Exploring Biased Agonism at FPR1 as a Means to Encode Danger Sensing †


Abstract
:1. Introduction
2. Materials and Methods
2.1. FPR1 Ligands and Reagents
2.2. FPR1-Encoding Plasmid, HeLa-FPR1- Cell Line, and Cell Culture Conditions
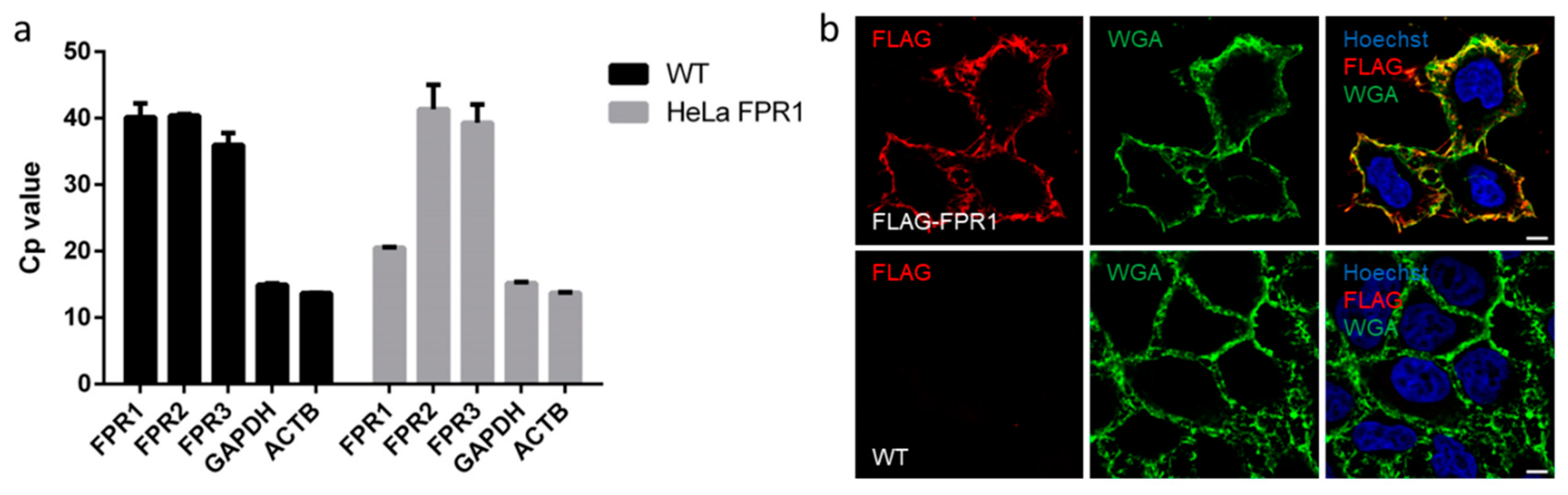
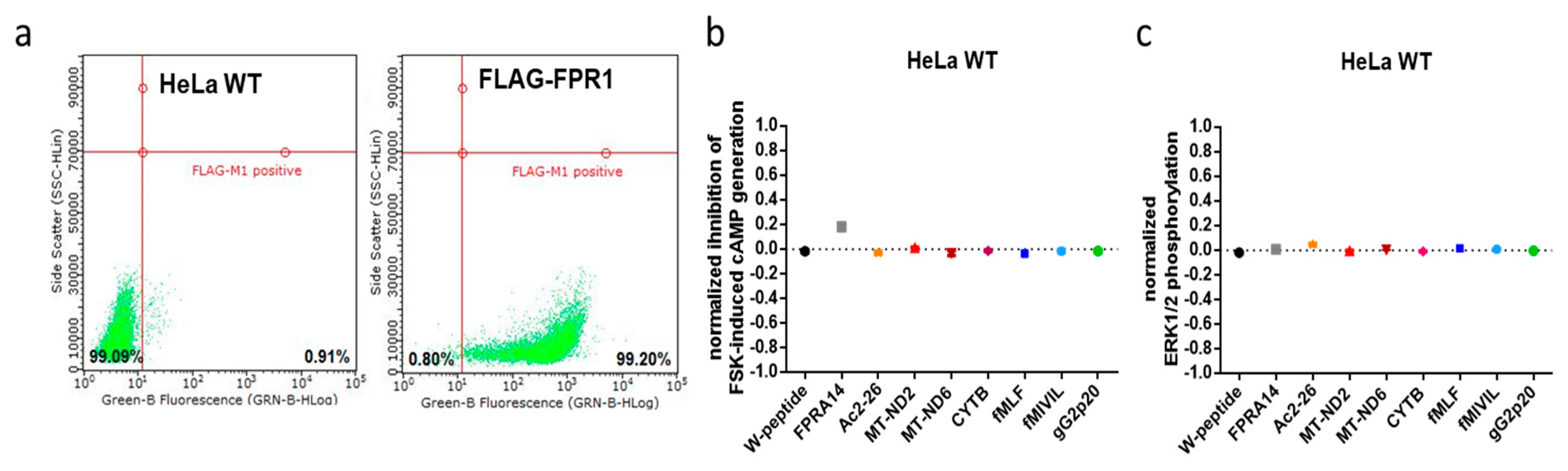
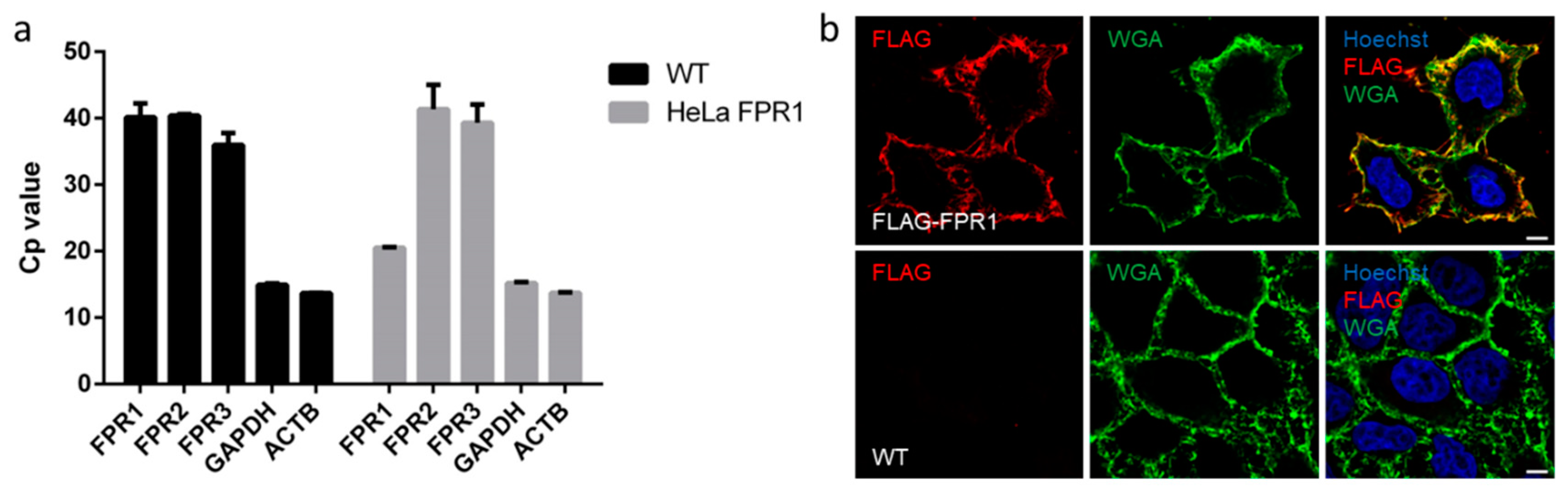
2.3. FPR1 Expression Analysis in Parental and Recombinant HeLa Cells by qPCR and Immunofluorescence Microscopy
2.4. Flow Cytometric Analysis of Agonist-Induced Receptor Internalization
2.5. HTRF-Based Quantification of cAMP Levels
2.6. HTRF-Based Quantification of MAPK/ERK Phosphorylation Levels
2.7. HTRF-Based Quantification of IP1 Levels
2.8. Curve Fitting
3. Results
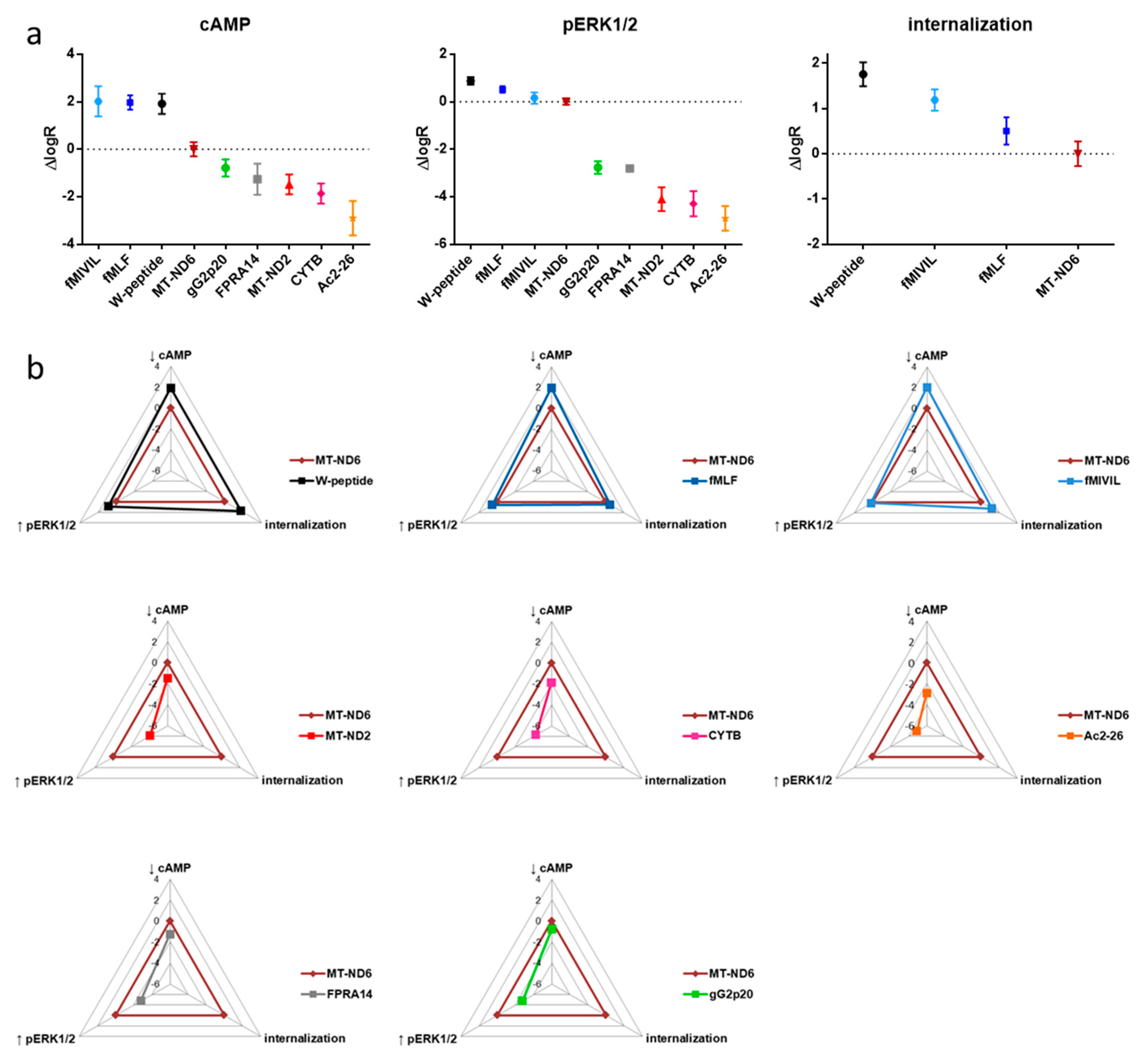
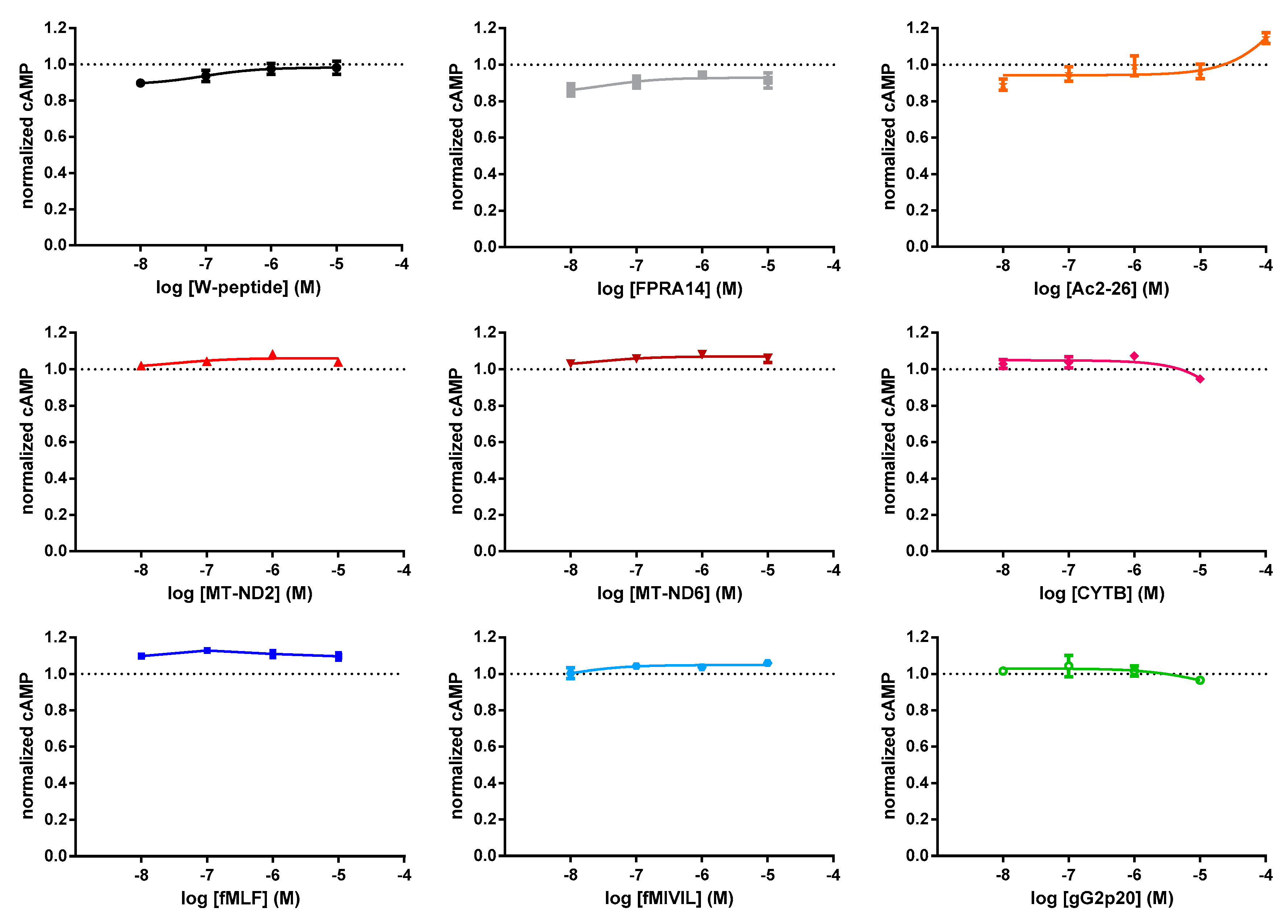
3.1. FRET-Based Analysis of Agonist-Induced Changes in cAMP Levels
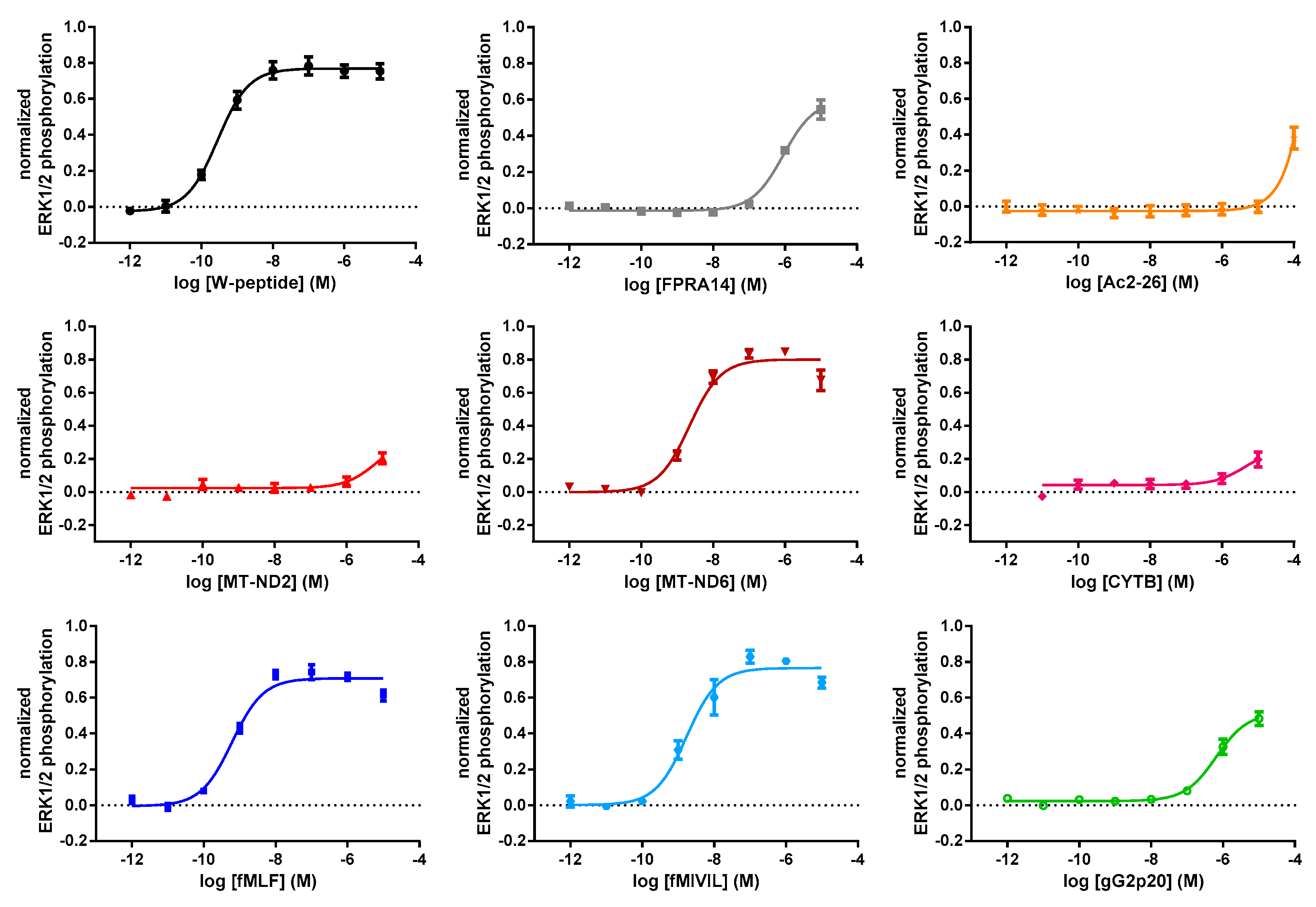
3.2. FRET-Based Analysis of Agonist-Induced Changes in MAPKinase Activation
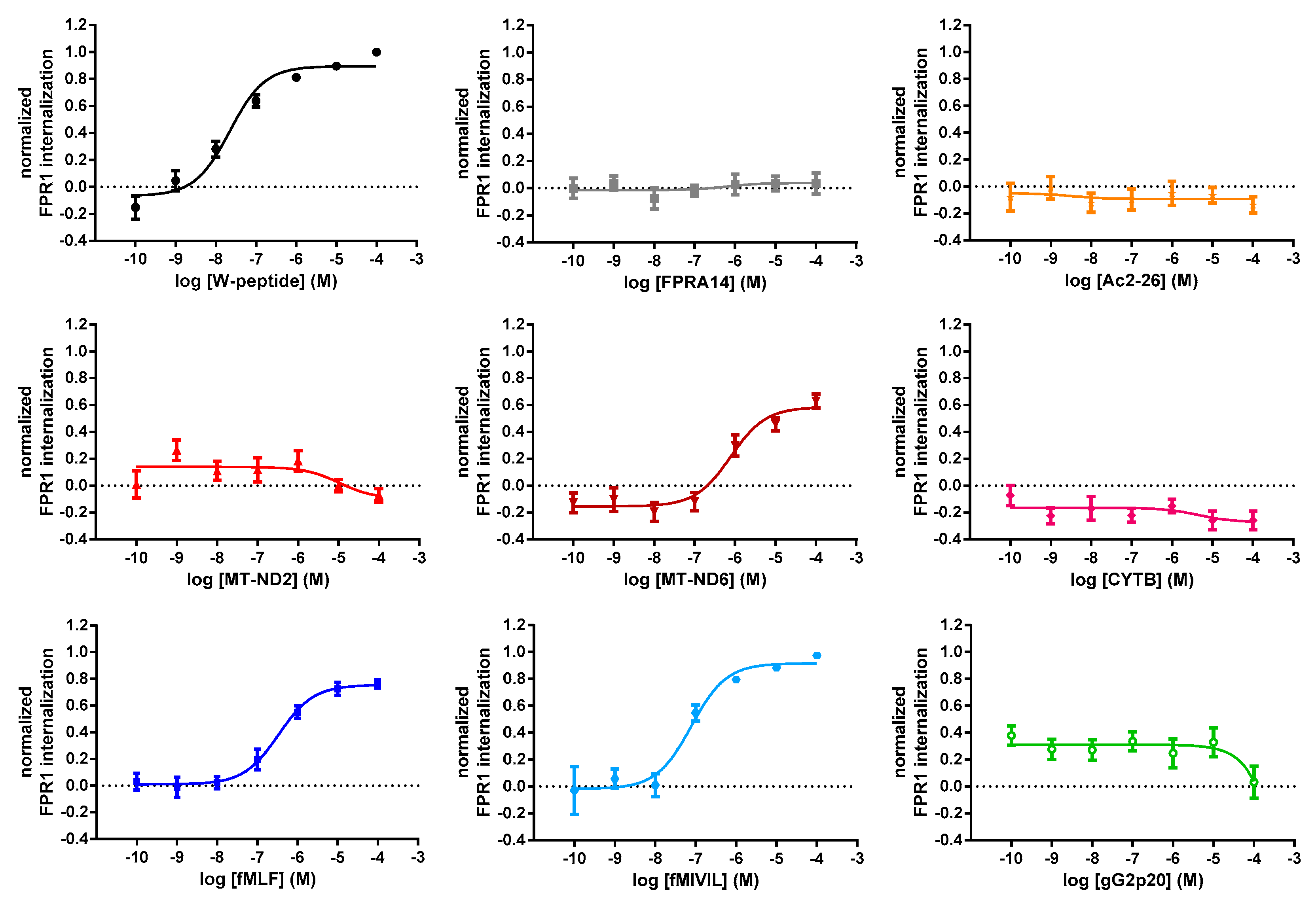
3.3. Agonist-Mediated Internalization of FPR1
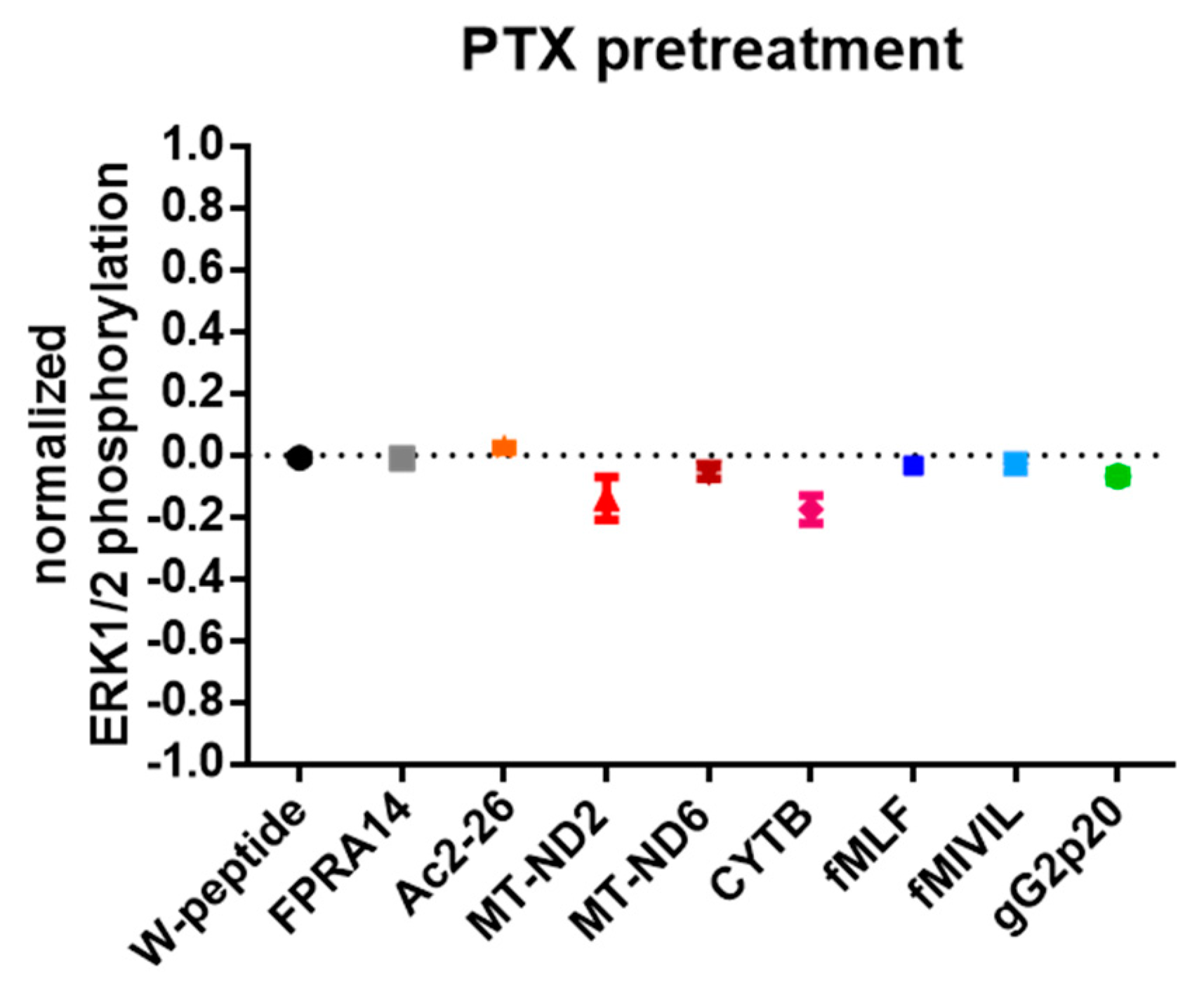
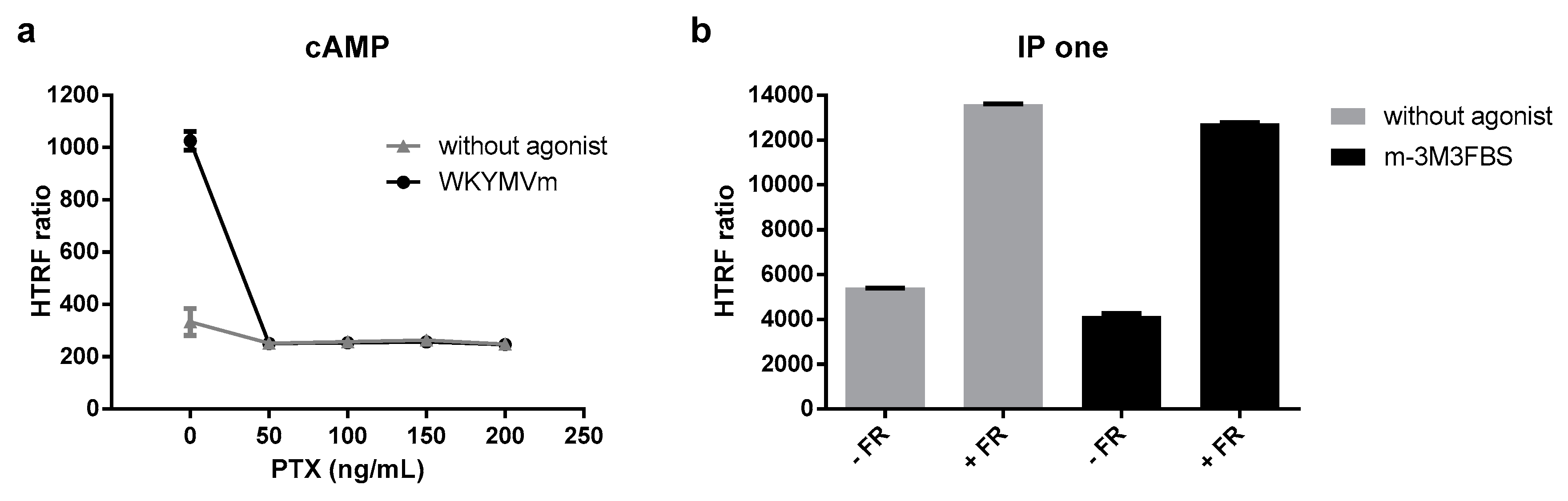
3.4. G protein Dependency of Agonist-Mediated Responses
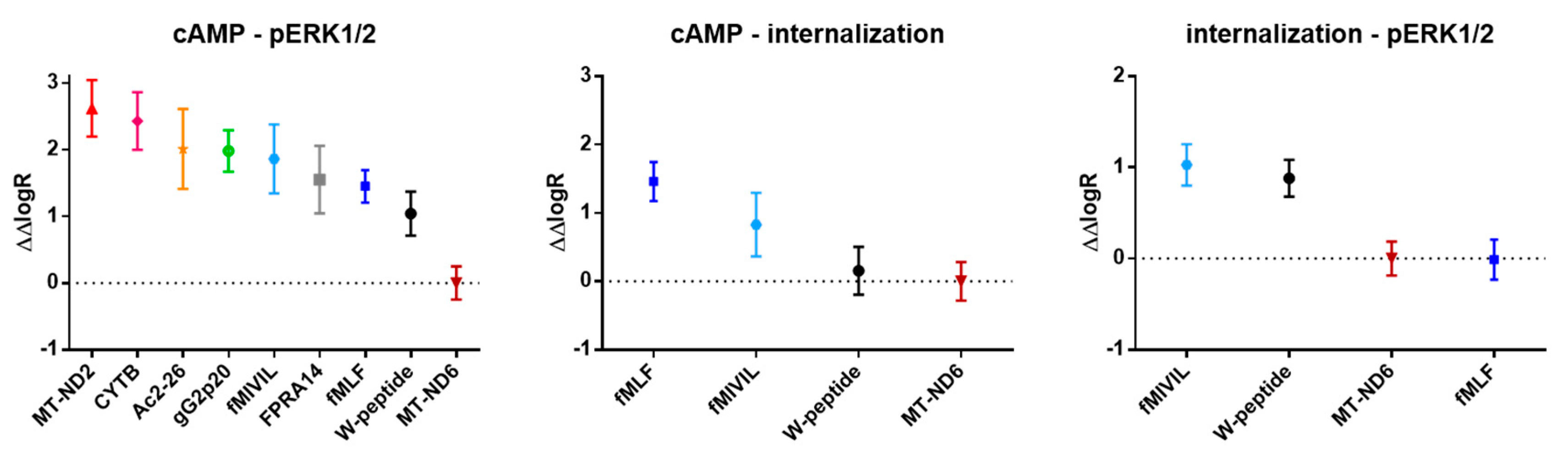
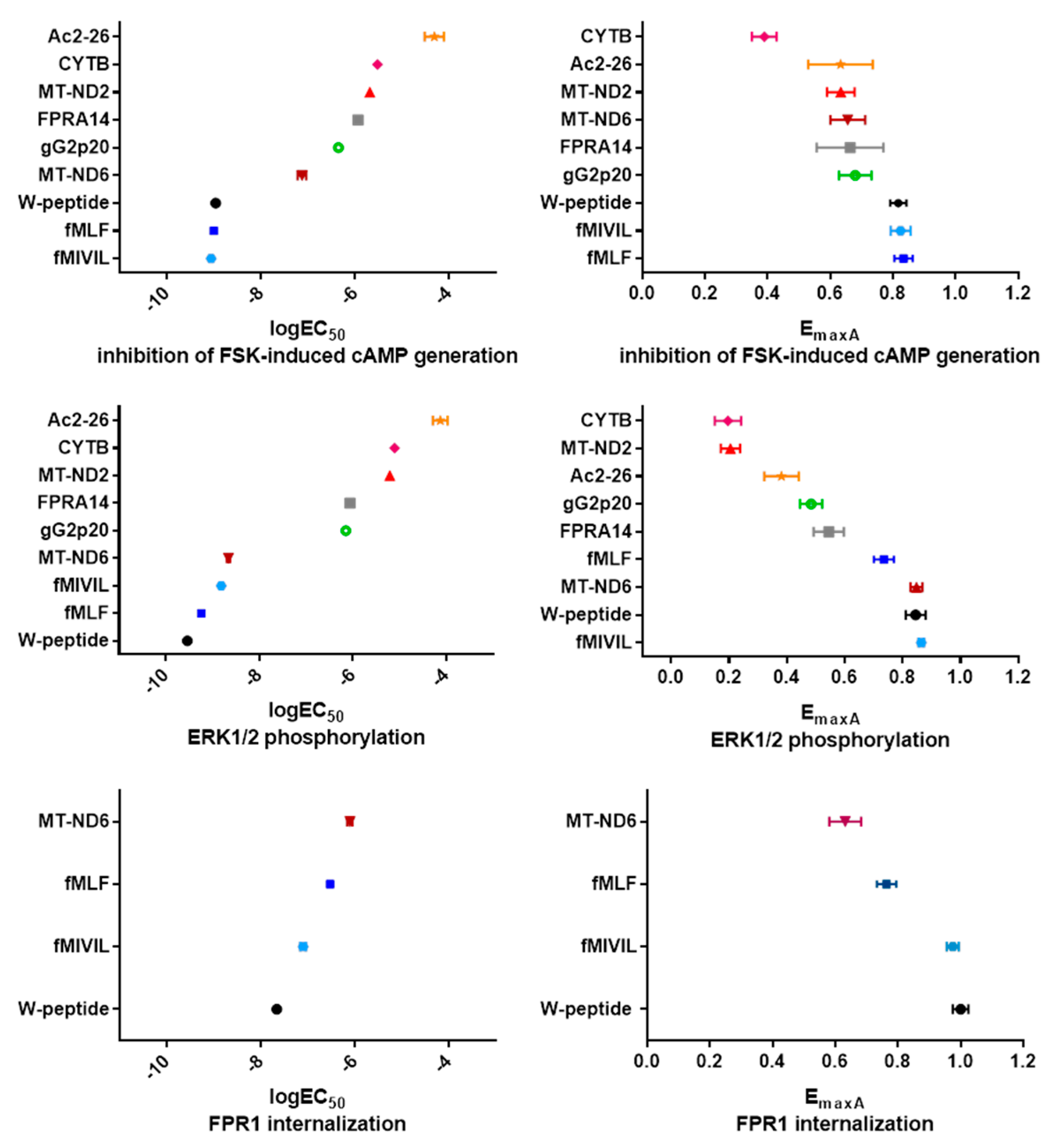
3.5. Analysis of Agonist Bias on FPR1
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Stock Concentration | Solvent | Solubility | M |
|---|---|---|---|---|
| Ac2-26 | 324 µM | PBS | 1 mg/mL | 3089.46 g/mol |
| CYTB | 1.0 mM | DMSO | 2 mg/mL | 1816.26 g/mol |
| fMIVIL | 3.25 mM | DMSO | 2 mg/mL | 615.83 g/mol |
| fMLF | 10 mM | DMSO | 4 mg/mL | 437.60 g/mol |
| FPRA14 | 100 mM | DMSO | 100 mM | 408.92 g/mol |
| gG2p20 | 1.36 mM | DMSO | 2 mg/mL | 1467.62 g/mol |
| MT-ND2 | 2.85 mM | DMSO | 2 mg/mL | 700.79 g/mol |
| MT-ND6 | 2.49 mM | DMSO | 2 mg/mL | 803.00 g/mol |
| WKYMVm | 2.3 mM | H2O | 2 mg/mL | 856.11 g/mol |
| Ligand | cAMP Formation | ERK1/2 Activation | Internalization | ||||
|---|---|---|---|---|---|---|---|
| logEC50 | EmaxA | logEC50 | EmaxA | logEC50 | EmaxA | ||
| synthetic | FPR14 | −5.94 0.19 | 0.66 0.11 | −6.06 0.05 | 0.55 0.05 | NR | NR |
| W-peptide | −8.96 0.15 | 0.82 0.03 | −9.54 0.06 | 0.85 0.04 | −7.66 0.08 | 1.00 0.03 | |
| endogenous | Ac2-26 | −4.31 0.20 | 0.63 0.10 | −4.13 0.16 | 0.38 0.06 | NR | NR |
| mitochondrial | CYTB | −5.52 0.11 | 0.39 0.04 | −5.11 0.24 | 0.20 0.05 | NR | NR |
| MT-ND2 | −5.69 0.13 | 0.63 0.04 | −5.21 0.16 | 0.21 0.03 | NR | NR | |
| MT-ND6 | −7.13 0.09 | 0.66 0.06 | −8.66 0.04 | 0.85 0.02 | −6.11 0.06 | 0.63 0.05 | |
| bacterial | fMIVIL | −9.06 0.24 | 0.82 0.03 | −8.81 0.11 | 0.86 0.01 | −7.10 0.06 | 0.97 0.02 |
| fMLF | −9.00 0.10 | 0.83 0.03 | −9.24 0.04 | 0.74 0.04 | −6.53 0.10 | 0.76 0.03 | |
| viral | gG2p20 | −6.36 0.12 | 0.68 0.05 | −6.15 0.08 | 0.48 0.04 | NR | NR |
References
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calfee, C.S.; Matthay, M.A. Clinical immunology: Culprits with evolutionary ties. Nature 2010, 464, 41–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science (New York N.Y.) 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- Boulay, F.; Tardif, M.; Brouchon, L.; Vignais, P. The human N-formylpeptide receptor. Characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry 1990, 29, 11123–11133. [Google Scholar] [CrossRef]
- Raabe, C.A.; Groper, J.; Rescher, U. Biased perspectives on formyl peptide receptors. Biochim. Et Biophys. Acta. Mol. Cell Res. 2019, 1866, 305–316. [Google Scholar] [CrossRef]
- Rankovic, Z.; Brust, T.F.; Bohn, L.M. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorganic Med. Chem. Lett. 2016, 26, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef] [PubMed]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Watson, U.; Vasudevan, L.; Saini, D.K. ERK Activation Pathways Downstream of GPCRs. Int. Rev. Cell Mol. Biol. 2018, 338, 79–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Han, J.; Hui, L. MAPK signaling in inflammation-associated cancer development. Protein Cell 2010, 1, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Calebiro, D.; Godbole, A. Internalization of G-protein-coupled receptors: Implication in receptor function, physiology and diseases. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Pavlos, N.J.; Friedman, P.A. GPCR Signaling and Trafficking: The Long and Short of It. Trends Endocrinol. Metab. 2017, 28, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Schrage, R.; Schmitz, A.L.; Gaffal, E.; Annala, S.; Kehraus, S.; Wenzel, D.; Bullesbach, K.M.; Bald, T.; Inoue, A.; Shinjo, Y.; et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 2015, 6, 10156. [Google Scholar] [CrossRef] [Green Version]
- Ernst, S.; Zobiack, N.; Boecker, K.; Gerke, V.; Rescher, U. Agonist-induced trafficking of the low-affinity formyl peptide receptor FPRL1. Cell. Mol. Life Sci. 2004, 61, 1684–1692. [Google Scholar] [CrossRef]
- Conche, C.; Boulla, G.; Trautmann, A.; Randriamampita, C. T cell adhesion primes antigen receptor-induced calcium responses through a transient rise in adenosine 3′,5′-cyclic monophosphate. Immunity 2009, 30, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Ruttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef]
- Gao, Z.G.; Jacobson, K.A. On the selectivity of the Galphaq inhibitor UBO-QIC: A comparison with the Galphai inhibitor pertussis toxin. Biochem. Pharmacol. 2016, 107, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression-a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlert, F.J. Analysis of allosterism in functional assays. J. Pharmacol. Exp. Ther. 2005, 315, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Tran, J.A.; Chang, A.; Matsui, M.; Ehlert, F.J. Estimation of relative microscopic affinity constants of agonists for the active state of the receptor in functional studies on M2 and M3 muscarinic receptors. Mol. Pharmacol. 2009, 75, 381–396. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, K.W.; Griffin, M.T.; Ehlert, F.J. Selectivity of agonists for the active state of M1 to M4 muscarinic receptor subtypes. J. Pharmacol. Exp. Ther. 2009, 328, 331–342. [Google Scholar] [CrossRef]
- Griffin, M.T.; Figueroa, K.W.; Liller, S.; Ehlert, F.J. Estimation of agonist activity at G protein-coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J. Pharmacol. Exp. Ther. 2007, 321, 1193–1207. [Google Scholar] [CrossRef] [Green Version]
- Muto, Y.; Guindon, S.; Umemura, T.; Kohidai, L.; Ueda, H. Adaptive evolution of formyl peptide receptors in mammals. J. Mol. Evol. 2015, 80, 130–141. [Google Scholar] [CrossRef]
- Tsu, R.C.; Lai, H.W.; Allen, R.A.; Wong, Y.H. Differential coupling of the formyl peptide receptor to adenylate cyclase and phospholipase C by the pertussis toxin-insensitive Gz protein. Biochem. J. 1995, 309 Pt 1, 331–339. [Google Scholar] [CrossRef]
- Seamon, K.B.; Padgett, W.; Daly, J.W. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3363–3367. [Google Scholar] [CrossRef] [Green Version]
- Marasco, W.A.; Phan, S.H.; Krutzsch, H.; Showell, H.J.; Feltner, D.E.; Nairn, R.; Becker, E.L.; Ward, P.A. Purification and identification of formyl-methionyl-leucyl-phenylalanine as the major peptide neutrophil chemotactic factor produced by Escherichia coli. J. Biol. Chem. 1984, 259, 5430–5439. [Google Scholar] [PubMed]
- Southgate, E.L.; He, R.L.; Gao, J.L.; Murphy, P.M.; Nanamori, M.; Ye, R.D. Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J. Immunol. (Baltim. Md. 1950) 2008, 181, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Kim, Y.; Park, J.C.; Suh, P.G.; Ryu, S.H. The synthetic chemoattractant peptide, Trp-Lys-Tyr-Met-Val-D-Met, enhances monocyte survival via PKC-dependent Akt activation. J. Leukoc. Biol. 2002, 71, 329–338. [Google Scholar] [PubMed]
- Katada, T.; Ui, M. Accelerated turnover of blood glucose in pertussis-sensitized rats due to combined actions of endogenous insulin and adrenergic beta-stimulation. Biochim. Et Biophys. Acta 1976, 421, 57–69. [Google Scholar] [CrossRef]
- Perretti, M.; Ahluwalia, A.; Harris, J.G.; Goulding, N.J.; Flower, R.J. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse. A qualitative comparison with an anti-CD11b monoclonal antibody. J. Immunol. (Baltim. Md. 1950) 1993, 151, 4306–4314. [Google Scholar]
- Bellner, L.; Thoren, F.; Nygren, E.; Liljeqvist, J.A.; Karlsson, A.; Eriksson, K. A proinflammatory peptide from herpes simplex virus type 2 glycoprotein G affects neutrophil, monocyte, and NK cell functions. J. Immunol. (Baltim. Md. 1950) 2005, 174, 2235–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cussell, P.J.G.; Howe, M.S.; Illingworth, T.A.; Gomez Escalada, M.; Milton, N.G.N.; Paterson, A.W.J. The formyl peptide receptor agonist FPRa14 induces differentiation of Neuro2a mouse neuroblastoma cells into multiple distinct morphologies which can be specifically inhibited with FPR antagonists and FPR knockdown using siRNA. PLoS ONE 2019, 14, e0217815. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Quinn, M.T. High-throughput screening for small-molecule activators of neutrophils: Identification of novel N-formyl peptide receptor agonists. Mol. Pharmacol. 2007, 71, 1061–1074. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ilasaca, M. Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP)-kinase cascades. Biochem. Pharmacol. 1998, 56, 269–277. [Google Scholar] [CrossRef]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G protein regulation of MAPK networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef] [Green Version]
- Gutkind, J.S. Regulation of mitogen-activated protein kinase signaling networks by G protein-coupled receptors. Sci. Stke Signal Transduct. Knowl. Environ. 2000, 2000, re1. [Google Scholar] [CrossRef] [PubMed]
- Vines, C.M.; Revankar, C.M.; Maestas, D.C.; LaRusch, L.L.; Cimino, D.F.; Kohout, T.A.; Lefkowitz, R.J.; Prossnitz, E.R. N-formyl peptide receptors internalize but do not recycle in the absence of arrestins. J. Biol. Chem. 2003, 278, 41581–41584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, B.M.; Marjon, N.A.; Prossnitz, E.R. Regulation of N-Formyl Peptide Receptor Signaling and Trafficking by Arrestin-Src Kinase Interaction. PLoS ONE 2016, 11, e0147442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bufe, B.; Zufall, F. The sensing of bacteria: Emerging principles for the detection of signal sequences by formyl peptide receptors. Biomol. Concepts 2016, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Mol. (BaselSwitz.) 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.; Lange, C.; Wilbers, A.; Goebeler, V.; Gerke, V.; Rescher, U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. (Baltim. Md. 1950) 2004, 172, 7669–7676. [Google Scholar] [CrossRef] [Green Version]
- Schloer, S.; Hubel, N.; Masemann, D.; Pajonczyk, D.; Brunotte, L.; Ehrhardt, C.; Brandenburg, L.O.; Ludwig, S.; Gerke, V.; Rescher, U. The annexin A1/FPR2 signaling axis expands alveolar macrophages, limits viral replication, and attenuates pathogenesis in the murine influenza A virus infection model. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 12188–12199. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T. Biased Receptor Signaling in Drug Discovery. Pharmacol. Rev. 2019, 71, 267–315. [Google Scholar] [CrossRef] [Green Version]
- Gesty-Palmer, D.; Luttrell, L.M. Refining efficacy: Exploiting functional selectivity for drug discovery. Adv. Pharmacol. (San DiegoCalif.) 2011, 62, 79–107. [Google Scholar] [CrossRef]
- Qin, C.X.; May, L.T.; Li, R.; Cao, N.; Rosli, S.; Deo, M.; Alexander, A.E.; Horlock, D.; Bourke, J.E.; Yang, Y.H.; et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017, 8, 14232. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Rabiet, M.J.; Huet, E.; Boulay, F. Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol. 2005, 35, 2486–2495. [Google Scholar] [CrossRef] [PubMed]
- Schrage, R.; De Min, A.; Hochheiser, K.; Kostenis, E.; Mohr, K. Superagonism at G protein-coupled receptors and beyond. Br. J. Pharmacol. 2016, 173, 3018–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.G.; DeVree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Eishingdrelo, H.; Kongsamut, S. Minireview: Targeting GPCR Activated ERK Pathways for Drug Discovery. Curr. Chem. Genom. Transl. Med. 2013, 7, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, D.; Shin, J.; Solnica-Krezel, L.; Raz, E. Spatio-temporal regulation of concurrent developmental processes by generic signaling downstream of chemokine receptors. eLife 2018, 7. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes Dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef] [Green Version]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.J.; Milligan, G. Allostery at G protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol. Rev. 2010, 62, 701–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, R.S.; Suzuki, K.G.; Prossnitz, E.R.; Koyama-Honda, I.; Nakada, C.; Fujiwara, T.K.; Kusumi, A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 2011, 192, 463–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gröper, J.; König, G.M.; Kostenis, E.; Gerke, V.; Raabe, C.A.; Rescher, U. Exploring Biased Agonism at FPR1 as a Means to Encode Danger Sensing. Cells 2020, 9, 1054. https://doi.org/10.3390/cells9041054
Gröper J, König GM, Kostenis E, Gerke V, Raabe CA, Rescher U. Exploring Biased Agonism at FPR1 as a Means to Encode Danger Sensing. Cells. 2020; 9(4):1054. https://doi.org/10.3390/cells9041054
Chicago/Turabian StyleGröper, Jieny, Gabriele M. König, Evi Kostenis, Volker Gerke, Carsten A. Raabe, and Ursula Rescher. 2020. "Exploring Biased Agonism at FPR1 as a Means to Encode Danger Sensing" Cells 9, no. 4: 1054. https://doi.org/10.3390/cells9041054
APA StyleGröper, J., König, G. M., Kostenis, E., Gerke, V., Raabe, C. A., & Rescher, U. (2020). Exploring Biased Agonism at FPR1 as a Means to Encode Danger Sensing. Cells, 9(4), 1054. https://doi.org/10.3390/cells9041054