Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells
2.3. Immunoblotting
2.4. Intracellular Doxorubicin Accumulation and Doxorubicin Kinetic Efflux
2.5. ATPases Activity
2.6. Caspase 3 Activity
2.7. Cell Viability
2.8. Proximity Ligation Assay
2.9. Confocal Microscope Analysis
2.10. Surface CRT Expression, ATP and HMGB1 Release
2.11. Phagocytosis and T Lymphocyte Activation
2.12. Generation of Pgp-Knocked out (KO) Clones
2.13. Statistical Analysis
3. Results
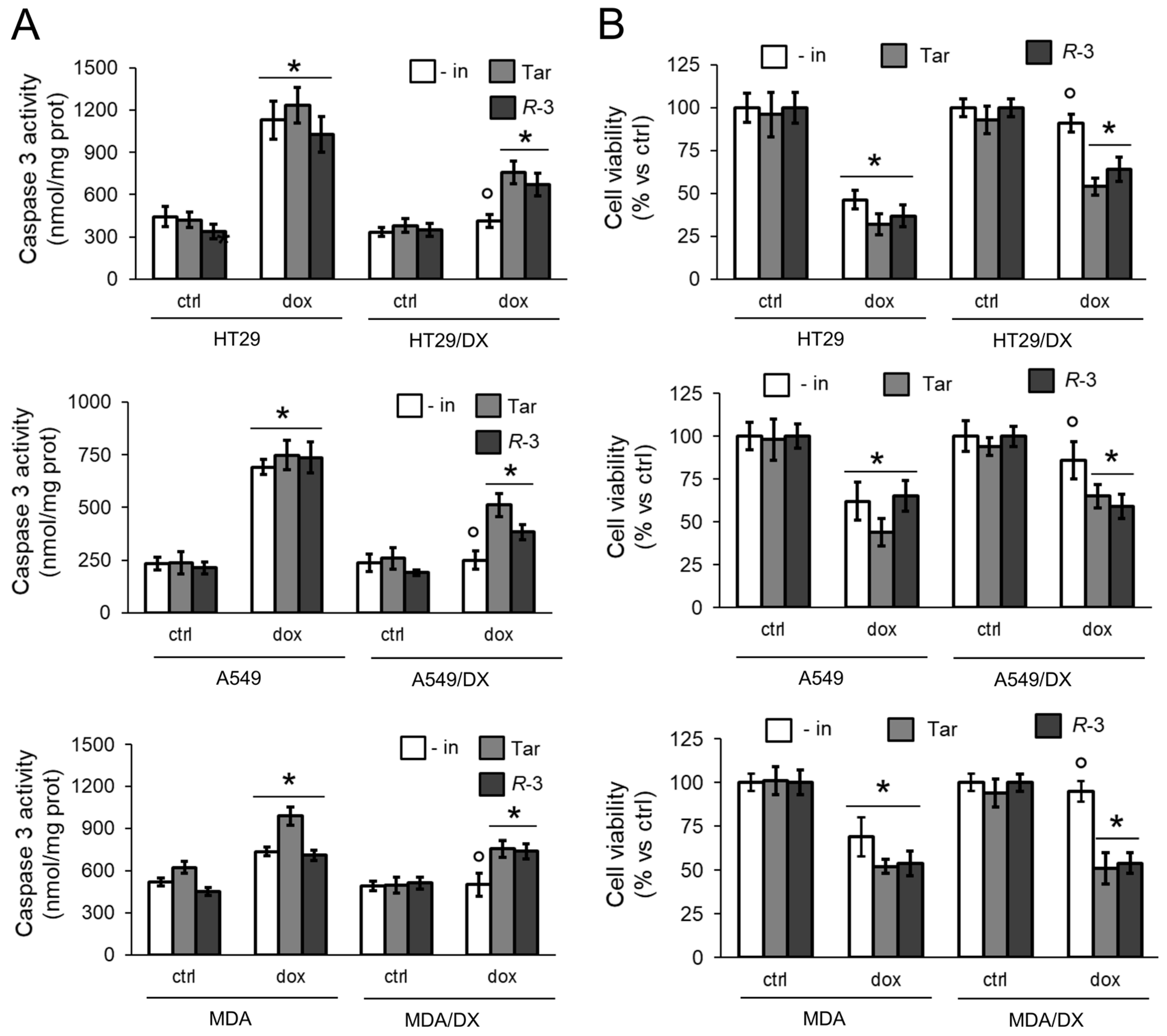
3.1. R-3 Increases Doxorubicin Intracellular Content and Cytotoxicity by Inhibiting Pgp Activity
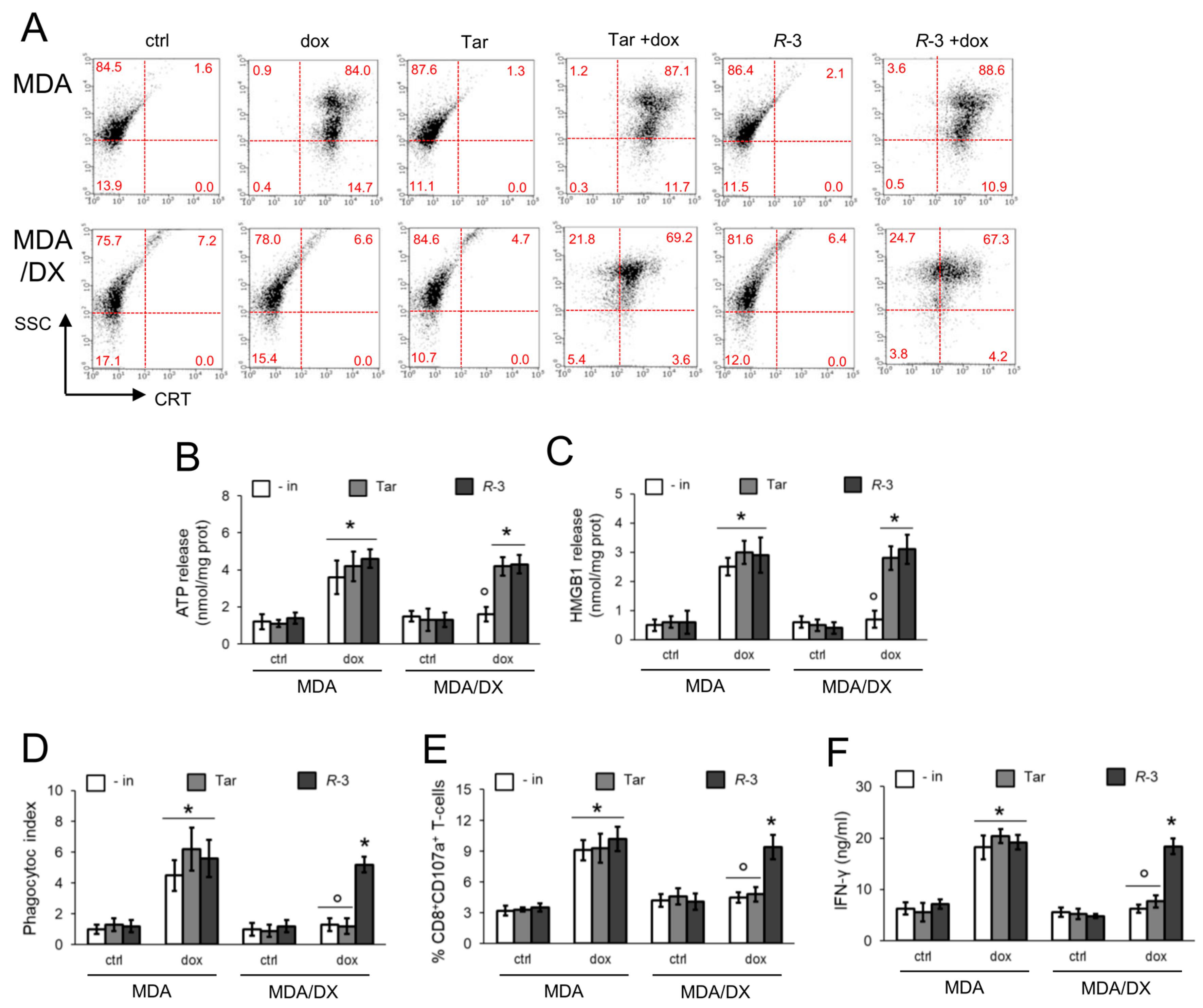
3.2. R-3 but Not Tariquidar Restores Doxorubicin-Induced Immunogenic Death in Resistant Cell Lines
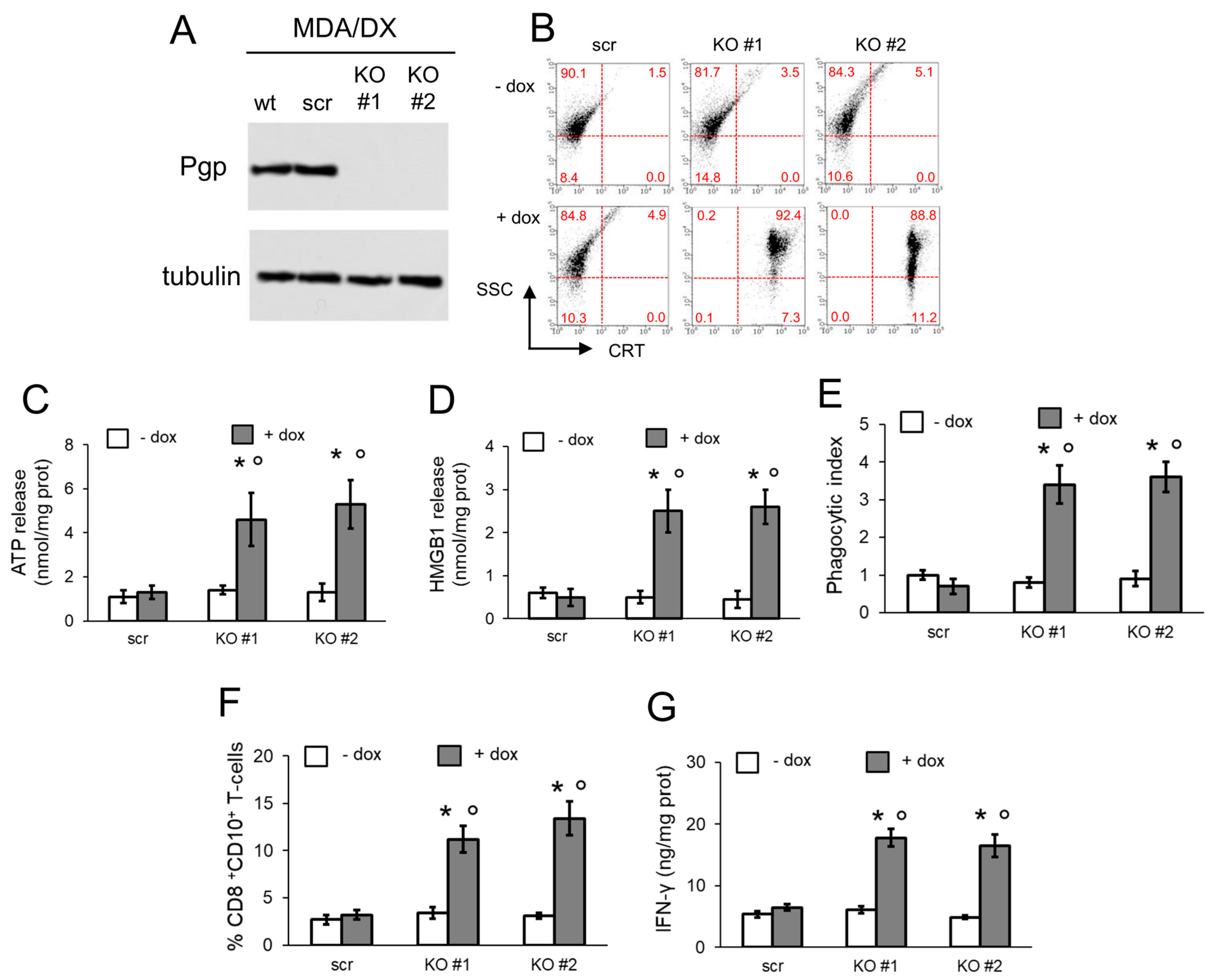
3.3. Pgp Internalization and Degradation Restores Immunogenic Cell Death
3.4. Pgp Removal is Necessary to Restore Immunogenic Cell Death in Doxorubicin-Resistant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. OncoImmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Buqué Martinez, A.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immuno Ther. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Michaud, M.; Sukkurwala, A.Q.; Martins, I.; Shen, S.; Zitvogel, L.; Kroemer, G. Subversion of the chemotherapy-induced anticancer immune response by the ecto-ATPase CD39. Oncoimmunology 2012, 1, 393–395. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.; Möller, A.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Hannani, D.; Poirier-Colame, V.; Ladoire, S.; Locher, C.; Sistigu, A.; Prada, N.; Adjemian, S.; Catani, J.P.; Freudenberg, M.; et al. Defective immunogenic cell death of HMGB1-deficient tumors: Compensatory therapy with TLR4 agonists. Cell Death Differ. 2014, 21, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radogna, F.; Dicato, M.; Diederich, M. Natural modulators of the hallmarks of immunogenic cell death. Biochem. Pharmacol. 2019, 162, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, e1654. [Google Scholar] [CrossRef] [Green Version]
- Marinello, J.; Delcuratolo, M.; Capranico, G. Anthracyclines as Topoisomerase II Poisons: From Early Studies to New Perspectives. Int. J. Mol. Sci. 2018, 19, e3480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boo, S.; Kopecka, J.; Brusa, D.; Gazzano, E.; Matera, L.; Ghigo, D.; Bosia, A.; Riganti, C. iNOS activity is necessary for the cytotoxic and immunogenic effects of doxorubicin in human colon cancer cells. Mol. Cancer 2009, 8, e108. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Castella, B.; Kopecka, J.; Campia, I.; Coscia, M.; Pescarmona, G.; Bosia, A.; Ghigo, D.; Massaia, M. Zoledronic acid restores doxorubicin chemosensitivity and immunogenic cell death in multidrug-resistant human cancer cells. PLoS ONE 2013, 8, 60975. [Google Scholar] [CrossRef] [Green Version]
- Gelsomino, G.; Corsetto, P.A.; Campia, I.; Montorfano, G.; Kopecka, J.; Castella, B.; Gazzano, E.; Ghigo, D.; Rizzo, A.M.; Riganti, C. Omega 3 fatty acids chemosensitize multidrug resistant colon cancer cells by down-regulating cholesterol synthesis and altering detergent resistant membranes composition. Mol. Cancer 2013, 12, 137. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Kopecka, J.; Campia, I.; Brusa, D.; Doublier, S.; Matera, L.; Ghigo, D.; Bosia, A.; Riganti, C. Nitric oxide and P-glycoprotein modulate the phagocytosis of colon cancer cells. J. Cell. Mol. Med. 2011, 15, 1492–1504. [Google Scholar] [CrossRef]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef]
- Dong, J.; Qin, Z.; Zhang, W.D.; Cheng, G.; Assaraf, A.G.; Ashby, C.R.; Chen, Z.S.; Cheng, X.D.; Qin, J.J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updates 2020, 49, 100681. [Google Scholar] [CrossRef]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef]
- Teodori, E.; Contino, M.; Riganti, C.; Bartolucci, G.; Braconi, L.; Manetti, D.; Romanelli, M.N.; Trezza, A.; Athanasios, A.; Spiga, O.; et al. Design, synthesis and biological evaluation of stereo- and regioisomers of amino aryl esters as multidrug resistance (MDR) reversers. Eur. J. Med. Chem. 2019, 182, e111655. [Google Scholar] [CrossRef]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar is an Inhibitor and Not a Substrate of Human and Mouse P-glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Kopecka, J.; Campia, I.; Jacobs, A.; Frei, A.P.; Ghigo, D.; Wollscheid, B.; Riganti, C. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 2015, 6, 6776–6793. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Miraglia, E.; Viarisio, D.; Costamagna, C.; Pescarmona, G.; Ghigo, D.; Bosia, A. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005, 65, 516–525. [Google Scholar]
- Kopecka, J.; Salzano, G.; Campia, I.; Lusa, S.; Ghigo, D.; De Rosa, G.; Riganti, C. Insights in the chemical components of liposomes responsible for P-glycoprotein inhibition. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 77–87. [Google Scholar] [CrossRef]
- Kopecka, J.; Salaroglio, I.C.; Righi, L.; Libener, R.; Orecchia, S.; Grosso, F.; Milosevic, V.; Ananthanarayanan, P.; Ricci, L.; Capelletto, E.; et al. Loss of C/EBP-β LIP drives cisplatin resistance in malignant pleural mesothelioma. Lung Cancer 2018, 120, 34–45. [Google Scholar] [CrossRef]
- Kopecka, J.; Rankin, G.M.; Salaroglio, I.C.; Poulsen, S.A.; Riganti, C. P-glycoprotein-mediated chemoresistance is reversed by carbonic anhydrase XII inhibitors. Oncotarget 2016, 7, 85861–85875. [Google Scholar] [CrossRef] [Green Version]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Riganti, C.; Kopecka, J.; Panada, E.; Barak, S.; Rubinstein, M. The role of C/EBP-β LIP in multidrug resistance. J. Natl. Cancer Inst. 2015, 107, e046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Q.; Sharom, F. Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of Pglycoprotein within the cytoplasmatic membrane leaflet. Biochemistry 2002, 41, 4744–4752. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Skovsgaard, T.; Stein, W.D.J. Pumping of drugs by Pglycoprotein: A two-step process? Pharm. Exp. Ther. 2003, 307, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajeva, I.K.; Wiese, M. Structure-activity relationships of tariquidar analogs as multidrug resistance modulators. AAPS J. 2009, 11, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.W.; Clarke, D.M. Mapping the Binding Site of the Inhibitor Tariquidar That Stabilizes the First Transmembrane Domain of P-glycoprotein. J. Biol. Chem. 2015, 290, 29389–29401. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [Green Version]
- Schcolnik-Cabrera, A.; Oldak, B.; Juárez, M.; Cruz-Rivera, M.; Flisser, A.; Mendlovic, F. Calreticulin in phagocytosis and cancer: Opposite roles in immune response outcomes. Apoptosis 2019, 24, 245–255. [Google Scholar] [CrossRef]
- Kim, H.; Barroso, M.; Samanta, R.; Greenberger, L.; Sztul, E. Experimentally induced changes in the endocytic traffic of P-glycoprotein alter drug resistance of cancer cells. Am. J. Physiol. 1997, 273, C687–C702. [Google Scholar] [CrossRef]
- Tome, M.E.; Schaefer, C.P.; Jacobs, L.M.; Zhang, Y.; Herndon, J.M.; Matty, F.O.; Davis, T.P. Identification of P-glycoprotein co-fractionating proteins and specific binding partners in rat brain microvessels. J. Neurochem. 2015, 134, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Hartz, A.M.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Aβ40 Reduces P-Glycoprotein at the Blood-Brain Barrier through the Ubiquitin-Proteasome Pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef]
- Al-Akra, L.; Bae, D.H.; Sahni, S.; Huang, M.L.H.; Park, K.C.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. Tumor stressors induce two mechanisms of intracellular P-glycoprotein-mediated resistance that are overcome by lysosomal-targeted thiosemicarbazones. J. Biol. Chem. 2018, 293, 3562–3587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buondonno, I.; Gazzano, E.; Tavanti, E.; Chegaev, K.; Kopecka, J.; Fanelli, M.; Rolando, B.; Fruttero, R.; Gasco, A.; Hattinger, C.; et al. Endoplasmic reticulum-targeting doxorubicin: A new tool effective against doxorubicin-resistant osteosarcoma. Cell. Mol. Life Sci. 2019, 76, 609–625. [Google Scholar] [CrossRef] [PubMed]
- Rolle, F.; Bincoletto, V.; Gazzano, E.; Rolando, B.; Lollo, G.; Stella, B.; Riganti, C.; Arpicco, S. Coencapsulation of disulfiram and doxorubicin in liposomes strongly reverses multidrug resistance in breast cancer cells. Int. J. Pharm. 2020, 119191. [Google Scholar] [CrossRef]
- Wei, N.; Sun, H.; Wang, F.; Liu, G. H1, a novel derivative of tetrandrine reverse P-glycoprotein-mediated multidrug resistance by inhibiting transport function and expression of P-glycoprotein. Cancer Chemother Pharmacol. 2011, 67, 1017–1025. [Google Scholar] [CrossRef]
- Székely, B.; Silber, A.L.; Pusztai, L. New therapeutic strategies for triple-negative breast cancer. Oncology (Williston Park) 2017, 31, 130–137. [Google Scholar]
- Aoto, K.; Mimura, K.; Okayama, H.; Saito, M.; Chida, S.; Noda, M.; Nakajima, T.; Saito, K.; Abe, N.; Ohki, S.; et al. Immunogenic tumor cell death induced by chemotherapy in patients with breast cancer and esophageal squamous cell carcinoma. Oncol. Rep. 2018, 39, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Di Somma, S.; Amato, J.; Iaccarino, N.; Pagano, B.; Randazzo, A.; Portella, G.; Malfitano, A.M. G-Quadruplex Binders Induce Immunogenic Cell Death Markers in Aggressive Breast Cancer Cells. Cancers (Basel) 2019, 11, e1797. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Condition | Km | Vmax |
|---|---|---|
| ctrl | 0.19 + 0.04 | 4.41 + 0.56 |
| + Tar | 0.94 + 0.15 * | 3.82 + 0.83 |
| + R-3 | 0.76 + 0.19 * | 1.32 + 0.42 *,1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopecka, J.; Godel, M.; Dei, S.; Giampietro, R.; Belisario, D.C.; Akman, M.; Contino, M.; Teodori, E.; Riganti, C. Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death. Cells 2020, 9, 1033. https://doi.org/10.3390/cells9041033
Kopecka J, Godel M, Dei S, Giampietro R, Belisario DC, Akman M, Contino M, Teodori E, Riganti C. Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death. Cells. 2020; 9(4):1033. https://doi.org/10.3390/cells9041033
Chicago/Turabian StyleKopecka, Joanna, Martina Godel, Silvia Dei, Roberta Giampietro, Dimas Carolina Belisario, Muhlis Akman, Marialessandra Contino, Elisabetta Teodori, and Chiara Riganti. 2020. "Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death" Cells 9, no. 4: 1033. https://doi.org/10.3390/cells9041033
APA StyleKopecka, J., Godel, M., Dei, S., Giampietro, R., Belisario, D. C., Akman, M., Contino, M., Teodori, E., & Riganti, C. (2020). Insights into P-Glycoprotein Inhibitors: New Inducers of Immunogenic Cell Death. Cells, 9(4), 1033. https://doi.org/10.3390/cells9041033