MeCP2 and Chromatin Compartmentalization


Abstract
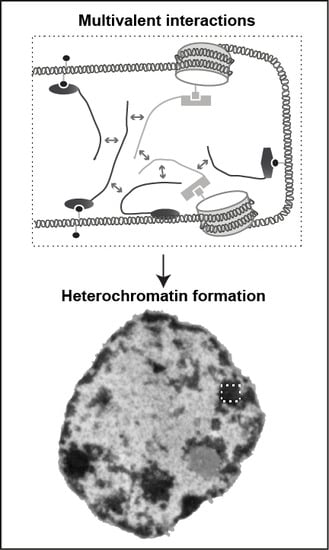
:
1. Introduction
2. MeCP2 Interactions, Modifications and Mutations
2.1. MeCP2 Isoforms and Domains
2.2. MeCP2 DNA Binding
2.3. MeCP2 Protein–Protein Interactions
2.4. MeCP2 Post-Translational Modifications
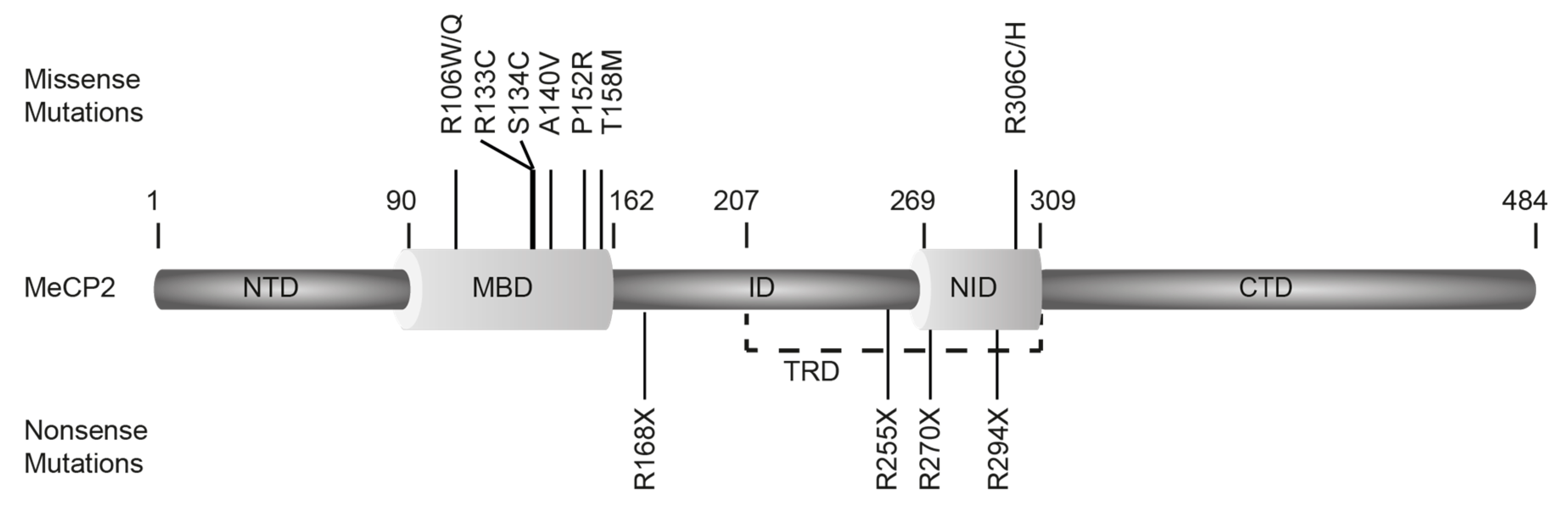
2.5. MeCP2 RTT Mutations
3. MeCP2 in Higher Order Chromatin Compartmentalization
3.1. MeCP2 and Chromatin Looping
3.2. MeCP2 and Heterochromatin Compartmentalization
3.3. Phase Separation and Heterochromatin Condensation
3.4. Model for MeCP2 Function in Chromocenter Clustering
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5caC | 5-carboxycytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| acet | acetylation |
| ATRX | a-thalassemia/mental retardation syndrome X linked |
| C | cytosine |
| CREB | cyclic AMP-responsive element-binding protein |
| CTCF | CCCTC-binding factor |
| CTD | C-terminal domain |
| (di)met | (di)methylation |
| gl | N-acetylglucosamine |
| HDAC | histone deacetylase |
| HP1 | heterochromatin protein 1 |
| ID | intervening domain |
| MBD | methyl-CpG-binding domain |
| MeCP2 | methyl-CpG binding protein 2 |
| NA | nucleosomal array |
| NID | NCoR/SMRT interaction domain |
| NTD | N-terminal domain |
| PAR | poly(ADP-ribosyl)ation |
| phos | phosphorylation |
| PTM | post-translational modification |
| RTT | Rett syndrome |
| TAD | topologically associated domain |
| TET | Ten-eleven translocation |
| TRD | transcriptional repression domain |
| ubi | ubiquitination |
| YB-1 | Y box-binding protein 1 |
References
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.; Hastert, F.D.; Cardoso, M.C. DNA Modification Readers and Writers and Their Interplay. J. Mol. Biol. 2019, 432, 1731–1746. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE 2014, 9, e90645. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.-m.; Feldman, D.A.; Bateup, H.S.; Gao, Q. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Fichou, Y.; Nectoux, J.; Bahi-Buisson, N.; Rosas-Vargas, H.; Girard, B.; Chelly, J.; Bienvenu, T. The first missense mutation causing Rett syndrome specifically affecting the MeCP2_e1 isoform. Neurogenetics 2009, 10, 127. [Google Scholar] [CrossRef]
- Saunders, C.J.; Minassian, B.E.; Chow, E.W.; Zhao, W.; Vincent, J.B. Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. Am. J. Med. Genet. Part A 2009, 149, 1019–1023. [Google Scholar] [CrossRef]
- Sheikh, T.I.; de Paz, A.M.; Akhtar, S.; Ausió, J.; Vincent, J.B. MeCP2_E1 N-terminal modifications affect its degradation rate and are disrupted by the Ala2Val Rett mutation. Hum. Mol. Genet. 2017, 26, 4132–4141. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, P.A.; Glass, C.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokura, K.; Kaul, S.C.; Wadhwa, R.; Nomura, T.; Khan, M.M.; Shinagawa, T.; Yasukawa, T.; Colmenares, C.; Ishii, S. The Ski protein family is required for MeCP2-mediated transcriptional repression. J. Biol. Chem. 2001, 276, 34115–34121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Yamada, T.; Kihara-Negishi, F.; Sakurai, T.; Oikawa, T. Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU.1-mediated transcriptional repression. Oncogene 2003, 22, 8688–8698. [Google Scholar] [CrossRef] [Green Version]
- Forlani, G.; Giarda, E.; Ala, U.; Di Cunto, F.; Salani, M.; Tupler, R.; Kilstrup-Nielsen, C.; Landsberger, N. The MeCP2/YY1 interaction regulates ANT1 expression at 4q35: Novel hints for Rett syndrome pathogenesis. Hum. Mol. Genet. 2010, 19, 3114–3123. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [Green Version]
- Leoh, L.S.; van Heertum, B.; De Rijck, J.; Filippova, M.; Rios-Colon, L.; Basu, A.; Martinez, S.R.; Tungteakkhun, S.S.; Filippov, V.; Christ, F.; et al. The stress oncoprotein LEDGF/p75 interacts with the methyl CpG binding protein MeCP2 and influences its transcriptional activity. Mol. Cancer Res. 2012, 10, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, M.L.; Adams, S.; Dunaway, K.W.; LaSalle, J.M. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 2012, 32, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.S.; Meehan, R.R.; Bird, A. Dissection of the Methyl-Cpg Binding Domain from the Chromosomal Protein Mecp2. Nucleic Acids Res. 1993, 21, 4886–4892. [Google Scholar] [CrossRef] [Green Version]
- Meehan, R.R.; Lewis, J.D.; Bird, A.P. Characterization of Mecp2, a Vertebrate DNA-Binding Protein with Affinity for Methylated DNA. Nucleic Acids Res. 1992, 20, 5085–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Woodcock, C.L. Unique Physical Properties and Interactions of the Domains of Methylated DNA Binding Protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitina, T.; Shi, X.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Woodcock, C.L. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 2007, 27, 864–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyst, M.J.; Connelly, J.; Merusi, C.; Bird, A. Sequence-specific DNA binding by AT-hook motifs in Me CP 2. FEBS Lett. 2016, 590, 2927–2933. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, R.I.; Smith, B.O.; Nan, X.; Free, A.; Soteriou, A.; Uhrin, D.; Bird, A.P.; Barlow, P.N. The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol. 1999, 291, 1055–1065. [Google Scholar] [CrossRef]
- Ho, K.L.; Mcnae, L.W.; Schmiedeberg, L.; Klose, R.J.; Bird, A.P.; Walkinshaw, M.D. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell 2008, 29, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.; Becker, A.; Jost, K.L.; Haase, S.; Thakur, B.K.; Brero, A.; Hardt, T.; Kudo, S.; Leonhardt, H.; Cardoso, M.C. MeCP2 Rett mutations affect large scale chromatin organization. Hum. Mol. Genet. 2011, 20, 4187–4195. [Google Scholar] [CrossRef] [Green Version]
- Casas-Delucchi, C.S.; Becker, A.; Bolius, J.J.; Cardoso, M.C. Targeted manipulation of heterochromatin rescues MeCP2 Rett mutants and re-establishes higher order chromatin organization. Nucleic Acids Res. 2012, 40, e176. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.C.; Wexler, B.B.; Rogers, D.J.; Hite, K.C.; Panchenko, T.; Ajith, S.; Black, B.E. DNA Binding Restricts the Intrinsic Conformational Flexibility of Methyl CpG Binding Protein 2 (MeCP2). J. Biol. Chem. 2011, 286, 18938–18948. [Google Scholar] [CrossRef] [Green Version]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; LaSalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, V.H.; McBryant, S.J.; Wade, P.A.; Woodcock, C.L.; Hansen, J.C. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J. Biol. Chem. 2007, 282, 15057–15064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagger, S.; Connelly, J.C.; Schweikert, G.; Webb, S.; Selfridge, J.; Ramsahoye, B.H.; Yu, M.; He, C.; Sanguinetti, G.; Sowers, L.C.; et al. MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet. 2017, 13, e1006793. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renthal, W.; Boxer, L.D.; Hrvatin, S.; Li, E.; Silberfeld, A.; Nagy, M.A.; Griffith, E.C.; Vierbuchen, T.; Greenberg, M.E. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018, 21, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.T.C.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic Readers for 5-(Hydroxy)Methylcytosine and Its Oxidized Derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, A.K.; Zhang, P.; Cardoso, M.C. Modifiers and Readers of DNA Modifications and Their Impact on Genome Structure, Expression, and Stability in Disease. Front. Genet. 2016, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Frauer, C.; Hoffmann, T.; Bultmann, S.; Casa, V.; Cardoso, M.C.; Antes, I.; Leonhardt, H. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS ONE 2011, 6, e21306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, K.; Lavery, L.A.; Baker, S.A.; Shaw, C.A.; Li, W.; Zoghbi, H.Y. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 5509–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, A.K.; Zhang, P.; Hastert, F.D.; Meyer, S.; Rausch, C.; Herce, H.D.; Muller, U.; Lehmkuhl, A.; Hellmann, I.; Trummer, C.; et al. Binding of MBD proteins to DNA blocks Tet1 function thereby modulating transcriptional noise. Nucleic Acids Res. 2017, 45, 2438–2457. [Google Scholar] [CrossRef] [Green Version]
- Muotri, A.R.; Marchetto, M.C.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef]
- Yu, F.; Zingler, N.; Schumann, G.; Stratling, W.H. Methyl-CpG-binding protein 2 represses LINE-1 expression and retrotransposition but not Alu transcription. Nucleic Acids Res. 2001, 29, 4493–4501. [Google Scholar] [CrossRef]
- Zhang, P.; Ludwig, A.K.; Hastert, F.D.; Rausch, C.; Lehmkuhl, A.; Hellmann, I.; Smets, M.; Leonhardt, H.; Cardoso, M.C. L1 retrotransposition is activated by Ten-eleven-translocation protein 1 and repressed by methyl-CpG binding proteins. Nucleus 2017, 8, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [Green Version]
- Lachner, M.; O’Carroll, N.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Agarwal, N.; Hardt, T.; Brero, A.; Nowak, D.; Rothbauer, U.; Becker, A.; Leonhardt, H.; Cardoso, M.C. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 2007, 35, 5402–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Sonoda, M. Self-interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem. Biophys. Res. Commun. 2003, 301, 287–292. [Google Scholar] [CrossRef]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Allmann, L.; Hofstatter, M.; Casa, V.; Weber, P.; Lehmkuhl, A.; Herce, H.D.; Cardoso, M.C. Direct homo- and hetero-interactions of MeCP2 and MBD2. PLoS ONE 2013, 8, e53730. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.T.; Chan, D.C.; Leder, P. FBP WW domains and the Abl SH3 domain bind to a specific class of proline-rich ligands. EMBO J. 1997, 16, 2376–2383. [Google Scholar] [CrossRef]
- Buschdorf, J.P.; Stratling, W.H. A WW domain binding region in methyl-CpG-binding protein MeCP2: Impact on Rett syndrome. J. Mol. Med. 2004, 82, 135–143. [Google Scholar] [CrossRef]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krithivas, A.; Fujimuro, M.; Weidner, M.; Young, D.B.; Hayward, S.D. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 2002, 76, 11596–11604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Jiang, Y.; Tremblay, D.C.; Bonvissuto, A.C.; Eubanks, J.H.; Mann, M.R.; Berube, N.G. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 2010, 18, 191–202. [Google Scholar] [CrossRef]
- Harikrishnan, K.N.; Chow, M.Z.; Baker, E.K.; Pal, S.; Bassal, S.; Brasacchio, D.; Wang, L.; Craig, J.M.; Jones, P.L.; Sif, S.; et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 2005, 37, 254–264. [Google Scholar] [CrossRef]
- Long, S.W.; Ooi, J.Y.; Yau, P.M.; Jones, P.L. A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci. Rep. 2011, 31, 333–343. [Google Scholar] [CrossRef]
- Jeffery, L.; Nakielny, S. Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 2004, 279, 49479–49487. [Google Scholar] [CrossRef] [Green Version]
- Bracaglia, G.; Conca, B.; Bergo, A.; Rusconi, L.; Zhou, Z.; Greenberg, M.E.; Landsberger, N.; Soddu, S.; Kilstrup-Nielsen, C. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009, 10, 1327–1333. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, L.M.; Zaghlula, M.; Sztainberg, Y.; Baker, S.A.; Klisch, T.J.; Tang, A.A.; Huang, E.J.; Zoghbi, H.Y. An RNA interference screen identifies druggable regulators of MeCP2 stability. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Zhang, P.; Allmann, L.; Meilinger, D.; Bertulat, B.; Eck, D.; Hofstaetter, M.; Bartolomei, G.; Hottiger, M.O.; Schreiber, V. Poly (ADP-ribosyl) ation of methyl CpG binding domain protein 2 regulates chromatin structure. J. Biol. Chem. 2016, 291, 4873–4881. [Google Scholar] [CrossRef] [Green Version]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Franco, B.; Rosner, M.R. CDKL5/Stk9 kinase inactivation is associated with neuronal developmental disorders. Hum. Mol. Genet. 2005, 14, 3775–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of Missing Proteins in the neXtProt Database and Unregistered Phosphopeptides in the PhosphoSitePlus Database as Part of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef]
- Sharma, K.; D’Souza, R.C.J.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Zanivan, S.; Gnad, F.; Wickstrom, S.A.; Geiger, T.; Macek, B.; Cox, J.; Fassler, R.; Mann, M. Solid tumor proteome and phosphoproteome analysis by high resolution mass spectrometry. J. Proteome Res. 2008, 7, 5314–5326. [Google Scholar] [CrossRef] [PubMed]
- Tweedie-Cullen, R.Y.; Reck, J.M.; Mansuy, I.M. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J. Proteome Res. 2009, 8, 4966–4982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.F.; Hu, K.P.; Chang, Q.; Wu, H.; Sherman, N.E.; Martinowich, K.; Klose, R.J.; Schanen, C.; Jaenisch, R.; Wang, W.D.; et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. USA 2009, 106, 4882–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, D.H.; Gabel, H.W.; Robinson, N.D.; Kastan, N.R.; Hu, L.S.; Cohen, S.; Navarro, A.J.; Lyst, M.J.; Ekiert, R.; Bird, A.P. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 2013, 499, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, V.; Guo, A.; Trudgian, D.; Thomas, B.; Acuto, O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015, 6, 6758. [Google Scholar] [CrossRef]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 2011, 4, rs5. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.Y.; Li, Y.; Wang, Y.; Chen, Y.; Zhao, Y.; Qin, J. Complications in the assignment of 14 and 28 Da mass shift detected by mass spectrometry as in vivo methylation from endogenous proteins. Anal. Chem. 2008, 80, 1721–1729. [Google Scholar] [CrossRef]
- Bergo, A.; Strollo, M.; Gai, M.; Barbiero, I.; Stefanelli, G.; Sertic, S.; Cobolli Gigli, C.; Di Cunto, F.; Kilstrup-Nielsen, C.; Landsberger, N. Methyl-CpG binding protein 2 (MeCP2) localizes at the centrosome and is required for proper mitotic spindle organization. J. Biol. Chem. 2015, 290, 3223–3237. [Google Scholar] [CrossRef] [Green Version]
- D’Annessa, I.; Gandaglia, A.; Brivio, E.; Stefanelli, G.; Frasca, A.; Landsberger, N.; Di Marino, D. Tyr120Asp mutation alters domain flexibility and dynamics of MeCP2 DNA binding domain leading to impaired DNA interaction: Atomistic characterization of a Rett syndrome causing mutation. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1180–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.L.; Gu, H.B.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.Y.; et al. Immunoaffinity Enrichment and Mass Spectrometry Analysis of Protein Methylation. Mol. Cell Proteom. 2014, 13, 372–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 2016, 9, rs9. [Google Scholar] [CrossRef] [PubMed]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanelli, G.; Gandaglia, A.; Costa, M.; Cheema, M.S.; Di Marino, D.; Barbiero, I.; Kilstrup-Nielsen, C.; Ausio, J.; Landsberger, N. Brain phosphorylation of MeCP2 at serine 164 is developmentally regulated and globally alters its chromatin association. Sci. Rep. 2016, 6, 28295. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Zhai, B.; Yu, Y.; Kiyotsugu, Y.; Raschle, T.; Etzkorn, M.; Seo, H.C.; Nagiec, M.; Luna, R.E.; Reinherz, E.L.; et al. Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2182–E2190. [Google Scholar] [CrossRef] [Green Version]
- Carrier, M.; Joint, M.; Lutzing, R.; Page, A.; Rochette-Egly, C. Phosphoproteome and Transcriptome of RA-Responsive and RA-Resistant Breast Cancer Cell Lines. PLoS ONE 2016, 11, e0157290. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Huang, M.; Zhu, Y.; Xin, Y.J.; Zhao, Y.K.; Huang, J.; Yu, J.X.; Zhou, W.H.; Qiu, Z.L. SUMOylation of MeCP2 is essential for transcriptional repression and hippocampal synapse development. J. Neurochem. 2014, 128, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.Q.; Yang, Q.K.; Lu, B.W.; Yi, W.; Cantin, G.; Chen, Y.L.; Fearns, C.; Yates, J.R., 3rd; Lee, J.D. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009, 69, 2663–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Cheng, Z.; Sun, M.; Wan, X.; Liu, P.; He, T.; Tan, M.; Zhao, Y. A chemical proteomics approach for global analysis of lysine monomethylome profiling. Mol. Cell. Proteom. 2015, 14, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Ni, J.J.; Huang, J.J.; Kou, Z.W.; Sun, F.Y. VEGF overexpression enhances the accumulation of phospho-S292 MeCP2 in reactive astrocytes in the adult rat striatum following cerebral ischemia. Brain Res. 2015, 1599, 32–43. [Google Scholar] [CrossRef]
- Parker, B.L.; Yang, G.; Humphrey, S.J.; Chaudhuri, R.; Ma, X.; Peterman, S.; James, D.E. Targeted phosphoproteomics of insulin signaling using data-independent acquisition mass spectrometry. Sci. Signal. 2015, 8, rs6. [Google Scholar] [CrossRef]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [Green Version]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine Succinylation Is a Frequently Occurring Modification in Prokaryotes and Eukaryotes and Extensively Overlaps with Acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem. Biol. 2011, 18, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X.; et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Stenoien, D.L.; Strittmatter, E.F.; Wang, J.; Ding, L.; Lipton, M.S.; Monroe, M.E.; Nicora, C.D.; Gristenko, M.A.; Tang, K.; et al. Phosphoproteome profiling of human skin fibroblast cells in response to low- and high-dose irradiation. J. Proteome Res. 2006, 5, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Grimsrud, P.A.; Carson, J.J.; Hebert, A.S.; Hubler, S.L.; Niemi, N.M.; Bailey, D.J.; Jochem, A.; Stapleton, D.S.; Keller, M.P.; Westphall, M.S.; et al. A Quantitative Map of the Liver Mitochondrial Phosphoproteome Reveals Posttranslational Control of Ketogenesis. Cell Metab. 2012, 16, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.V.; Rodriguiz, R.M.; Hutchinson, A.N.; Kim, I.H.; Wetsel, W.C.; West, A.E. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat. Neurosci. 2010, 13, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, X.; Chau, K.F.; Williams, E.C.; Chang, Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat. Neurosci. 2011, 14, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Udeshi, N.D.; O’Malley, M.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteom. 2010, 9, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [Green Version]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteom. 2012, 11, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteom. 2013, 91, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Feodorova, Y.; Guy, J.; Peichl, L.; Jost, K.L.; Kimura, H.; Cardoso, M.C.; Bird, A.; Leonhardt, H.; Joffe, B.; et al. DNA methylation reader MECP2: Cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin 2014, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.L.; et al. Genome-Wide Activity-Dependent MeCP2 Phosphorylation Regulates Nervous System Development and Function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inui, K.; Akagi, M.; Ono, J.; Tsukamoto, H.; Shimono, K.; Mano, T.; Imai, K.; Yamada, M.; Muramatsu, T.; Sakai, N.; et al. Mutational analysis of MECP2 in Japanese patients with atypical Rett syndrome. Brain Dev. 2001, 23, 212–215. [Google Scholar] [CrossRef]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A Progressive Syndrome of Autism, Dementia, Ataxia, and Loss of Purposeful Hand Use in Girls—Retts Syndrome—Report of 35 Cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Yusufzai, T.M.; Wolffe, A.P. Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 2000, 39, 7100–7106. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding analysis of methyl-CpG binding domain of MeCP2 and Rett syndrome mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef]
- Brown, K.; Selfridge, J.; Lagger, S.; Connelly, J.; De Sousa, D.; Kerr, A.; Webb, S.; Guy, J.; Merusi, C.; Koerner, M.V. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum. Mol. Genet. 2015, 25, 558–570. [Google Scholar] [CrossRef]
- Venkateswaran, S.; McMillan, H.J.; Doja, A.; Humphreys, P. Adolescent onset cognitive regression and neuropsychiatric symptoms associated with the A140V MECP 2 mutation. Dev. Med. Child Neurol. 2014, 56, 91–94. [Google Scholar] [CrossRef] [Green Version]
- Jentarra, G.M.; Olfers, S.L.; Rice, S.G.; Srivastava, N.; Homanics, G.E.; Blue, M.; Naidu, S.; Narayanan, V. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.Y.; Wu, C.; Jin, Y.; Gao, M.; Li, G.H.; Turner, D.; Shen, J.X.; Zhang, S.J.; Narayanan, V.; Jentarra, G. Electrophysiological Phenotypes of Me CP 2 A140V Mutant Mouse Model. CNS Neurosci. Ther. 2014, 20, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Sampathkumar, R.; Shannon, O.; Brittany, G.; Hilbert, A.; Sean, S.; Vinodh, N. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Research 2016, 5, 2269. [Google Scholar] [CrossRef]
- Lundvall, M.; Samuelsson, L.; Kyllerman, M. Male Rett phenotypes in T158M and R294X MeCP2-mutations. Neuropediatrics 2006, 37, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Sher, A.A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M. MECP2 Mutation Interrupts Nucleolin–mTOR–P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, Q.; Wang, A.; Hamzah, H.; Waldman, A.; Jiang, K.; Dong, Q.; Li, R.; Kim, J.; Turner, D.; Chang, Q. CREB signaling is involved in Rett syndrome pathogenesis. J. Neurosci. 2017, 37, 3671–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapleau, C.A.; Calfa, G.D.; Lane, M.C.; Albertson, A.J.; Larimore, J.L.; Kudo, S.; Armstrong, D.L.; Percy, A.K.; Pozzo-Miller, L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol. Dis. 2009, 35, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Lawson-Yuen, A.; Liu, D.; Han, L.; Jiang, Z.I.; Tsai, G.E.; Basu, A.C.; Picker, J.; Feng, J.; Coyle, J.T. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007, 1180, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schaevitz, L.; Gomez, N.; Zhen, D.; Berger-Sweeney, J. MeCP2 R168X male and female mutant mice exhibit Rett-like behavioral deficits. Genes Brain Behav. 2013, 12, 732–740. [Google Scholar] [CrossRef]
- Bissonnette, J.M.; Schaevitz, L.R.; Knopp, S.J.; Zhou, Z. Respiratory phenotypes are distinctly affected in mice with common Rett syndrome mutations MeCP2 T158A and R168X. Neuroscience 2014, 267, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Georgel, P.T.; Horowitz-Scherer, R.A.; Adkins, N.; Woodcock, C.L.; Wade, P.A.; Hansen, J.C. Chromatin compaction by human MeCP2 assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 2003, 278, 32181–32188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusufzai, T.M.; Wolffe, A.P. Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res. 2000, 28, 4172–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitcher, M.R.; Herrera, J.A.; Buffington, S.A.; Kochukov, M.Y.; Merritt, J.K.; Fisher, A.R.; Schanen, N.C.; Costa-Mattioli, M.; Neul, J.L. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum. Mol. Genet. 2015, 24, 2662–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villard, L. MECP2 mutations in males. J. Med. Genet. 2007, 44, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.A.; Chen, L.; Wilkins, A.D.; Yu, P.; Lichtarge, O.; Zoghbi, H.Y. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 2013, 152, 984–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, D.; Allen, M.; Zhang, L.; Amorim, M.; Wang, I.-T.J.; Reyes, A.-R.S.; Mercado-Berton, A.; Ong, C.; Cohen, S.; Hu, L. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2012, 15, 274. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, R.; Bird, A. The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. 2019, 432, 1602–1623. [Google Scholar] [CrossRef]
- Heckman, L.D.; Chahrour, M.H.; Zoghbi, H.Y. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 2014, 3, e02676. [Google Scholar] [CrossRef]
- Ravn, K.; Nielsen, J.; Uldall, P.; Hansen, F.; Schwartz, M. No correlation between phenotype and genotype in boys with a truncating MECP2 mutation. J. Med. Genet. 2003, 40, e5. [Google Scholar] [CrossRef] [Green Version]
- Galvao, T.C.; Thomas, J.O. Structure-specific binding of MeCP2 to four-way junction DNA through its methyl CpG-binding domain. Nucleic Acids Res. 2005, 33, 6603–6609. [Google Scholar] [CrossRef] [Green Version]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanborn, A.L.; Rao, S.S.; Huang, S.-C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Vernimmen, D.; Gloor, G.B.; Berube, N.G. Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 2014, 42, 8356–8368. [Google Scholar] [CrossRef] [PubMed]
- Murrell, A.; Heeson, S.; Reik, W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nat. Genet. 2004, 36, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Wendt, K.S.; Ito, Y.; Huddleston, J.E.; Uribe-Lewis, S.; Woodfine, K.; Krueger, C.; Reik, W.; Peters, J.-M.; Murrell, A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009, 5, e1000739. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.Z.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sandstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Witcher, M.; Emerson, B.M. Epigenetic Silencing of the p16(INK4a) Tumor Suppressor Is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Soto-Reyes, E.; Recillas-Targa, F. Epigenetic regulation of the human p53 gene promoter by the CTCF transcription factor in transformed cell lines. Oncogene 2010, 29, 2217–2227. [Google Scholar] [CrossRef] [Green Version]
- Marina, R.J.; Sturgill, D.; Bailly, M.A.; Thenoz, M.; Varma, G.; Prigge, M.F.; Nanan, K.K.; Shukla, S.; Haque, N.; Oberdoerffer, S. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. EMBO J. 2016, 35, 335–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanan, K.K.; Sturgill, D.M.; Prigge, M.F.; Thenoz, M.; Dillman, A.A.; Mandler, M.D.; Oberdoerffer, S. TET-catalyzed 5-carboxylcytosine promotes CTCF binding to suboptimal sequences genome-wide. iScience 2019, 19, 326–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barutcu, A.R.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Imbalzano, A.N. The connection between BRG1, CTCF and topoisomerases at TAD boundaries. Nucleus 2017, 8, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Pearson, E.C.; Bates, D.L.; Prospero, T.D.; Thomas, J.O. Neuronal nuclei and glial nuclei from mammalian cerebral cortex: Nucleosome repeat lengths, DNA contents and H1 contents. Eur. J. Biochem. 1984, 144, 353–360. [Google Scholar] [CrossRef]
- Ghosh, R.P.; Horowitz-Scherer, R.A.; Nikitina, T.; Shlyakhtenko, L.S.; Woodcock, C.L. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol. Cell. Biol. 2010, 30, 4656–4670. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kamboj, S.; Malone, B.M.; Kudo, S.; Twiss, J.L.; Czymmek, K.J.; LaSalle, J.M.; Schanen, N.C. Analysis of protein domains and Rett syndrome mutations indicate that multiple regions influence chromatin-binding dynamics of the chromatin-associated protein MECP2 in vivo. J. Cell Sci. 2008, 121, 1128–1137. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T.; Gunjan, A.; Hock, R.; Bustin, M.; Brown, D.T. Dynamic binding of histone H1 to chromatin in living cells. Nature 2000, 408, 877. [Google Scholar] [CrossRef]
- Nan, X.; Campoy, F.J.; Bird, A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, F.; Li, Z.; Cao, Q.; Huang, L.; Chen, S. MeCP2-mediated epigenetic regulation in senescent endothelial progenitor cells. Stem Cell Res. Ther. 2018, 9, 87. [Google Scholar] [CrossRef]
- Pandey, S.; Simmons, G.E., Jr.; Malyarchuk, S.; Calhoun, T.N.; Pruitt, K. A novel MeCP2 acetylation site regulates interaction with ATRX and HDAC1. Genes Cancer 2015, 6, 408–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.R. The mammalian centromere: Its molecular architecture. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 1996, 372, 153–162. [Google Scholar] [CrossRef]
- Baccarini, P. Sulle Cinesi Vegetative del Cynomorium coccineum L. N Giorn Bot. Ital. N Ser. 1908, 15, 189–203. [Google Scholar]
- Brero, A.; Easwaran, H.P.; Nowak, D.; Grunewald, I.; Cremer, T.; Leonhardt, H.; Cardoso, M.C. Methyl CpG–binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 2005, 169, 733–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertulat, B.; De Bonis, M.L.; Della Ragione, F.; Lehmkuhl, A.; Milden, M.; Storm, C.; Jost, K.L.; Scala, S.; Hendrich, B.; D’Esposito, M. MeCP2 dependent heterochromatin reorganization during neural differentiation of a novel Mecp2-deficient embryonic stem cell reporter line. PLoS ONE 2012, 7, e47848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitina, T.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Grigoryev, S.A.; Woodcock, C.L. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 2007, 282, 28237–28245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protter, D.S.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Brown, J. Nucleoli: Composition, function, and dynamics. Plant Physiol. 2012, 158, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Jülicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L. Protein phase separation: A new phase in cell biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Dubochet, J.; Adrian, M.; Schultz, P.; Oudet, P. Cryo-electron microscopy of vitrified SV40 minichromosomes: The liquid drop model. EMBO J. 1986, 5, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Rogge, R.; Tamura, S.; Joti, Y.; Hikima, T.; Szerlong, H.; Krause, C.; Herman, J.; Seidel, E.; DeLuca, J. Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. 2016, 35, 1115–1132. [Google Scholar] [CrossRef]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of chromatin by intrinsic and regulated phase separation. Cell 2019, 179, 470–484.e421. [Google Scholar] [CrossRef]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase separation drives heterochromatin domain formation. Nature 2017, 547, 241245. [Google Scholar] [CrossRef]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef] [Green Version]

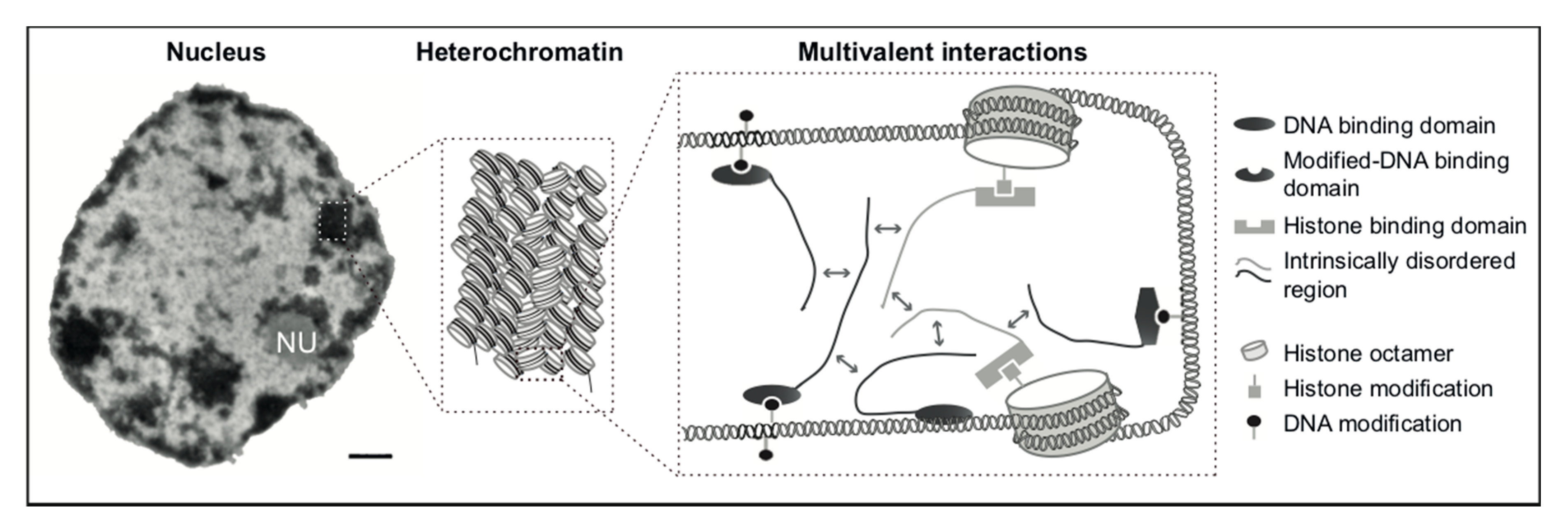

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interactor | MeCP2 Function Upon Interaction | References | |
|---|---|---|---|
| Transcriptional repression | HP.1 | repression, formation of subcellular silencing compartments | Agarwal et al., 2007 [51] |
| PU.1 | formation of repression complex, possibly recruitment of mSin3A-HDAC | Suzuki et al., 2003 [15] | |
| Dnmt1 | association with MeCP2 contributes to maintenance methylation | Kimura & Shiota 2003 [53] | |
| LANA | MeCP2 directs LANA to chromocenters, might contribute to LANA-mediated repression | Matsumura et al., 2010, Krithivas et al., 2002 [62,63] | |
| ATRX | targeting to heterochromatic regions in mature neurons, silencing of imprinted genes; possibly control of nucleosome positioning | Nan et al., 2007, Kernohan et al., 2010 [57,64] | |
| Sin3A | transcriptional repression, corepression complex with HDAC and MeCP2 | Nan et al., 1998, Jones et al., 1998 [11,12] | |
| YY1 | cooperation in repression | Forlani et al., 2010 [16] | |
| c-Ski | transcriptional repression | Kokura et al., 2001 [14] | |
| MBD2 | heterointeractions, might increase heterochromatin clustering | Becker et al., 2013 [56] | |
| MeCP2 | homointeractions, might increase heterochromatin clustering | Becker et al., 2013 [56] | |
| N-CoR | recruitment of N-CoR/SMRT to methylated DNA, bridge function of MeCP2 | Kokura et al., 2001, Lyst et al., 2013 [14,20] | |
| Brahma | transcriptional repression | Harikrishnan et al., 2005 [65] | |
| CoREST | transcriptional repression possibly involving REST, CoREST, MeCP2, SUV39H1 and HP1 | Lunyak et al., 2002 [13] | |
| CREB | transcriptional activation | Chahrour et al., 2008 [17] | |
| LEDGF/p75 | might differentially influence gene activation | Leoh et al., 2012 [18] | |
| SMC1, SMC3 | interaction with MeCP2, ATRX, might promote repression by loop formation | Kernohan et al., 2010, Gonzales et al., 2012 [19,64] | |
| RNA interaction | Prpf3 | RNA binding, possibly involved in splicing | Long et al., 2011 [66] |
| mRNA, siRNA | not known | Jeffrey et al., 2004 [67] | |
| YB-1 | RNA-dependent complex, regulation of splicing | Young et al., 2005 [60] | |
| Sdccag1 | not known | Long et al., 2011 [66] | |
| FBP11 | not known | Buschdorf & Stratling 2004, Bedford et al., 1997 [58,59] | |
| HYPC | not known | Buschdorf & Stratling 2004 [59] | |
| post-translational modifiers | H3K9 MT | targeting of histone methylation to methylated DNA | Fuks et al., 2003, Lunyak et al., 2002 [13,48] |
| SUV39H1 | association with MeCP2 might contribute to silencing by methylation of H3K9, creating HP1 binding sites | Lunyak et al., 2002 [13] | |
| HDAC 1/2 | histone deacetylases form corepression complex with MeCP2 and Sin3A | Nan et al., 1998, Jones et al., 1998 [11,12] | |
| HIPK2, HIPK1 | kinases might phosphorylate MeCP2 on S80 and S216 | Bracaglia et al., 2009, Lombardi et al., 2017 [68,69] | |
| PARP | poly(ADP-ribosyl)ation reduces MeCP2 heterochromatin clustering ability | Becker et al., 2016 [70] | |
| CDKL5 | association in vitro, phosphorylation of MeCP2 by CDKL5 unclear (opposing results in the two publications) | Mari et al., 2005, Lin et al., 2005 [71,72] |
| Residue* | Modification | Species | MS/Other Methods | References** | |
|---|---|---|---|---|---|
| NTD | K12 | ubi | human | x/- | Gonzales et al., 2012 [19] |
| S13 | phos | human, mouse | x/- | Gonzales et al., 2012, Humphrey et al., 2013, Shiromizu et al., 2013 [19,74,75] | |
| S53 | phos | human | x/- | Shiromizu et al., 2013, Bian et al., 2014, Sharma et al., 2014 [75,76,77] | |
| S68 | phos | mouse | x/- | Huttlin et al., 2010 [78] | |
| S70 | phos | mouse, human | x/- | Huttlin et al., 2010, Mertins et al., 2016 [78,79] | |
| S78 | phos | human, mouse, rat | x/- | Dephoure et al., 2008, Zanivan et al., 2008, Tweedie-Cullen et al., 2009 [80,81,82] | |
| S80 | phos | human, mouse, rat | x/x | Zhou et al., 2006, Tao et al., 2009, Bracaglia et al., 2009 [68,83,84] | |
| K82 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| S86 | phos | mouse, human | x/x | Ebert et al., 2013, Mertins et al., 2014 [85,86] | |
| MBD | R115 | met | human | x/- | Geoghegan et al., 2015 [87] |
| S116 | phos | human | x/- | Dephoure et al., 2008, Kettenbach et al., 2011, Sharma et al., 2014 [77,80,88] | |
| K119 | ubi, dimet | human | x/- | Gonzales et al., 2012, Jung et al., 2008 [19,89] | |
| Y120 | phos | human, mouse | x/x | Dephoure et al., 2008, Bergo et al., 2015, D’Annessa et al., 2018 [80,90,91] | |
| K130 | ubi | human | x/- | Wagner et al., 2011, Gonzales et al., 2012 [19,92] | |
| K135 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| T148 | phos | mouse | x/- | Tao et al., 2009 [84] | |
| S149 | phos | mouse, human | x/- | Tao et al., 2009, Olsen et al., 2010, Kettenbach et al., 2011 [84,88,93] | |
| T160 | phos | mouse | x/- | Tweedie-Cullen et al., 2009 [82] | |
| R162 | met | mouse, human | x/- | Guo et al., 2014, Larsen et al., 2016 [94,95] | |
| ID | 163–206 | PAR | human, mouse, rat | x/x | Jungmichel et al., 2013, Becker et al., 2016 [70,96] |
| S164 | phos | mouse | x/x | Tao et al., 2009, Tweedie-Cullen et al., 2009, Stefanelli et al., 2016 [82,84,97] | |
| S166 | phos | mouse, human | x/- | Huttlin et al., 2010, Yi et al., 2014, Mertins et al., 2014 [78,86,98] | |
| S178 | phos | human | x/- | Shiromizu et al., 2013 [75] | |
| T184 | phos | human, mouse | x/- | Mertins et al., 2014 [86] | |
| T203 | phos | human | x/- | Carrier et al., 2016 [99] | |
| S204 | phos | human | x/- | Carrier et al., 2016 [99] | |
| K210 | dimet | human | x/- | Jung et al., 2008 [89] | |
| S216 | phos | human (mouse, rat) | x/x | Olsen et al., 2010, Kettenbach et al., 2011, Lombardi et al., 2017 [69,88,93] | |
| K219 | acet | rat | x/- | Lundby et al., 2012 [100] | |
| K223 | ubi | human | x/- | Akimov et al., 2018 [101] | |
| K223 | SUMO | mouse | -/x | Cheng et al., 2014 [102] | |
| T228*** | phos | human | x/- | Mertins et al., 2014 [86] | |
| S229 | phos | human, rat (mouse) | x/x | Zhou et al., 2006, Chen et al., 2009, Gonzales et al., 2012 [19,83,103] | |
| K233 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| 244–275 | PAR | human, mouse, rat | x/x | Jungmichel et al., 2013, Becker et al., 2016 [70,96] | |
| K249 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| K256 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| K267 | met | human | x/- | Wu et al., 2015 [104] | |
| NID | K271 | ubi | human | x/- | Gonzales et al., 2012 [19] |
| S274 | phos | mouse (human) | x/x | Tweedie-Cullen et al., 2009, Humphrey et al., 2013, Ebert et al., 2013 [74,82,85] | |
| S292 | phos | mouse, rat | x/x | Humphrey et al., 2013, Liu et al., 2015 [74,105] | |
| S295 | phos | mouse | x/- | Humphrey et al., 2013 [74] | |
| K305 | ubi | human | x/- | Gonzales et al., 2012 [19] | |
| K307 | ubi, acet | human | x/- | Gonzales et al., 2012 [19] | |
| T308 | phos | mouse | -/x | Ebert et al., 2013 [85] | |
| CTD | T311 | phos | mouse, human | x/- | Huttlin et al., 2010, Mertins et al., 2014, Parker et al., 2015 [78,86,106] |
| S313 | phos | human, mouse | x/- | Bian et al., 2014, Sharma et al., 2014, Parker et al., 2015 [76,77,106] | |
| K321 | acet, ubi | human, mouse | x/- | Gonzales et al., 2012, Beli et al., 2012, Weinert et al., 2013 [19,107,108] | |
| T327 | phos | human | x/- | Shiromizu et al., 2013 [75] | |
| S341 | phos | mouse | x/- | Humphrey et al., 2013 [74] | |
| K347 | met | human | x/x | Dhayalan et al., 2011, Wu et al., 2015 [109,110] | |
| S357 | phos | human | x/- | Yang et al., 2006 [111] | |
| S359 | phos | human | x/- | Yang et al., 2006, Bian et al., 2014 [76,111] | |
| S360 | phos | human, mouse | x/- | Yang et al., 2006, Grimsrud et al., 2012, Humphrey et al., 2013 [74,111,112] | |
| S393 | phos | human | x/- | Bian et al., 2014 [76] | |
| S399 | phos | mouse, rat, human | x/- | Tao et al., 2009, Gonzales et al., 2012 [19,84] | |
| S421 | phos | mouse, rat (human) | x/x | Zhou et al., 2006, Tao et al., 2009, Deng et al., 2010 [83,84,113] | |
| S424 | phos | human, rat, mouse | x/x | Dephoure et al., 2008, Tao et al., 2009, Li et al., 2011 [80,84,114] | |
| T434 | gl | rat, mouse | x/- | Wang et al., 2010, Alfaro et al., 2012, Trinidad et al., 2012 [115,116,117] | |
| T441 | gl | mouse | x/- | Alfaro et al., 2012 [116] | |
| T443/T444*** | gl | rat | x/- | Wang et al., 2010 [115] | |
| K447 | acet | human | x/- | Choudhary et al., 2009, Beli et al., 2012, Wu et al., 2015 [104,107,118] | |
| T477 | phos | human | x/- | Sharma et al., 2014 [77] | |
| S484 | phos | human, mouse | x/- | Kettenbach et al., 2011, Schweppe et al., 2013, Mertins et al., 2014 [86,88,119] |
| Mutation | Frequency | Effect on: Mice, Cell, Protein | References | |
|---|---|---|---|---|
| MBD | R106W | 132 | Protein: Abolished methyl-DNA binding ability. | Ballestar et al., 2000 [126] |
| R106Q | 21 | Protein: Reduced methyl-DNA binding ability. | Yang et al., 2016 [127] | |
| R133C | 217 | Mice: Decreased life span of 42 weeks and body weight. Protein: Reduced chromatin binding ability. | Brown et al., 2015 [128] | |
| S134C | 21 | Protein: Decreased stability and folding, reduced methyl-DNA binding. | Yang et al., 2016 [127] | |
| A140V | 28 | Mice: Late onset cognitive regression, pyramidal symptoms, parkinsonism, and bipolar symptoms. Increased cell packing density, abnormal dendritic branching of neurons. Life span: >14 months. Cell: Smaller neuron size. Down-regulation of the mTOR signaling pathway. Protein: Increased folding stability. | Venkateswaran et al., 2014 Jentarra et al., 2010 Ma et al., 2014 Sampathkumar et al., 2016 Yang et al., 2016 [127,129,130,131,132] | |
| P152R | 71 | Protein: Decreased stability and folding, reduced methyl-DNA binding. | Yang et al., 2016 [127] | |
| T158M | 419 | Mice: Decreased life span of 13 weeks and body weight. Disturbed nucleolin subcellular localization. Cell: Reduced neurite outgrowth, reduced dendritic complexity, and impaired mitochondrial health in forebrain neurons, reduced CREB and phosphorylated CREB levels. Protein: Decreased protein stability and methyl-DNA binding ability. | Lundvall et al., 2006 Olson et al., 2018 Bu et al., 2017 Chapleau et al., 2009 Brown et al., 2015 [128,133,134,135,136] | |
| ID | R168X | 364 | Mice: Breathing dysfunction, hind limb clasping and atrophy, hypoactivity. Decreased life span of ~12 weeks. Male mice: Impaired motor and cognitive function and reduced anxiety, abnormal hypoxic and hypercapnic responses, apnea incidence, irregular breath cycle and decreased breathing rate, enriched outside chromocenters. Protein: Decreased chromatin compaction ability, decreased methyl-DNA binding. | Lawson-Yuen et al., 2007 Schaevitz et al., 2013 Bissonnette et al., 2014 Georgel et al., 2003 Yusufzai et al., 2000 [137,138,139,140,141] |
| R255X | 313 | Mice: Decreased brain weight, increased breathing, incidence of arrhythmia, anxiety, motor and learning impairments. Cell: mTORC1 pathway abnormalities, decreased nucleolin level, increased phosphorylation of mTORC2 (S2481) and mTORC1 (S2448). Protein: Decreased methyl-DNA binding. | Pitcher et al., 2015 Olson et al., 2018 Yusufzai et al., 2000 [134,141,142] | |
| NID | R270X | 274 | Male: Severe neonatal encephalopathy and death before 4 years of age. Mice: Median life span of 85 days, increased body weight, decreased brain weight. Cell: Less athalassemia/mental retardation syndrome X linked (ATRX) foci. Protein: Decreased methyl-DNA binding, failed to form a higher order structure with nucleic acids and reduced activity to oligomerize nucleic acids. | Villard et al., 2007 Baker et al., 2013 Yusufzai et al., 2000 [141,143,144] |
| R294X | 237 | Cell: Induce caspase mediated apoptosis, rescued by FoxG1. Protein: Decreased methyl-DNA binding; decreased stability. | Lundvall et al., 2006 Yusufzai et al., 2000 [133,141] | |
| R306C | 245 | Mice: Hind limb clasping, impaired mobility and motor coordination, reduced brain weight and size. Cell: Loss of interaction with NCoR/SMRT. Protein: Loss of T308 phosphorylation. | Lyst et al., 2013 [20] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, A.; Zhang, H.; Cardoso, M.C. MeCP2 and Chromatin Compartmentalization. Cells 2020, 9, 878. https://doi.org/10.3390/cells9040878
Schmidt A, Zhang H, Cardoso MC. MeCP2 and Chromatin Compartmentalization. Cells. 2020; 9(4):878. https://doi.org/10.3390/cells9040878
Chicago/Turabian StyleSchmidt, Annika, Hui Zhang, and M. Cristina Cardoso. 2020. "MeCP2 and Chromatin Compartmentalization" Cells 9, no. 4: 878. https://doi.org/10.3390/cells9040878
APA StyleSchmidt, A., Zhang, H., & Cardoso, M. C. (2020). MeCP2 and Chromatin Compartmentalization. Cells, 9(4), 878. https://doi.org/10.3390/cells9040878