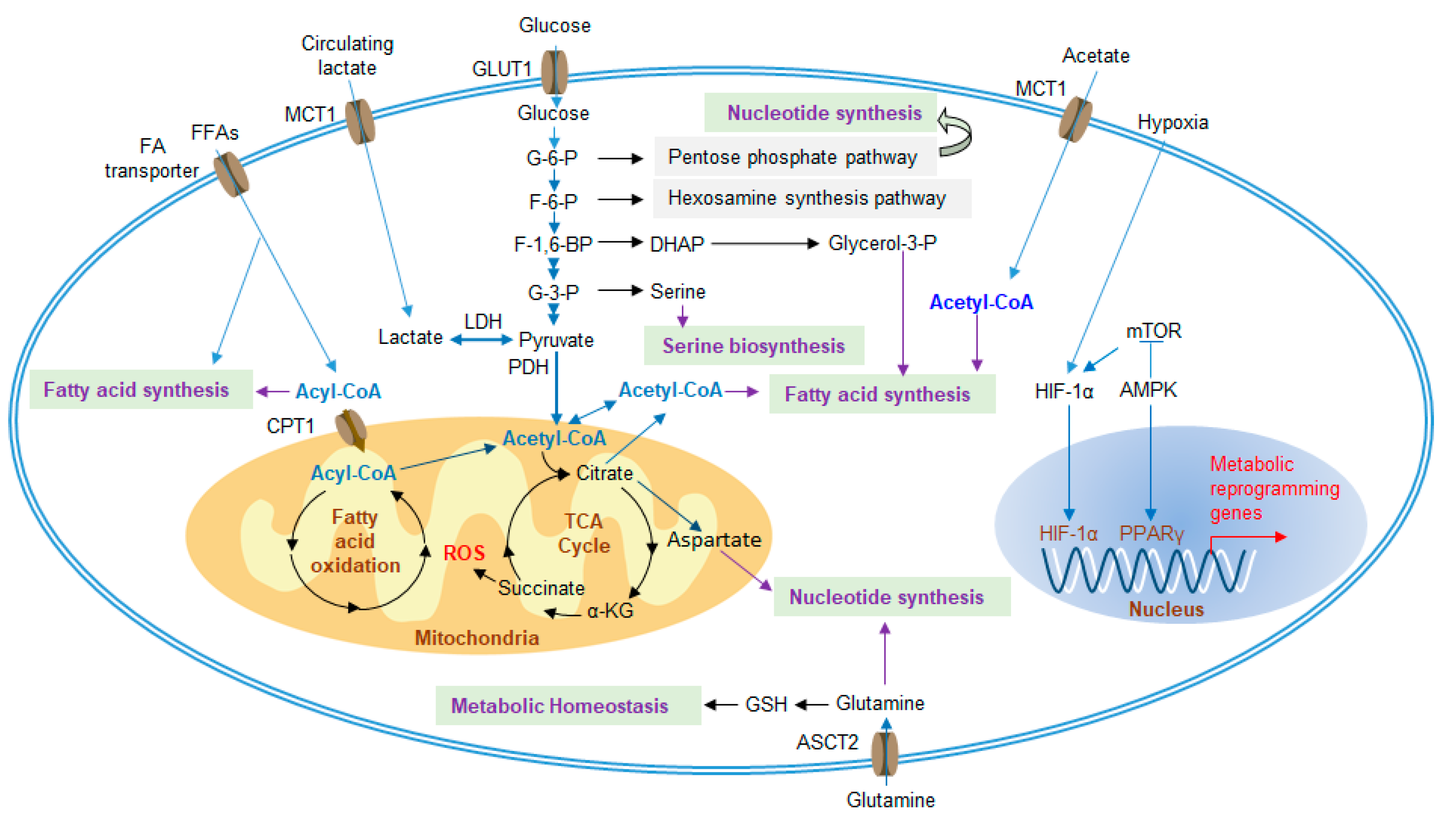
3. Metabolic Reprogramming of MDSC Functions
3.1. Glycolysis and Lactate
3.2. Amino Acid Metabolism
3.3. Glutamine Metabolism
3.4. Lipid Metabolism
3.5. Extracellular Adenosine
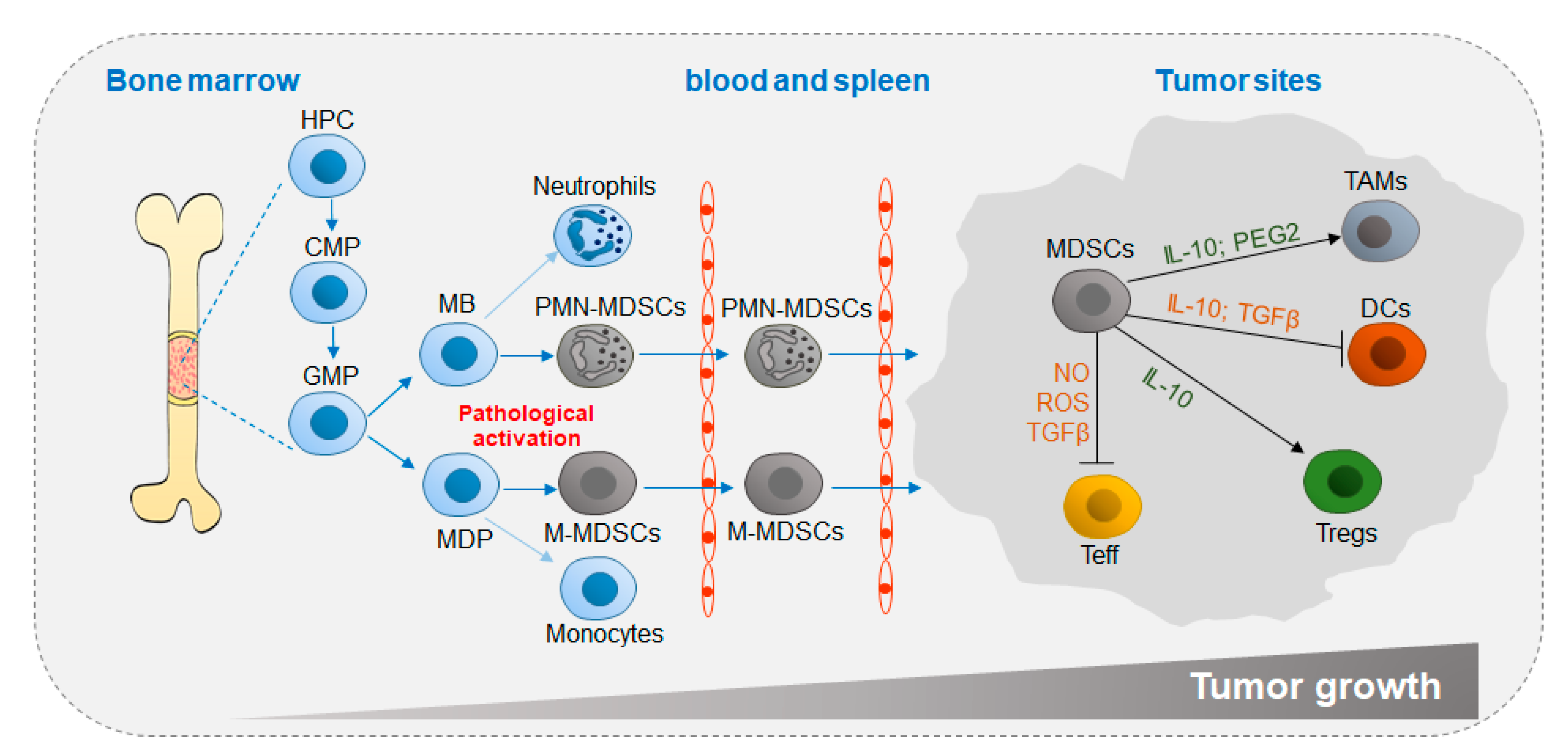
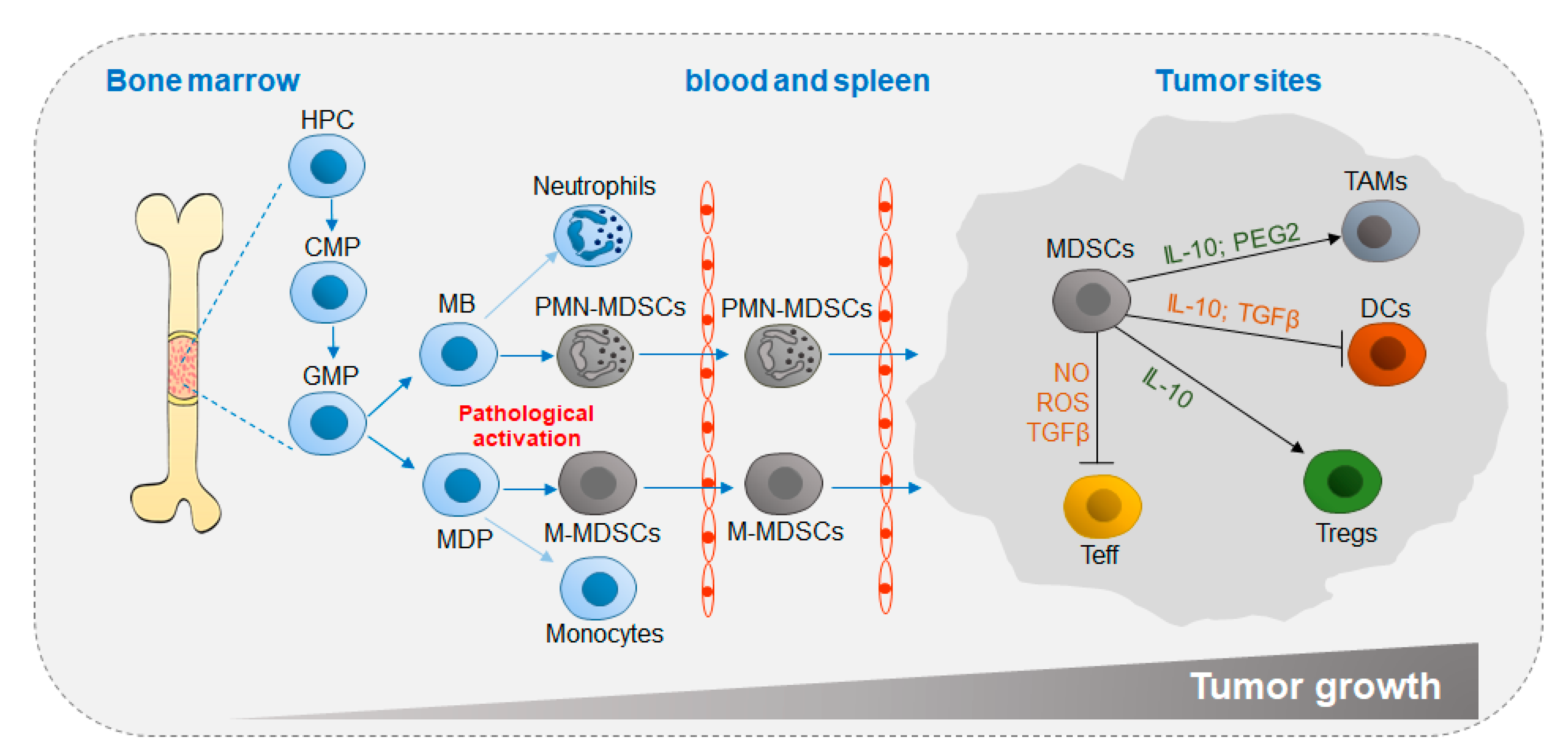
4. Metabolic Activity of MDSCs in Cancer
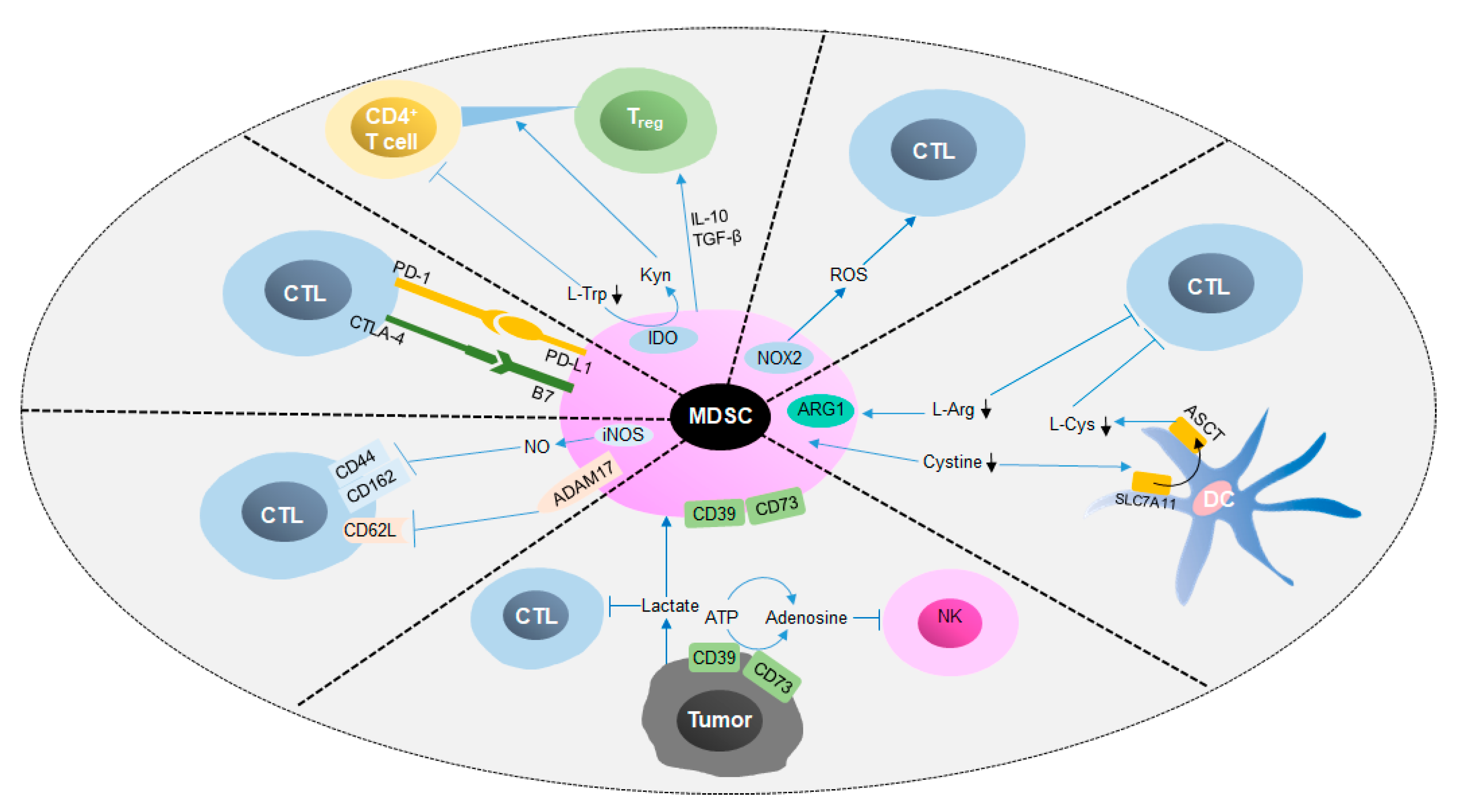
4.1. Immunoregulatory Effects of MDSCs in Cancer
4.2. Metabolic Regulatory Mechanisms of MDSC Functions in Cancer
5. The Therapeutic Effects of Targeting MDSCs
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Schmidt, K.; Zilio, S.; Schmollinger, J.C.; Bronte, V.; Blankenstein, T.; Willimsky, G. Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood 2013, 121, 1740–1748. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef]
- Malek, E.; de Lima, M.; Letterio, J.J.; Kim, B.G.; Finke, J.H.; Driscoll, J.J.; Giralt, S.A. Myeloid-derived suppressor cells: The green light for myeloma immune escape. Blood Rev. 2016, 30, 341–348. [Google Scholar] [CrossRef]
- Wegiel, B.; Vuerich, M.; Daneshmandi, S.; Seth, P. Metabolic Switch in the Tumor Microenvironment Determines Immune Responses to Anti-cancer Therapy. Front. Oncol. 2018, 8, 284. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Trovato, R.; Canè, S.; Petrova, V.; Sartoris, S.; Ugel, S.; De Sanctis, F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Yuen, V.W.; Wong, C.C. Hypoxia and the Metastatic Niche. Adv. Exp. Med. Biol. 2019, 1136, 97–112. [Google Scholar] [CrossRef]
- Pastaki Khoshbin, A.; Eskian, M.; Keshavarz-Fathi, M.; Rezaei, N. Roles of Myeloid-Derived Suppressor Cells in Cancer Metastasis: Immunosuppression and Beyond. Arch. Immunol. Ther. Exp. (Warsz.) 2019, 67, 89–102. [Google Scholar] [CrossRef]
- Principi, E.; Raffaghello, L. The role of the P2X7 receptor in myeloid-derived suppressor cells and immunosuppression. Curr. Opin. Pharmacol. 2019, 47, 82–89. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Mandruzzato, S.; Brandau, S.; Britten, C.M.; Bronte, V.; Damuzzo, V.; Gouttefangeas, C.; Maurer, D.; Ottensmeier, C.; van der Burg, S.H.; Welters, M.J.; et al. Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: Results from an interim study. Cancer Immunol. Immunother. 2016, 65, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Scalea, J.R.; Lee, Y.S.; Davila, E.; Bromberg, J.S. Myeloid-Derived Suppressor Cells and Their Potential Application in Transplantation. Transplantation 2018, 102, 359–367. [Google Scholar] [CrossRef]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin; Ahmad, S. The receptor for advanced glycation end products: A fuel to pancreatic cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Hoing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer. Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A.; Chen, J. Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu. Rev. Immunol. 2017, 35, 533–550. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Lee, J.K.; Won, C.; Yi, E.H.; Seok, S.H.; Kim, M.H.; Kim, S.J.; Chung, M.H.; Lee, H.G.; Ikuta, K.; Ye, S.K. Signal transducer and activator of transcription 3 (Stat3) contributes to T-cell homeostasis by regulating pro-survival Bcl-2 family genes. Immunology 2013, 140, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer 2019, 144, 933–946. [Google Scholar] [CrossRef]
- Al-Khami, A.A.; Rodriguez, P.C.; Ochoa, A.C. Energy metabolic pathways control the fate and function of myeloid immune cells. J. Leukoc. Biol. 2017, 102, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Hix, L.M.; Karavitis, J.; Khan, M.W.; Shi, Y.H.; Khazaie, K.; Zhang, M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J. Biol. Chem. 2013, 288, 11676–11688. [Google Scholar] [CrossRef] [Green Version]
- Varikuti, S.; Oghumu, S.; Elbaz, M.; Volpedo, G.; Ahirwar, D.K.; Alarcon, P.C.; Sperling, R.H.; Moretti, E.; Pioso, M.S.; Kimble, J.; et al. STAT1 gene deficient mice develop accelerated breast cancer growth and metastasis which is reduced by IL-17 blockade. Oncoimmunology 2017, 6, e1361088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- De Sanctis, F.; Solito, S.; Ugel, S.; Molon, B.; Bronte, V.; Marigo, I. MDSCs in cancer: Conceiving new prognostic and therapeutic targets. Biochim. Biophys. Acta 2016, 1865, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Marigo, I.; Bosio, E.; Solito, S.; Mesa, C.; Fernandez, A.; Dolcetti, L.; Ugel, S.; Sonda, N.; Bicciato, S.; Falisi, E.; et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010, 32, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Thevenot, P.T.; Sierra, R.A.; Raber, P.L.; Al-Khami, A.A.; Trillo-Tinoco, J.; Zarreii, P.; Ochoa, A.C.; Cui, Y.; Del Valle, L.; Rodriguez, P.C. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 2014, 41, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, W.; Tang, Z.; Gao, Y.; Qi, H.; Su, X.; Zhang, Y.; Yang, R. LncRNA RNCR3 promotes Chop expression by sponging miR-185-5p during MDSC differentiation. Oncotarget 2017, 8, 111754–111769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.E.; Gan, J.; Zhang, R.D.; Cheng, Y.R.; Huang, G.J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand. J. Gastroenterol. 2011, 46, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weller, C.; Weber, R.; Groth, C.; Riester, Z.; Huser, L.; Sun, Q.; Nagibin, V.; Kirschning, C.; et al. Melanoma Extracellular Vesicles Generate Immunosuppressive Myeloid Cells by Upregulating PD-L1 via TLR4 Signaling. Cancer Res. 2019, 79, 4715–4728. [Google Scholar] [CrossRef]
- Wang, J.; Shirota, Y.; Bayik, D.; Shirota, H.; Tross, D.; Gulley, J.L.; Wood, L.V.; Berzofsky, J.A.; Klinman, D.M. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J. Immunol. 2015, 194, 4215–4221. [Google Scholar] [CrossRef] [Green Version]
- Hossain, D.M.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer. Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [Green Version]
- Shirota, Y.; Shirota, H.; Klinman, D.M. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J. Immunol. 2012, 188, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Li, F.; Qin, Z. TNF Receptor 2 Makes Tumor Necrosis Factor a Friend of Tumors. Front. Immunol. 2018, 9, 1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Payen, V.L.; Mina, E.; Van Hee, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, S.L.; Chen, W.W.; Su, Y.C.; Su, Y.W.; Chuang, T.H.; Hsu, S.C.; Huang, L.R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Yang, J.; Luo, F.; Qian, J.; Liu, R.; Zhang, D.; Yu, H.; Chu, Y. mTOR-mediated glycolysis contributes to the enhanced suppressive function of murine tumor-infiltrating monocytic myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2018, 67, 1355–1364. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, Y.; Wang, H.; Li, Y.; Shao, L.; Wang, R.; Lu, J.; Yang, Z.; Wang, J.; Zhao, Y. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci. Rep. 2016, 6, 20250. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Pang, B.; Lin, G.; Zhen, Y.; Yi, H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Szefel, J.; Danielak, A.; Kruszewski, W.J. Metabolic pathways of L-arginine and therapeutic consequences in tumors. Adv. Med. Sci. 2019, 64, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Cimen Bozkus, C.; Elzey, B.D.; Crist, S.A.; Ellies, L.G.; Ratliff, T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015, 195, 5237–5250. [Google Scholar] [CrossRef]
- Won, W.J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin. Cancer Res. 2007, 13, 5243–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Sica, A.; Strauss, L.; Consonni, F.M.; Travelli, C.; Genazzani, A.; Porta, C. Metabolic regulation of suppressive myeloid cells in cancer. Cytokine Growth Factor Rev. 2017, 35, 27–35. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhen, Y.; Ma, Z.; Li, H.; Yu, J.; Xu, Z.G.; Wang, X.Y.; Yi, H.; Yang, Y.G. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci. Transl. Med. 2016, 8, 331ra340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, P.; Angelini, G.; Delfino, L.; Matucci, A.; Rubartelli, A. The thiol redox state of lymphoid organs is modified by immunization: Role of different immune cell populations. Eur. J. Immunol. 2008, 38, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Grohmann, U.; Puccetti, P. The Coevolution of IDO1 and AhR in the Emergence of Regulatory T-Cells in Mammals. Front. Immunol. 2015, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Buijs, N.; Luttikhold, J.; Houdijk, A.P.; van Leeuwen, P.A.M. The role of a disturbed arginine/NO metabolism in the onset of cancer cachexia: A working hypothesis. Curr. Med. Chem. 2012, 19, 5278–5286. [Google Scholar] [CrossRef]
- Hammami, I.; Chen, J.; Bronte, V.; DeCrescenzo, G.; Jolicoeur, M. L-glutamine is a key parameter in the immunosuppression phenomenon. Biochem. Biophys. Res. Commun. 2012, 425, 724–729. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Brunner, F.J.; Waldeyer, C.; Ojeda, F.; Salomaa, V.; Kee, F.; Sans, S.; Thorand, B.; Giampaoli, S.; Brambilla, P.; Tunstall-Pedoe, H.; et al. Application of non-HDL cholesterol for population-based cardiovascular risk stratification: Results from the Multinational Cardiovascular Risk Consortium. Lancet 2019. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Swirski, F.K. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol. Metab. 2013, 24, 129–136. [Google Scholar] [CrossRef] [Green Version]
- George, A.B. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Li, Z.; Kang, Y. Lipid Metabolism Fuels Cancer’s Spread. Cell Metab. 2017, 25, 228–230. [Google Scholar] [CrossRef] [Green Version]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e818. [Google Scholar] [CrossRef]
- LXR Agonism Depletes MDSCs to Promote Antitumor Immunity. Cancer Discov. 2018, 8, 263. [CrossRef] [Green Version]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Boison, D.; Yegutkin, G.G. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, J.; Cekic, C. Regulation of lymphocyte function by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia 2013, 15, 1400–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, S.; Miele, L. Targeting the adenosine A2b receptor in the tumor microenvironment overcomes local immunosuppression by myeloid-derived suppressor cells. Oncoimmunology 2014, 3, e27989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Ku, A.W.; Muhitch, J.B.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H.; et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. Elife 2016, 5. [Google Scholar] [CrossRef]
- Schouppe, E.; Mommer, C.; Movahedi, K.; Laoui, D.; Morias, Y.; Gysemans, C.; Luyckx, A.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur. J. Immunol. 2013, 43, 2930–2942. [Google Scholar] [CrossRef]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef] [Green Version]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chretien, M.L.; Guillerey, C.; Putz, E.M.; Bald, T.; Forster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648.e635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, N.; Tachibana, M.; Ago, Y.; Goda, H.; Sakurai, F.; Mizuguchi, H. LY341495, an mGluR2/3 Antagonist, Regulates the Immunosuppressive Function of Myeloid-Derived Suppressor Cells and Inhibits Melanoma Tumor Growth. Biol. Pharm. Bull. 2018, 41, 1866–1869. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Wang, K.; Huang, X.J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef] [Green Version]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.L.; Ye, S.B.; Ouyang, L.Y.; Chen, Y.S.; He, J.; Huang, H.Q.; Zeng, Y.X.; Zhang, X.S.; Li, J. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: A novel prognostic indicator. Cancer Immunol. Immunother. 2015, 64, 1587–1599. [Google Scholar] [CrossRef] [Green Version]
- Xiu, B.; Lin, Y.; Grote, D.M.; Ziesmer, S.C.; Gustafson, M.P.; Maas, M.L.; Zhang, Z.; Dietz, A.B.; Porrata, L.F.; Novak, A.J.; et al. IL-10 induces the development of immunosuppressive CD14(+)HLA-DR(low/-) monocytes in B-cell non-Hodgkin lymphoma. Blood Cancer J. 2015, 5, e328. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, K.A.; Badawy, H.M.; Radwan, W.M.; Shehata, M.A.; Bassuoni, M.A. CD14(+) HLA-DR low/(-) monocytes as indicator of disease aggressiveness in B-cell non-Hodgkin lymphoma. Int. J. Lab. Hematol. 2014, 36, 650–655. [Google Scholar] [CrossRef]
- Calabretta, E.; d’Amore, F.; Carlo-Stella, C. Immune and Inflammatory Cells of the Tumor Microenvironment Represent Novel Therapeutic Targets in Classical Hodgkin Lymphoma. Int. J. Mol. Sci. 2019, 20, 5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Xu, H.; Wang, S. Immunosuppressive Role of Myeloid-Derived Suppressor Cells and Therapeutic Targeting in Lung Cancer. J. Immunol. Res. 2018, 2018, 6319649. [Google Scholar] [CrossRef] [PubMed]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.S.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.H.; Lee, K.Y.; Chang, Y.L.; Chan, Y.F.; Kuo, L.W.; Lin, T.Y.; Chung, F.T.; Kuo, C.S.; Yu, C.T.; Lin, S.M.; et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am. J. Respir. Crit. Care Med. 2012, 186, 1025–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Deng, Z.; Peng, Y.; Han, L.; Liu, J.; Wang, L.; Li, B.; Zhao, J.; Jiao, S.; Wei, H. Ascites-derived IL-6 and IL-10 synergistically expand CD14HLA-DR myeloid-derived suppressor cells in ovarian cancer patients. Oncotarget 2017, 8, 76843–76856. [Google Scholar] [CrossRef] [Green Version]
- Idorn, M.; Kollgaard, T.; Kongsted, P.; Sengelov, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Urakawa, S.; Yamasaki, M.; Goto, K.; Haruna, M.; Hirata, M.; Morimoto-Okazawa, A.; Kawashima, A.; Iwahori, K.; Makino, T.; Kurokawa, Y.; et al. Peri-operative monocyte count is a marker of poor prognosis in gastric cancer: Increased monocytes are a characteristic of myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2019, 68, 1341–1350. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Hennart, B.; Allorge, D.; van Geel, N.; Van Gele, M.; et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology 2015, 4, e982382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells-An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015, 4, e954829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, A.; de Rosny, C.; Kieu, T.L.; Perrey, S.; Berger, H.; Fluckiger, A.; Muller, T.; Pais de Barros, J.P.; Pichon, L.; Hichami, A.; et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1beta secretion in 5-fluorouracil-treated MDSC: Implication in cancer treatment. Cell Death Dis. 2019, 10, 485. [Google Scholar] [CrossRef]
- Kao, J.; Ko, E.C.; Eisenstein, S.; Sikora, A.G.; Fu, S.; Chen, S.-H. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit. Rev. Oncol. Hematol. 2011, 77, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Wang, J.; Wang, L.; Wen, J.; Guo, Y.; Qiao, W.; Zhou, J.; Xu, G.; Zhi, F. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am. J. Cancer Res. 2017, 7, 41–52. [Google Scholar]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Su, Z.; Heiser, A.; Dannull, J.; Eruslanov, E.; Kubler, H.; Yancey, D.; Dahm, P.; Vieweg, J. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2008, 14, 8270–8278. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Seo, J.H.; Kim, Y.J.; Kim, Y.S.; Ko, H.J.; Kang, C.Y. The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int. J. Cancer 2012, 131, 741–751. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; DuBois, R.N. CXCR2-Expressing Myeloid-Derived Suppressor Cells Are Essential to Promote Colitis-Associated Tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Youn, J.I. Role of myeloid-derived suppressor cells in immune checkpoint inhibitor therapy in cancer. Arch. Pharm. Res. 2019, 42, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Eissler, N.; Blanc, K.L.; Johnsen, J.I.; Kogner, P.; Kiessling, R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin. Cancer Res. 2016, 22, 3849–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M-MDSC | PMN-MDSC | |
|---|---|---|
| CD11b+Ly6G−Ly6Chi | CD11b+Ly6GhiLy6Clo | |
| Extracellular ROS | + | ++ |
| NO | ++ | − |
| ARG1 | + | + |
| iNOS | + | − |
| PGE2 | + | ++ |
| IL-10 | + | + |
| M-MDSC | PMN-MDSC | e-MDSC | |
|---|---|---|---|
| CD11b+CD14+CD15−CD33+ HLA-DR− | CD11b+CD14+ CD15+ (or CD66b+) CD33+LOX-1+ | Lin−HLA-DR−CD33+ | |
| Extracellular ROS | + | ++ | + |
| NO | ++ | − | + |
| ARG1 | + | ++ | + |
| iNOS | ++ | + | − |
| PGE2 | + | ++ | N/A |
| IL-10 | + | + | N/A |
| Pro-Tumor Targeting in MDSCs | Cancer Type | Metabolic Regulation Mechanism | Ref. |
|---|---|---|---|
| ARG1 | Multiple myeloma | PMN-MDSCs are induced by multiple myeloma-related mesenchymal stem cells to have high expression of ARG1 and exert immunosuppressive function in tumor | [103] |
| Multiple myeloma | PMN-MDSCs limit the anti-tumor response of T cells by increase the expression of ARG1 and other suppressive molecules, which is correlated with the expression of IL-18 | [104] | |
| Head and neck cancer and urological cancers | Higher expression and activity of ARG1 in PMN-MDSCs, compared to M-MDSCs and e-MDSCs, contribute to potent pro-tumoral functions | [18] | |
| CPT1 | Renal cell carcinoma; Breast cancer; Colon cancer | PMN-MDSCs suppress immune response by increasing the expression of CPT1 and uptake of FA to promote FAO in tumors | [77] |
| mGluR2/3 | Melanoma | PMN-MDSCs promote melanoma growth and inhibit proliferation of T cell via overexpressing metabotropic glutamate receptor (mGluR) 2/3 | [105] |
| LOX-1 | Non-small cell lung cancer (NSCLC); Head and neck cancer (HNC); Colon cancer | Lectin-type oxidized LDL receptor 1 (LOX-1), encoded at high levels in PMN-MDSCs, is related to ER stress and lipid metabolism in tumor | [82] |
| FATP2 | EL4 lymphoma; Lewis lung carcinoma; CT26 colon carcinoma; Pancreatic cancer | Overexpression of fatty acid transport protein 2 (FATP2) in PMN-MDSCs is conductive to tumor growth by the synthesis of PGE2 | [29] |
| Pro-Tumor Targeting in MDSCs | Cancer Type | Metabolic Regulation Mechanism | Ref. |
|---|---|---|---|
| mTOR | 3LL Lewis lung carcinoma | Tumor-infiltrating M-MDSCs are associated with increased glycolysis induced by mTOR and display strong inhibitory abilities | [49] |
| iNOS | Non-small cell lung cancer | MDSCs with high expression of iNOS inhibit T cell functions, which leads to poor response to chemotherapy | [114] |
| Ovarian cancer | Compared with healthy donors, the number of M-MDSCs increased in ovarian cancer, and the overexpression of iNOS was induced by STAT3 | [115] | |
| Prostate cancer | High levels of iNOS overexpressing MDSCs are positive correlated with the number of Tregs | [116] | |
| IDO | Gastric cancer | M-MDSCs (not PMN-MDSCs) produce IDO and blunt anti-tumor response of T cells | [117] |
| Chronic lymphocytic leukemia | M-MDSCs suppress the activity of T cells and induce Tregs by increasing IDO activity | [118] | |
| Melanoma | IDO was highly expressed in M-MDSCs rather than PMN-MDSCs, and the IDO activity is positively correlated with tumor growth | [119] | |
| CPT1 | Renal cell carcinoma; Breast cancer; Colon cancer | M-MDSCs inhibit immune response by increasing the expression of CPT1 and uptake of FA to promote FAO in tumor | [77] |
| mGluR2/3 | Melanoma | M-MDSCs promotes melanoma growth and inhibits the proliferation of T cells via metabotropic glutamate receptor (mGluR) 2/3 | [105] |
| Target | Agent | Combination Therapy | Cancer Type | Phase | Clinical Trial | |
|---|---|---|---|---|---|---|
| Depletion | MDSCs | Gemcitabine | Nivolumab | Non-small cell lung cancer | II | NCT03302247 |
| 5-Fluorouracil | Bevacizumab | Glioblastoma | Recruiting | NCT02669173 | ||
| Cyclophosphamide/ Decitabine/Carboplatin/ Paclitaxel/Doxorubicin | Pembrolizumab | Breast cancer | II | NCT02957968 | ||
| RGX-104 | Nivolumab | Advanced Solid Malignancies and Lymphoma | I | NCT02922764 | ||
| Ibrutinib | Nivolumab | Metastatic Malignant Solid Neoplasm | I | NCT03525925 | ||
| Blocking recruitment and expansion | CCR5 | Vicriviroc | Pembrolizumab | Advanced/metastatic microsatellite stable colorectal cancer | II | NCT03631407 |
| Leronlimab | Carboplatin | Metastatic Triple Negative Breast Cancer | I/II | NCT03838367 | ||
| CXCR2 | SX-682 | Pembrolizumab | Melanoma | Recruiting | NCT03161431 | |
| AZD5069 | Metastatic castration resistant prostate cancer | I | NCT03177187 | |||
| c-Kit | Imatinib | Chronic myeloid leukemia | II | NCT00852566 | ||
| Inhibition of suppressive function | STAT3 | AZD9150 | Ovarian cancer and gastrointestinal cancer | II | NCT02417753 | |
| COX2 | Celecoxib | Cisplatin | Ovarian cancer | Recruiting | NCT02432378 | |
| PDE5 | Tadalafil | Head and Neck Squamous Cell Carcinoma | I | NCT02544880 | ||
| IDO | BMS-986205 | Nivolumab | Glioblastoma | Recruiting | NCT04047706 | |
| TLR7 | Imiquimod | Paclitaxel | Breast cancer | II | NCT00821964 | |
| Promotion of differentiation | MDSCs | ATRA | Ipilimumab | Melanoma | II | NCT02403778 |
| ATRA | Pembrolizumab | Melanoma | Recruiting | NCT03200847 | ||
| Vitamin D | CLL | |||||
| Epigenetic therapy | HDAC | Entinostat | Nivolumab | Metastatic Cholangiocarcinoma and Pancreatic Cancer | Recruiting | NCT03250273 |
| Entinostat | Ipilimumab/ Nivolumab | Breast cancer | I | NCT02453620 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Jia, A.; Bi, Y.; Wang, Y.; Liu, G. Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells 2020, 9, 1011. https://doi.org/10.3390/cells9041011
Wang Y, Jia A, Bi Y, Wang Y, Liu G. Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells. 2020; 9(4):1011. https://doi.org/10.3390/cells9041011
Chicago/Turabian StyleWang, Yufei, Anna Jia, Yujing Bi, Yuexin Wang, and Guangwei Liu. 2020. "Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer" Cells 9, no. 4: 1011. https://doi.org/10.3390/cells9041011
APA StyleWang, Y., Jia, A., Bi, Y., Wang, Y., & Liu, G. (2020). Metabolic Regulation of Myeloid-Derived Suppressor Cell Function in Cancer. Cells, 9(4), 1011. https://doi.org/10.3390/cells9041011