Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Background
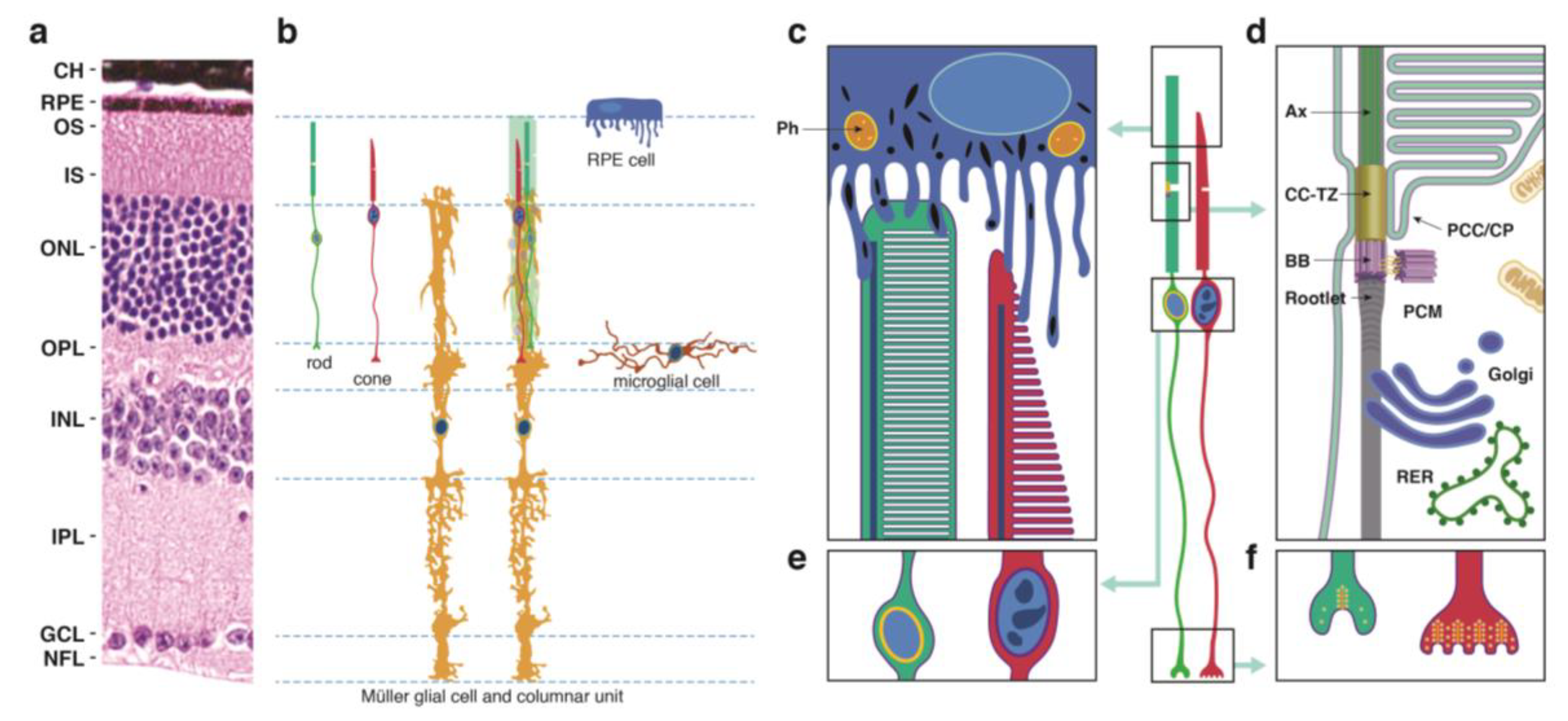
2.1. Photoreceptor (PR) Cell Structure
- The outer segment (OS), which is cylindrical in rod PR cells and tapered in cones, contains phototransduction proteins that sense light and amplify the ensuing signal, culminating in PR cell hyperpolarization (Figure 1c). Much of the phototransduction apparatus is localized to double-bilayer discs formed by evagination of the plasma membrane at the base of the OS. These discs are largely internalized in rods except at the base of the OS, but remain contiguous with the plasma membrane in cones to yield a highly convoluted OS surface [23].
- The OS is stabilized by a ciliary axoneme, which runs through much of its length (Figure 1d; Ax). At the proximal end of the axoneme, the connecting cilium, analogous to the transition zone in other cilia (Figure 1d; CC-TZ), serves as a conduit through which all membrane and protein components destined for the OS are thought to pass. At the base of the connecting cilium lies the basal body (Figure 1d; BB), a cylindrical organelle derived from the mother centriole. Altogether these structures represent a modified primary cilium that encompasses an extensive network of protein complexes that transport proteins and lipids and shares characteristics with primary cilia in many other cell types. The ciliary networks also function to prevent the flow of OS components to other parts of the cell and may associate with the intracellular trafficking apparatus to ensure the directed movement of needed components to the OS.
- The inner segment (IS) contains the biosynthetic machinery and energy sources needed to produce and assemble newly synthesized phototransduction proteins and their associated membranes (Figure 1d). The capacity of this cellular factory is impressive, as up to 10% of the OS is shed daily and removed via phagocytosis by the retinal pigment epithelium (see below) and must be renewed. Most protein and lipid components are synthesized de novo, but the IS also has an extensive recycling machinery that can reassemble components provided from outside the cell.
- The cell body or soma includes the nucleus, which is highly condensed in rod PR cells, but is larger in cones and includes patches of heterochromatin (Figure 1e). To increase the density of rod and cone OSs in the retina, the somas are stacked in columns within the outer nuclear layer (ONL). This arrangement necessitates thin cell extensions reaching from the soma to the IS or to the synapse. PR cell loss is measured by counting ONL nuclei, which are prominently stained in retinal sections (Figure 1a), or in the case of rods, which are more abundant than cones, by measuring ONL thickness from micrographs or by OCT.
- The PR cell terminus contains ribbon synapses close to the presynaptic membrane loaded with vesicles containing the excitatory neurotransmitter glutamate (Figure 1f). In the dark, a steady-state level of glutamate is released at the synapse, which is reduced when the cells are hyperpolarized in the light. Changes in glutamate levels at the synapse signal postsynaptic secondary neurons in the inner nuclear layer, which communicate with ganglion cells on the vitreal surface of the retina that connect through long axons to the visual cortex of the brain.
2.2. Neighboring Cells
2.3. Inherited Diseases that Cause PR Cell Loss
3. Methods
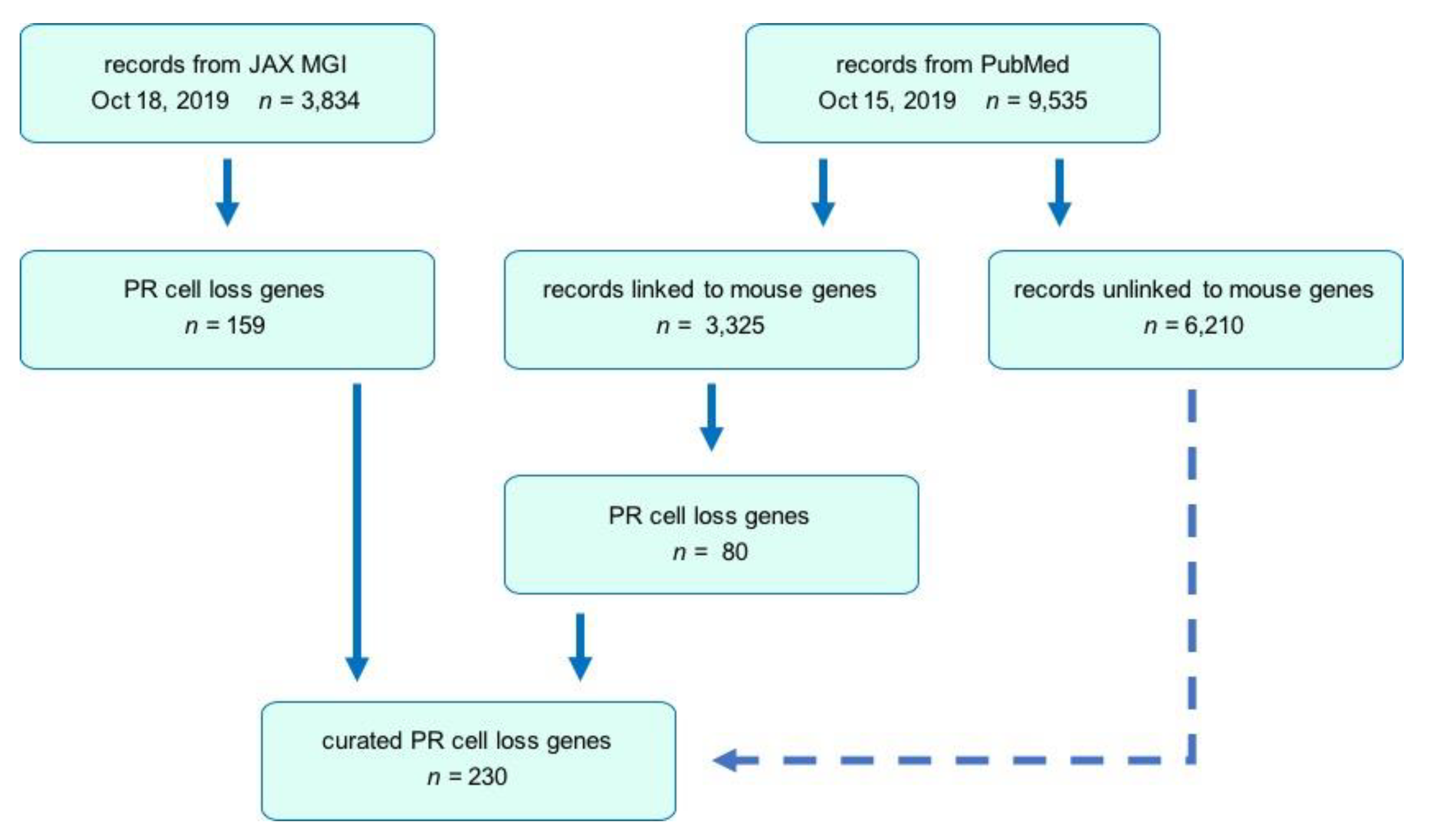
3.1. Public Database and Literature Searches
3.2. Search Strategy
3.3. Comparative Analysis and Updating the MGI Database
3.4. Inclusion/Exclusion Criteria
3.5. Heterogeneity of Data
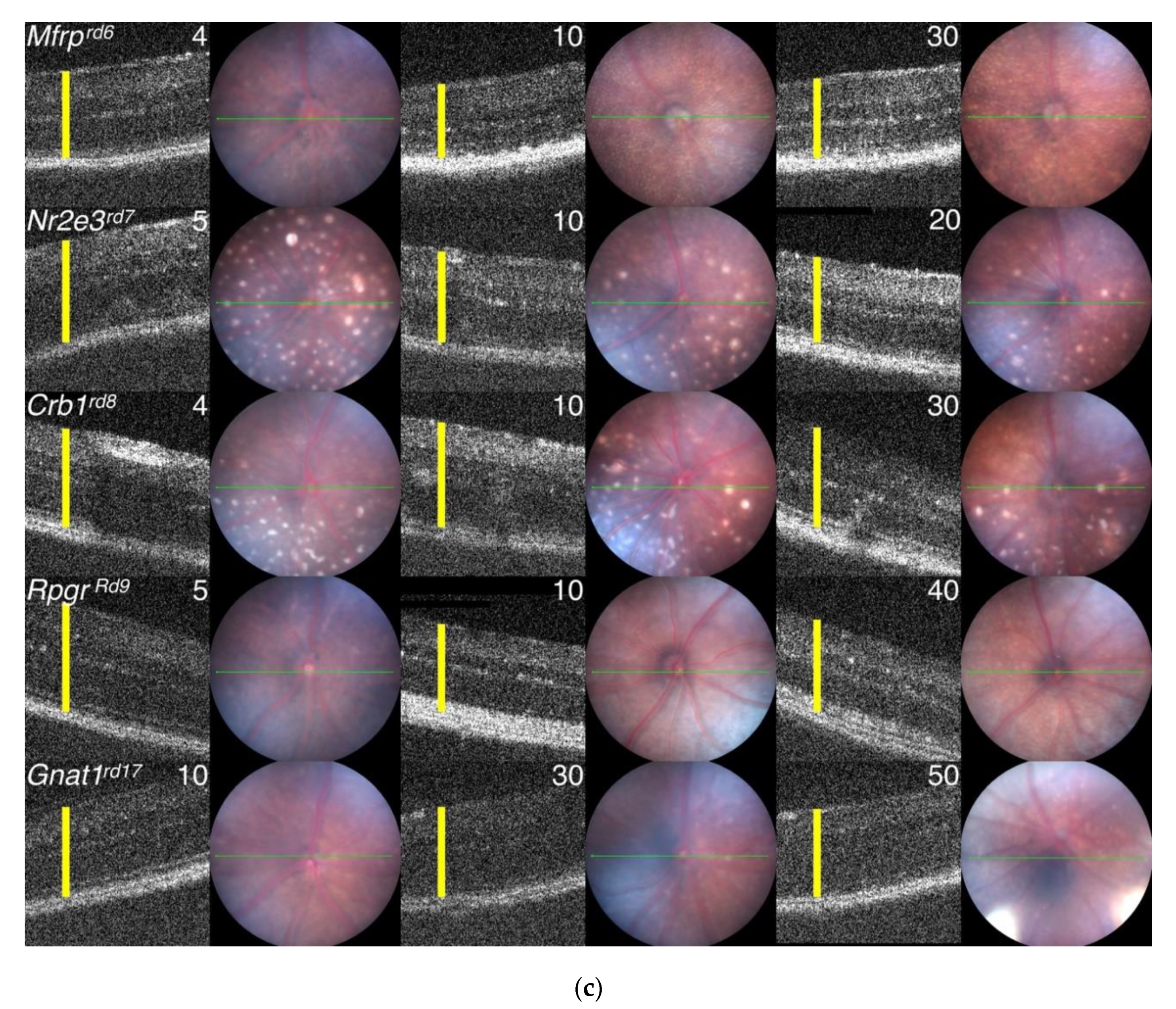
3.6. Comparison of Progressive PR Cell Loss
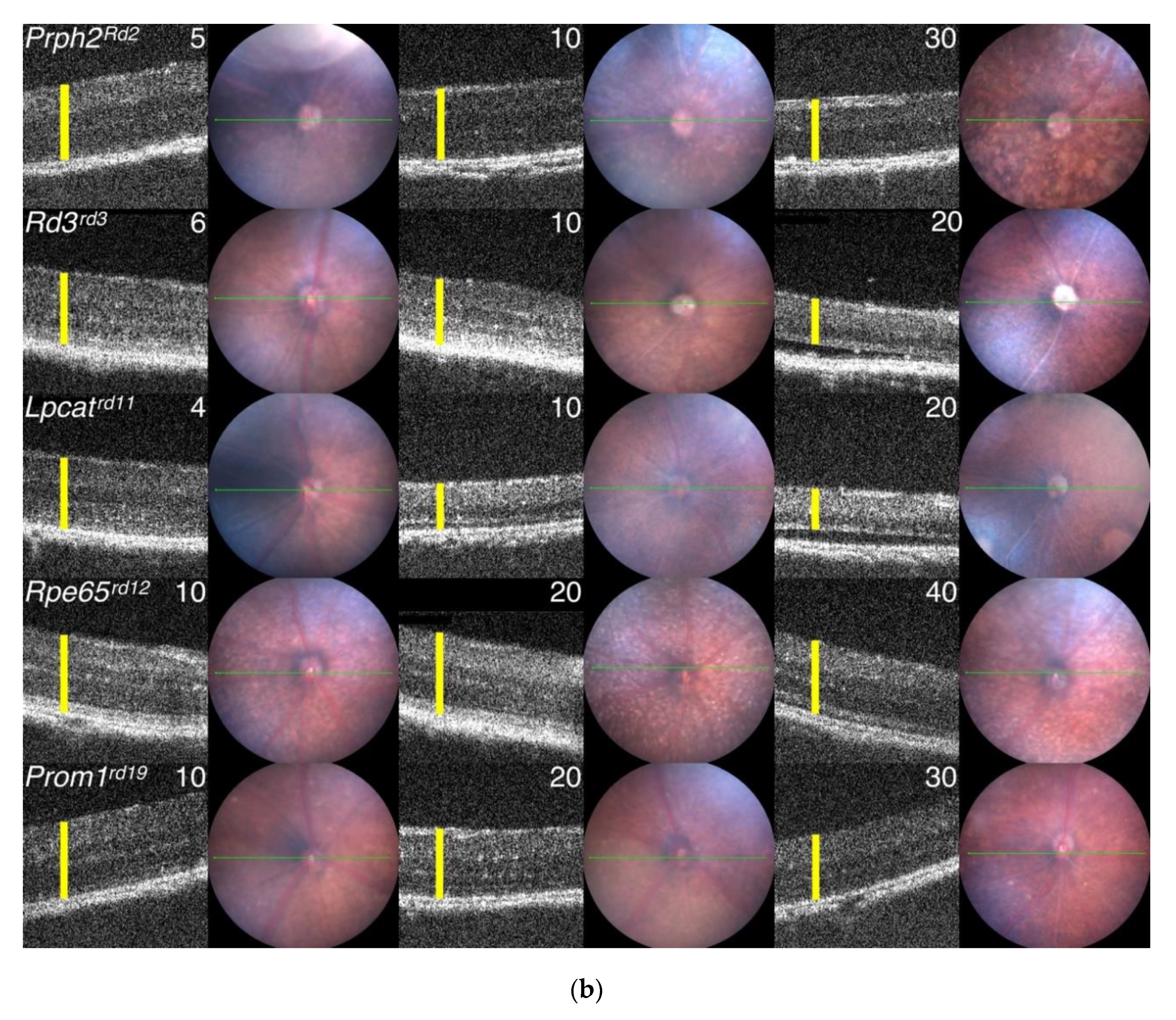
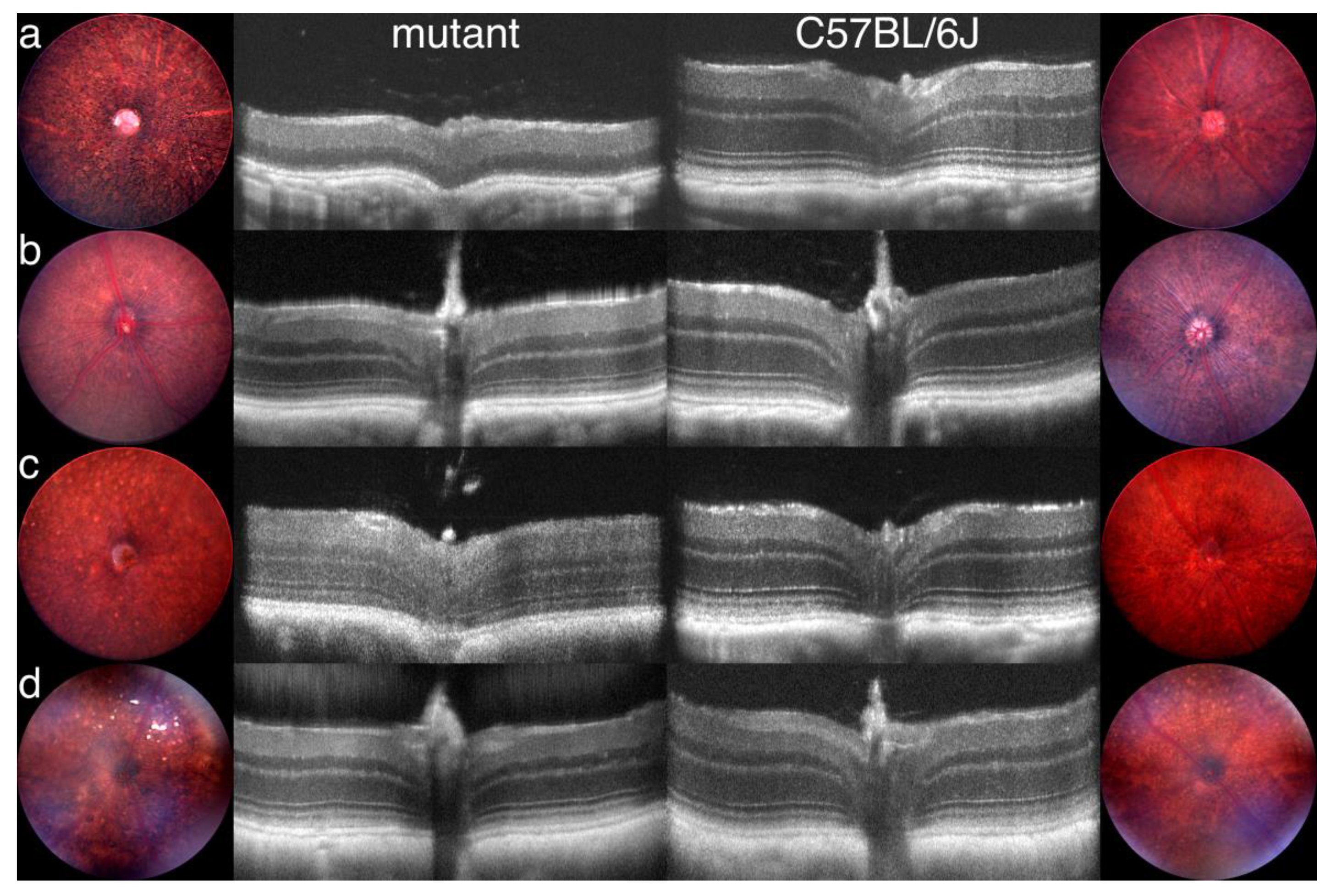
3.7. Generation of Primary Data Using Fundus Imaging and OCT Scans
4. Results
4.1. Summary of Studies that Report PR Cell Loss
4.1.1. PR Cell Loss Models
4.1.2. Mouse Models from Phenotyping Programs
5. Analysis
5.1. Progression of PR Cell Loss
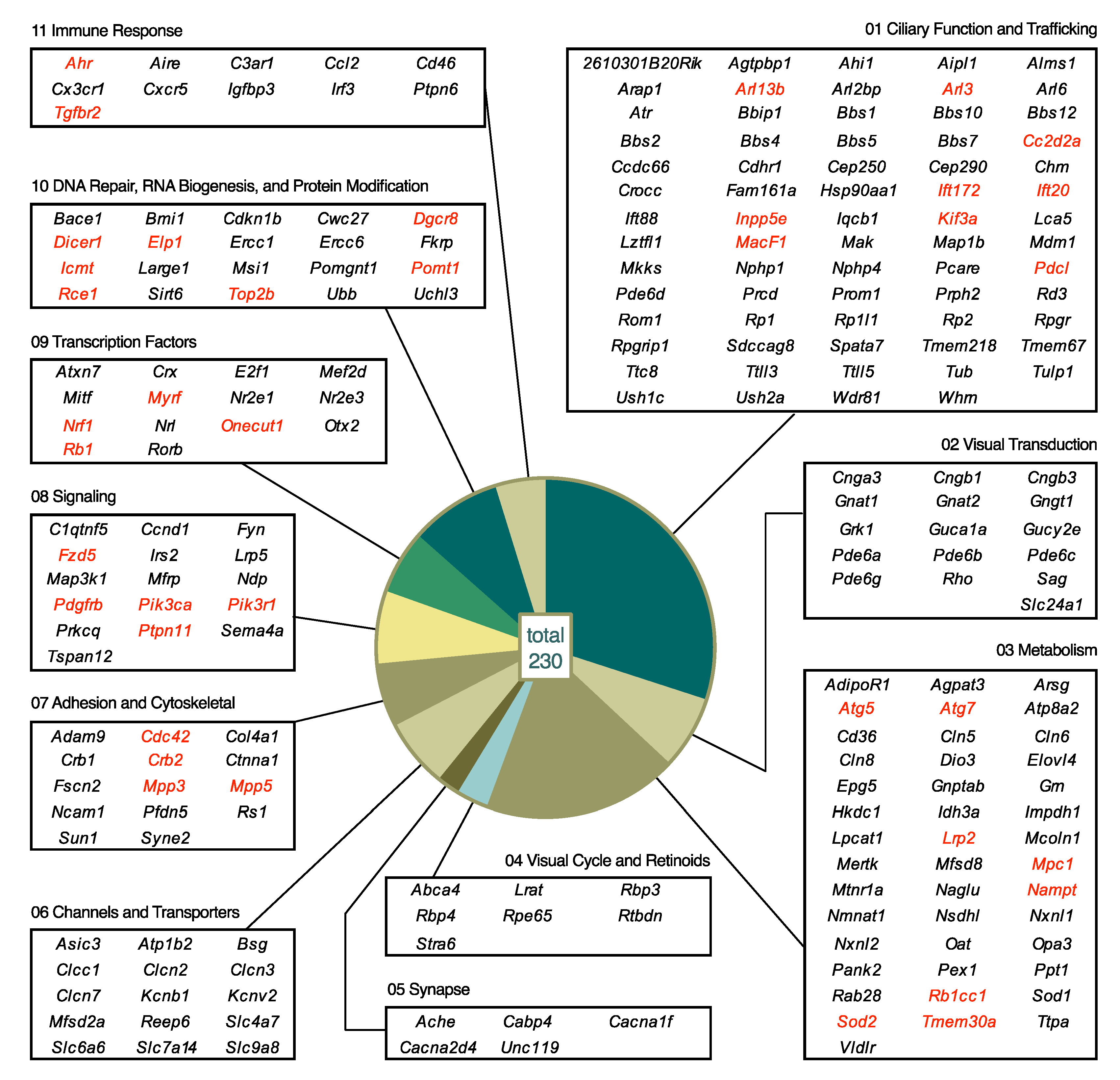
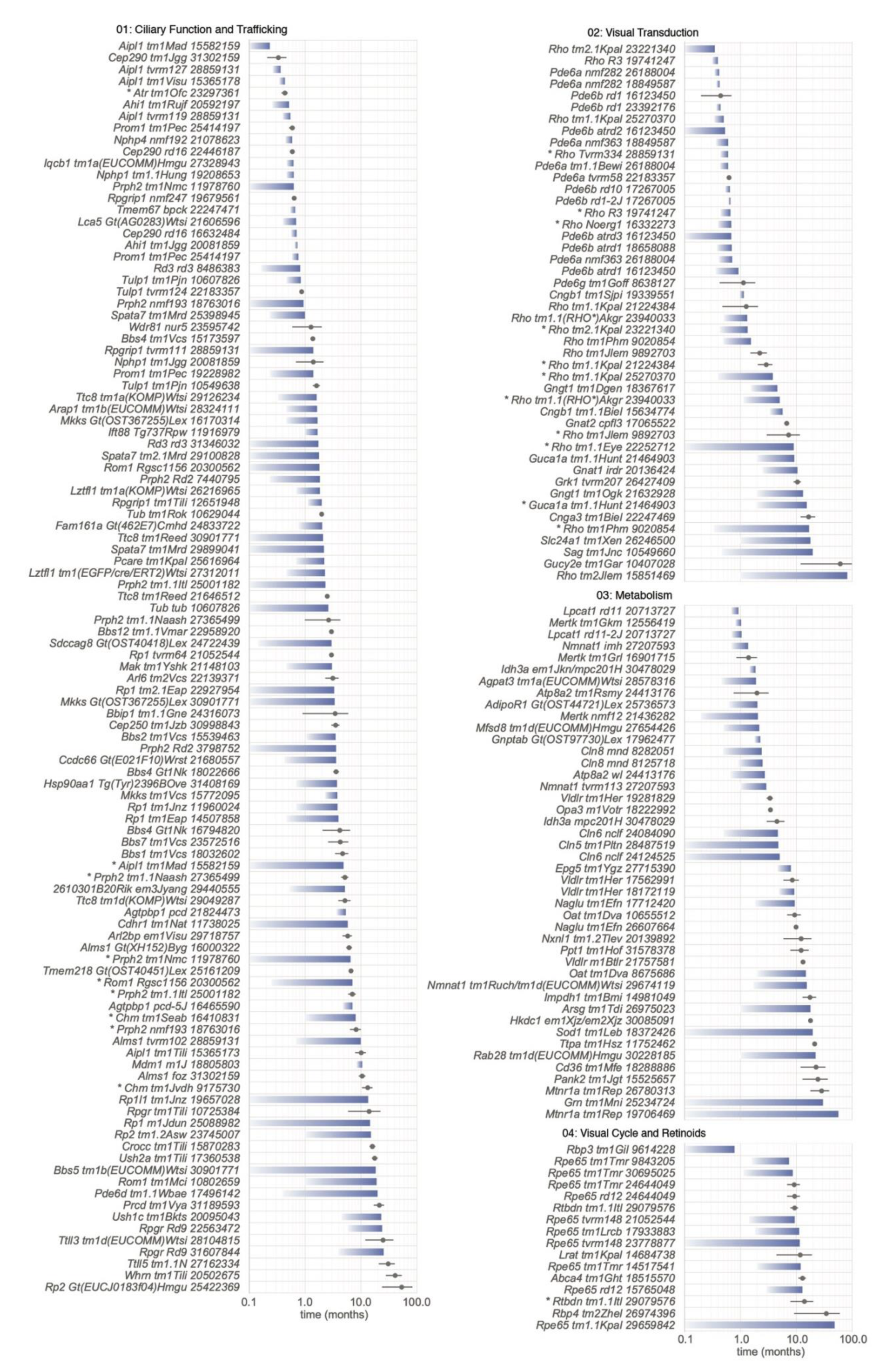
5.2. Biological Processes Affected by Mutations
5.2.1. Category 01: Ciliary Function and Trafficking
5.2.2. Category 02: Visual Transduction
5.2.3. Category 03: Metabolism
5.2.4. Category 04: Visual Cycle and Retinoids
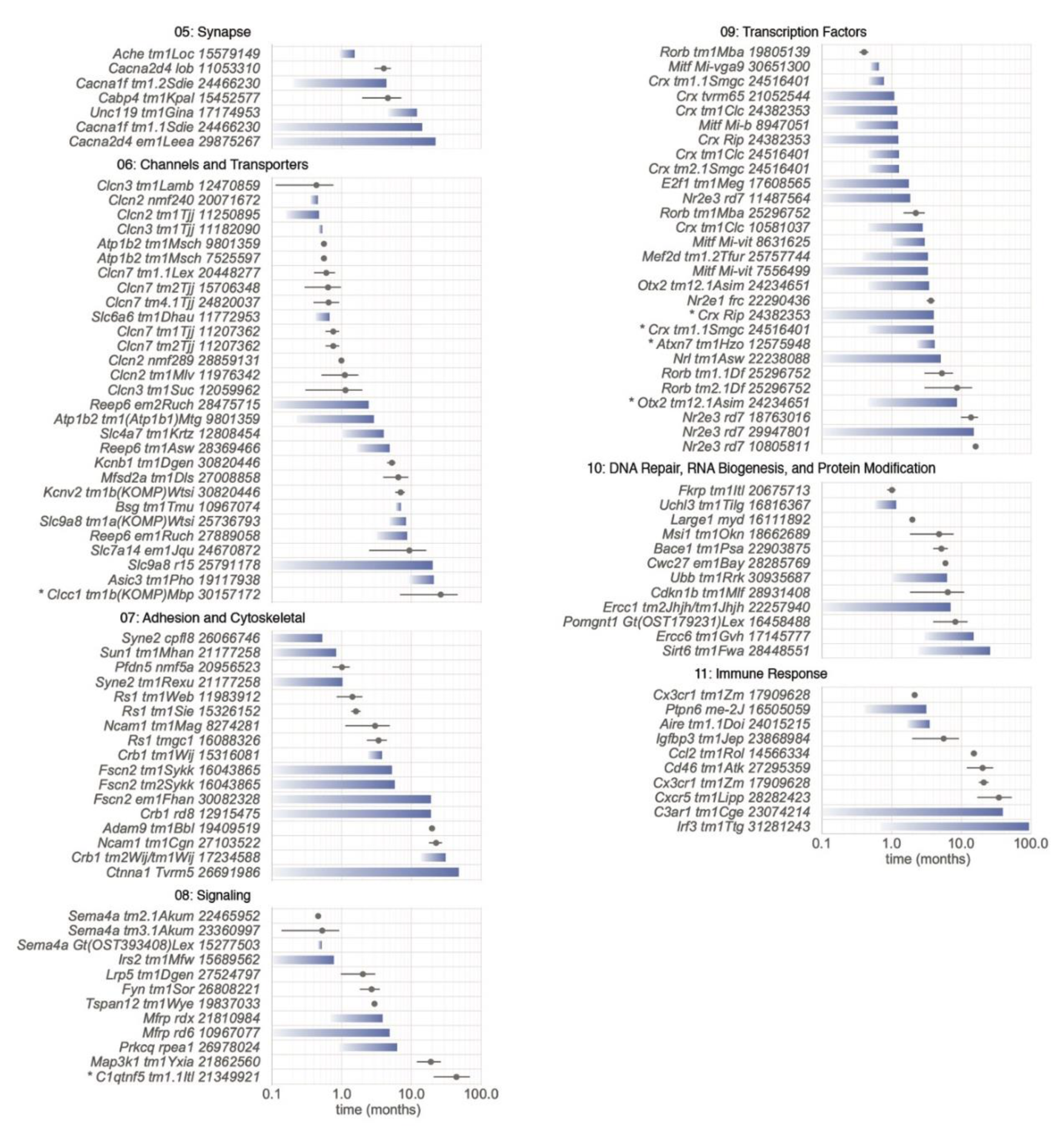
5.2.5. Category 05: Synapse
5.2.6. Category 06: Channels and Transporters
5.2.7. Category 07: Adhesion and Cytoskeletal
5.2.8. Category 08: Signaling
5.2.9. Category 09: Transcription Factors
5.2.10. Category 10: DNA Repair, RNA Biogenesis, and Protein Modification
5.2.11. Category 11: Immune Response
5.3. Omitted Models with PR Abnormalities that May be of Interest
5.4. Factors Leading to Phenotypic Variability
5.4.1. Effects of Allelic Heterogeneity
5.4.2. Effects of Genetic Interactions
5.4.3. Effects of Environment on PR Degeneration
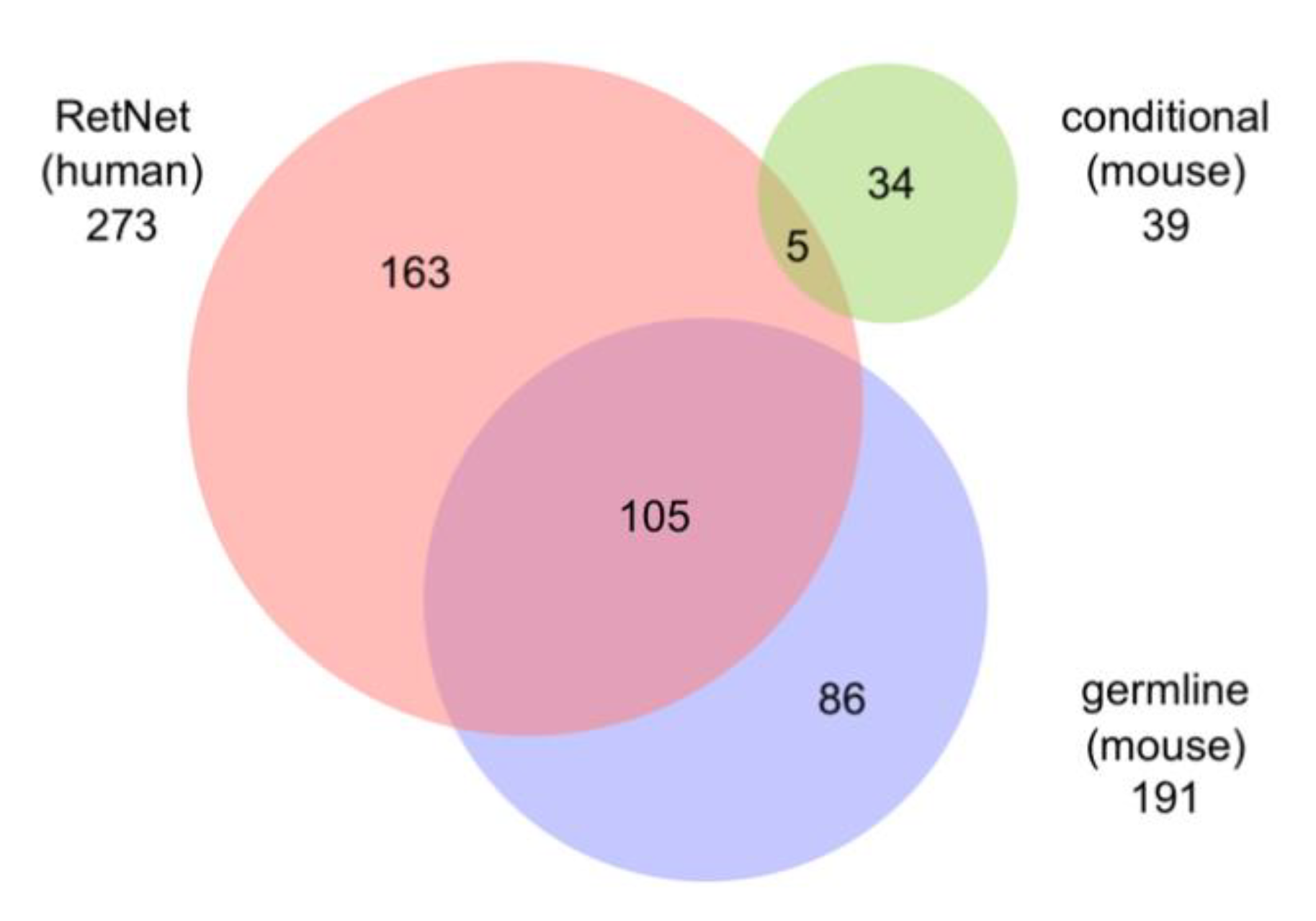
5.5. Relationship to Human Disease Genes
6. Discussion
6.1. Variability in Measuring PR Cell Loss
6.2. Correlation of PR Cell Loss with Gene Function
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cremers, F.P.M.; Boon, C.J.F.; Bujakowska, K.; Zeitz, C. Special Issue Introduction: Inherited Retinal Disease: Novel Candidate Genes, Genotype-Phenotype Correlations, and Inheritance Models. Genes (Basel) 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- RetNet—Retinal Information Network. Available online: https://sph.uth.edu/retnet/home.htm (accessed on 8 December 2019).
- Picaud, S.; Dalkara, D.; Marazova, K.; Goureau, O.; Roska, B.; Sahel, J.A. The primate model for understanding and restoring vision. Proc. Natl. Acad. Sci. USA 2019, 116, 26280–26287. [Google Scholar] [CrossRef] [PubMed]
- Bunel, M.; Chaudieu, G.; Hamel, C.; Lagoutte, L.; Manes, G.; Botherel, N.; Brabet, P.; Pilorge, P.; Andre, C.; Quignon, P. Natural models for retinitis pigmentosa: Progressive retinal atrophy in dog breeds. Hum. Genet. 2019, 138, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L.; Jobling, A.I.; Vessey, K.A.; Luu, C.; Guymer, R.H.; Baird, P.N. Animal models of retinal disease. Prog. Mol. Biol. Transl. Sci. 2011, 100, 211–286. [Google Scholar] [CrossRef]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Model. Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef]
- Angueyra, J.M.; Kindt, K.S. Leveraging Zebrafish to Study Retinal Degenerations. Front. Cell Dev. Biol. 2018, 6, 110. [Google Scholar] [CrossRef]
- Lehmann, M.; Knust, E.; Hebbar, S. Drosophila melanogaster: A Valuable Genetic Model Organism to Elucidate the Biology of Retinitis Pigmentosa. Methods Mol. Biol. 2019, 1834, 221–249. [Google Scholar] [CrossRef]
- Travis, G.H.; Brennan, M.B.; Danielson, P.E.; Kozak, C.A.; Sutcliffe, J.G. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature 1989, 338, 70–73. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.C.; Applebury, M.L.; Farber, D.B. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Parry, D.A.; Johnson, C.A. The role of primary cilia in the development and disease of the retina. Organogenesis 2014, 10, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Baehr, W.; Hanke-Gogokhia, C.; Sharif, A.; Reed, M.; Dahl, T.; Frederick, J.M.; Ying, G. Insights into photoreceptor ciliogenesis revealed by animal models. Prog. Retin. Eye Res. 2019, 71, 26–56. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vis. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Wang, J.; Howell, D.; Davisson, M.T.; Roderick, T.H.; Nusinowitz, S.; Heckenlively, J.R. Mouse models of ocular diseases. Vis. NeuroSci. 2005, 22, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Shi, L.Y.; Hicks, W.; Wang, J.; Hurd, R.; Naggert, J.K.; Chang, B.; Nishina, P.M. Mouse model resources for vision research. J. Ophthalmol. 2011, 2011, 391384. [Google Scholar] [CrossRef]
- Won, J.; Shi, L.Y.; Hicks, W.; Wang, J.; Naggert, J.K.; Nishina, P.M. Translational vision research models program. Adv. Exp. Med. Biol. 2012, 723, 391–397. [Google Scholar] [CrossRef]
- Chang, B. Mouse models for studies of retinal degeneration and diseases. Methods Mol. Biol. 2013, 935, 27–39. [Google Scholar] [CrossRef]
- Chang, B. Mouse Models as Tools to Identify Genetic Pathways for Retinal Degeneration, as Exemplified by Leber’s Congenital Amaurosis. Methods Mol. Biol. 2016, 1438, 417–430. [Google Scholar] [CrossRef]
- Krebs, M.P.; Collin, G.B.; Hicks, W.L.; Yu, M.; Charette, J.R.; Shi, L.Y.; Wang, J.; Naggert, J.K.; Peachey, N.S.; Nishina, P.M. Mouse models of human ocular disease for translational research. PLoS ONE 2017, 12, e0183837. [Google Scholar] [CrossRef]
- Do, M.T.; Yau, K.W. Intrinsically photosensitive retinal ganglion cells. Physiol. Rev. 2010, 90, 1547–1581. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.A.; Li, T.; Swaroop, A. Photoreceptor sensory cilia and ciliopathies: Focus on CEP290, RPGR and their interacting proteins. Cilia 2012, 1, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; O’Sullivan, M.L.; Mukherjee, D.; Punal, V.M.; Farsiu, S.; Kay, J.N. Anatomy and spatial organization of Muller glia in mouse retina. J. Comp. Neurol. 2017, 525, 1759–1777. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Glia of the human retina. Glia 2019, 68, 768–796. [Google Scholar] [CrossRef]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef]
- Suspitsin, E.N.; Imyanitov, E.N. Bardet-Biedl Syndrome. Mol. Syndromol. 2016, 7, 62–71. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Database (MGD) at the Mouse Genome Informatics website, The Jackson Laboratory, Bar Harbor, Maine. Available online: http://www.informatics.jax.org (accessed on 18 October 2019).
- PubMed [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/pubmed/ (accessed on 15 October 2019).
- Clarke, G.; Collins, R.A.; Leavitt, B.R.; Andrews, D.F.; Hayden, M.R.; Lumsden, C.J.; McInnes, R.R. A one-hit model of cell death in inherited neuronal degenerations. Nature 2000, 406, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.P.; Xiao, M.; Sheppard, K.; Hicks, W.; Nishina, P.M. Bright-Field Imaging and Optical Coherence Tomography of the Mouse Posterior Eye. Methods Mol. Biol. 2016, 1438, 395–415. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.P. Using Vascular Landmarks to Orient 3D Optical Coherence Tomography Images of the Mouse Eye. Curr. Protoc. Mouse Biol. 2017, 7, 176–190. [Google Scholar] [CrossRef]
- Dyer, M.A.; Donovan, S.L.; Zhang, J.; Gray, J.; Ortiz, A.; Tenney, R.; Kong, J.; Allikmets, R.; Sohocki, M.M. Retinal degeneration in Aipl1-deficient mice: A new genetic model of Leber congenital amaurosis. Brain Res. Mol. Brain Res. 2004, 132, 208–220. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Wen, X.H.; Woodruff, M.L.; Pawlyk, B.; Yang, J.; Fain, G.L.; Sandberg, M.A.; Makino, C.L.; Li, T. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13903–13908. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Hanke-Gogokhia, C.; Wu, Z.; Gerstner, C.D.; Frederick, J.M.; Zhang, H.; Baehr, W. Arf-like Protein 3 (ARL3) Regulates Protein Trafficking and Ciliogenesis in Mouse Photoreceptors. J. Biol. Chem. 2016, 291, 7142–7155. [Google Scholar] [CrossRef]
- International Mouse Phenotyping Consortium: Home—IMPC. Available online: https://www.mousephenotype.org/ (accessed on 22 December 2019).
- Bujakowska, K.M.; Liu, Q.; Pierce, E.A. Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028274. [Google Scholar] [CrossRef]
- Gilliam, J.C.; Chang, J.T.; Sandoval, I.M.; Zhang, Y.; Li, T.; Pittler, S.J.; Chiu, W.; Wensel, T.G. Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 2012, 151, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Rohlich, P. The sensory cilium of retinal rods is analogous to the transitional zone of motile cilia. Cell Tiss. Res. 1975, 161, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Datta, P. Photoreceptor outer segment as a sink for membrane proteins: Hypothesis and implications in retinal ciliopathies. Hum. Mol. Genet. 2017, 26, R75–R82. [Google Scholar] [CrossRef]
- Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674–686. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef]
- Sedmak, T.; Wolfrum, U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell 2011, 103, 449–466. [Google Scholar] [CrossRef]
- Salinas, R.Y.; Pearring, J.N.; Ding, J.D.; Spencer, W.J.; Hao, Y.; Arshavsky, V.Y. Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J. Cell Biol. 2017, 216, 1489–1499. [Google Scholar] [CrossRef]
- LaVail, M.M. Kinetics of rod outer segment renewal in the developing mouse retina. J. Cell Biol. 1973, 58, 650–661. [Google Scholar] [CrossRef]
- De Robertis, E. Morphogenesis of the retinal rods; an electron microscope study. J. Biophys. Biochem. Cytol. 1956, 2, 209–218. [Google Scholar] [CrossRef]
- Insinna, C.; Besharse, J.C. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev. Dyn. 2008, 237, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, J.L.; Cole, D.G.; Diener, D.R. Intraflagellar transport: The eyes have it. J. Cell Biol. 1999, 144, 385–388. [Google Scholar] [CrossRef]
- Chuang, J.Z.; Zhao, Y.; Sung, C.H. SARA-regulated vesicular targeting underlies formation of the light-sensing organelle in mammalian rods. Cell 2007, 130, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Z.; Hsu, Y.C.; Sung, C.H. Ultrastructural visualization of trans-ciliary rhodopsin cargoes in mammalian rods. Cilia 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, R.H.; Fisher, S.K.; Anderson, D.H. Disc morphogenesis in vertebrate photoreceptors. J. Comp. Neurol. 1980, 190, 501–508. [Google Scholar] [CrossRef]
- Ding, J.D.; Salinas, R.Y.; Arshavsky, V.Y. Discs of mammalian rod photoreceptors form through the membrane evagination mechanism. J. Cell Biol. 2015, 211, 495–502. [Google Scholar] [CrossRef]
- Burgoyne, T.; Meschede, I.P.; Burden, J.J.; Bailly, M.; Seabra, M.C.; Futter, C.E. Rod disc renewal occurs by evagination of the ciliary plasma membrane that makes cadherin-based contacts with the inner segment. Proc. Natl. Acad. Sci. USA 2015, 112, 15922–15927. [Google Scholar] [CrossRef]
- Young, R.W. The renewal of photoreceptor cell outer segments. J. Cell Biol. 1967, 33, 61–72. [Google Scholar] [CrossRef]
- Jin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef]
- Liew, G.M.; Ye, F.; Nager, A.R.; Murphy, J.P.; Lee, J.S.; Aguiar, M.; Breslow, D.K.; Gygi, S.P.; Nachury, M.V. The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 2014, 31, 265–278. [Google Scholar] [CrossRef]
- Taub, D.G.; Liu, Q. The Role of Intraflagellar Transport in the Photoreceptor Sensory Cilium. Adv. Exp. Med. Biol. 2016, 854, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Baker, S.A.; Deane, J.A.; Cole, D.G.; Dickert, B.L.; Rosenbaum, J.L.; Witman, G.B.; Besharse, J.C. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 2002, 157, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jensen, V.L.; Leroux, M.R. Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr. Opin. Cell Biol. 2017, 47, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Baehr, W. Membrane protein transport in photoreceptors: The function of PDEdelta: The Proctor lecture. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8653–8666. [Google Scholar] [CrossRef]
- Hsu, Y.; Garrison, J.E.; Kim, G.; Schmitz, A.R.; Searby, C.C.; Zhang, Q.; Datta, P.; Nishimura, D.Y.; Seo, S.; Sheffield, V.C. BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet. 2017, 13, e1007057. [Google Scholar] [CrossRef]
- Jiang, L.; Wei, Y.; Ronquillo, C.C.; Marc, R.E.; Yoder, B.K.; Frederick, J.M.; Baehr, W. Heterotrimeric kinesin-2 (KIF3) mediates transition zone and axoneme formation of mouse photoreceptors. J. Biol. Chem. 2015, 290, 12765–12778. [Google Scholar] [CrossRef]
- Ronquillo, C.C.; Hanke-Gogokhia, C.; Revelo, M.P.; Frederick, J.M.; Jiang, L.; Baehr, W. Ciliopathy-associated IQCB1/NPHP5 protein is required for mouse photoreceptor outer segment formation. FASEB J. 2016, 30, 3400–3412. [Google Scholar] [CrossRef]
- Hanke-Gogokhia, C.; Wu, Z.; Sharif, A.; Yazigi, H.; Frederick, J.M.; Baehr, W. The guanine nucleotide exchange factor Arf-like protein 13b is essential for assembly of the mouse photoreceptor transition zone and outer segment. J. Biol. Chem. 2017, 292, 21442–21456. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attie-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef]
- Caspary, T.; Larkins, C.E.; Anderson, K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 2007, 12, 767–778. [Google Scholar] [CrossRef]
- Eblimit, A.; Agrawal, S.A.; Thomas, K.; Anastassov, I.A.; Abulikemu, T.; Moayedi, Y.; Mardon, G.; Chen, R. Conditional loss of Spata7 in photoreceptors causes progressive retinal degeneration in mice. Exp. Eye Res. 2018, 166, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Singla, V.; Reiter, J.F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef]
- Garcia-Gonzalo, F.R.; Reiter, J.F. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb. Perspect. Biol. 2017, 9, a028134. [Google Scholar] [CrossRef]
- Shi, X.; Garcia, G., 3rd; Van De Weghe, J.C.; McGorty, R.; Pazour, G.J.; Doherty, D.; Huang, B.; Reiter, J.F. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat. Cell Biol. 2017, 19, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Li, C.; Kida, K.; Inglis, P.N.; Mohan, S.; Semenec, L.; Bialas, N.J.; Stupay, R.M.; Chen, N.; Blacque, O.E.; et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J. Cell Biol. 2011, 192, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Knorz, V.J.; Spalluto, C.; Lessard, M.; Purvis, T.L.; Adigun, F.F.; Collin, G.B.; Hanley, N.A.; Wilson, D.I.; Hearn, T. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol. Biol. Cell 2010, 21, 3617–3629. [Google Scholar] [CrossRef] [PubMed]
- Hearn, T.; Renforth, G.L.; Spalluto, C.; Hanley, N.A.; Piper, K.; Brickwood, S.; White, C.; Connolly, V.; Taylor, J.F.; Russell-Eggitt, I.; et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat. Genet. 2002, 31, 79–83. [Google Scholar] [CrossRef]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat. Genet. 2002, 31, 74–78. [Google Scholar] [CrossRef]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Nishina, P.M.; et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef]
- Brun, A.; Yu, X.; Obringer, C.; Ajoy, D.; Haser, E.; Stoetzel, C.; Roux, M.J.; Messaddeq, N.; Dollfus, H.; Marion, V. In vivo phenotypic and molecular characterization of retinal degeneration in mouse models of three ciliopathies. Exp. Eye Res. 2019, 186, 107721. [Google Scholar] [CrossRef]
- May-Simera, H.L.; Gumerson, J.D.; Gao, C.; Campos, M.; Cologna, S.M.; Beyer, T.; Boldt, K.; Kaya, K.D.; Patel, N.; Kretschmer, F.; et al. Loss of MACF1 Abolishes Ciliogenesis and Disrupts Apicobasal Polarity Establishment in the Retina. Cell Rep. 2016, 17, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Gerding, W.M.; Schreiber, S.; Schulte-Middelmann, T.; de Castro Marques, A.; Atorf, J.; Akkad, D.A.; Dekomien, G.; Kremers, J.; Dermietzel, R.; Gal, A.; et al. Ccdc66 null mutation causes retinal degeneration and dysfunction. Hum. Mol. Genet. 2011, 20, 3620–3631. [Google Scholar] [CrossRef] [PubMed]
- Insolera, R.; Shao, W.; Airik, R.; Hildebrandt, F.; Shi, S.H. SDCCAG8 regulates pericentriolar material recruitment and neuronal migration in the developing cortex. Neuron 2014, 83, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Veleri, S.; Manjunath, S.H.; Fariss, R.N.; May-Simera, H.; Brooks, M.; Foskett, T.A.; Gao, C.; Longo, T.A.; Liu, P.; Nagashima, K.; et al. Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis. Nat. Commun. 2014, 5, 4207. [Google Scholar] [CrossRef]
- Tallila, J.; Jakkula, E.; Peltonen, L.; Salonen, R.; Kestila, M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet. 2008, 82, 1361–1367. [Google Scholar] [CrossRef]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.; Letteboer, S.J.; van Beersum, S.E.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef]
- Mejecase, C.; Hummel, A.; Mohand-Said, S.; Andrieu, C.; El Shamieh, S.; Antonio, A.; Condroyer, C.; Boyard, F.; Foussard, M.; Blanchard, S.; et al. Whole exome sequencing resolves complex phenotype and identifies CC2D2A mutations underlying non-syndromic rod-cone dystrophy. Clin. Genet. 2019, 95, 329–333. [Google Scholar] [CrossRef]
- Lewis, W.R.; Bales, K.L.; Revell, D.Z.; Croyle, M.J.; Engle, S.E.; Song, C.J.; Malarkey, E.B.; Uytingco, C.R.; Shan, D.; Antonellis, P.J.; et al. Mks6 mutations reveal tissue- and cell type-specific roles for the cilia transition zone. FASEB J. 2019, 33, 1440–1455. [Google Scholar] [CrossRef]
- Sorusch, N.; Bauss, K.; Plutniok, J.; Samanta, A.; Knapp, B.; Nagel-Wolfrum, K.; Wolfrum, U. Characterization of the ternary Usher syndrome SANS/ush2a/whirlin protein complex. Hum. Mol. Genet. 2017, 26, 1157–1172. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Zhao, Y.; Adamian, M.; Pawlyk, B.; Sun, X.; McMillan, D.R.; Liberman, M.C.; Li, T. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 2010, 6, e1000955. [Google Scholar] [CrossRef] [PubMed]
- Khattree, N.; Ritter, L.M.; Goldberg, A.F. Membrane curvature generation by a C-terminal amphipathic helix in peripherin-2/rds, a tetraspanin required for photoreceptor sensory cilium morphogenesis. J. Cell Sci. 2013, 126, 4659–4670. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Hicks, D.; Molday, L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Investig. Ophthalmol. Vis. Sci. 1987, 28, 50–61. [Google Scholar] [PubMed]
- Wood, C.R.; Huang, K.; Diener, D.R.; Rosenbaum, J.L. The cilium secretes bioactive ectosomes. Curr. Biol. 2013, 23, 906–911. [Google Scholar] [CrossRef]
- Wood, C.R.; Rosenbaum, J.L. Ciliary ectosomes: Transmissions from the cell’s antenna. Trends Cell Biol. 2015, 25, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum. Mol. Genet. 2014, 23, 6260–6274. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; Zulliger, R.; Naash, M.I. The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Hum. Mol. Genet. 2016, 25, 3500–3514. [Google Scholar] [CrossRef]
- Sanyal, S.; De Ruiter, A.; Hawkins, R.K. Development and degeneration of retina in rds mutant mice: Light microscopy. J. Comp. Neurol. 1980, 194, 193–207. [Google Scholar] [CrossRef]
- Sanyal, S.; Hawkins, R.K. Development and degeneration of retina in rds mutant mice: Effects of light on the rate of degeneration in albino and pigmented homozygous and heterozygous mutant and normal mice. Vis. Res. 1986, 26, 1177–1185. [Google Scholar] [CrossRef]
- McNally, N.; Kenna, P.F.; Rancourt, D.; Ahmed, T.; Stitt, A.; Colledge, W.H.; Lloyd, D.G.; Palfi, A.; O’Neill, B.; Humphries, M.M.; et al. Murine model of autosomal dominant retinitis pigmentosa generated by targeted deletion at codon 307 of the rds-peripherin gene. Hum. Mol. Genet. 2002, 11, 1005–1016. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; Al-Ubaidi, M.R.; Naash, M.I. Initiation of rod outer segment disc formation requires RDS. PLoS ONE 2014, 9, e98939. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Goldberg, A.F.; Vidgen, D.; Collins, L.; Ploder, L.; Schwarz, L.; Molday, L.L.; Rossant, J.; Szel, A.; Molday, R.S.; et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet. 2000, 25, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Suzuki, T.; Ikeda, K.; Masuya, H.; Sezutsu, H.; Kaneda, H.; Kobayashi, K.; Miura, I.; Kurihara, Y.; Yokokura, S.; et al. A monogenic dominant mutation in Rom1 generated by N-ethyl-N-nitrosourea mutagenesis causes retinal degeneration in mice. Mol. Vis. 2010, 16, 378–391. [Google Scholar]
- Spencer, W.J.; Pearring, J.N.; Salinas, R.Y.; Loiselle, D.R.; Skiba, N.P.; Arshavsky, V.Y. Progressive Rod-Cone Degeneration (PRCD) Protein Requires N-Terminal S-Acylation and Rhodopsin Binding for Photoreceptor Outer Segment Localization and Maintaining Intracellular Stability. Biochemistry 2016, 55, 5028–5037. [Google Scholar] [CrossRef]
- Allon, G.; Mann, I.; Remez, L.; Sehn, E.; Rizel, L.; Nevet, M.J.; Perlman, I.; Wolfrum, U.; Ben-Yosef, T. PRCD is Concentrated at the Base of Photoreceptor Outer Segments and is Involved in Outer Segment Disc Formation. Hum. Mol. Genet. 2019, 28, 4078–4088. [Google Scholar] [CrossRef]
- Zangerl, B.; Goldstein, O.; Philp, A.R.; Lindauer, S.J.; Pearce-Kelling, S.E.; Mullins, R.F.; Graphodatsky, A.S.; Ripoll, D.; Felix, J.S.; Stone, E.M.; et al. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 2006, 88, 551–563. [Google Scholar] [CrossRef]
- Spencer, W.J.; Ding, J.D.; Lewis, T.R.; Yu, C.; Phan, S.; Pearring, J.N.; Kim, K.Y.; Thor, A.; Mathew, R.; Kalnitsky, J.; et al. PRCD is essential for high-fidelity photoreceptor disc formation. Proc. Natl. Acad. Sci. USA 2019, 116, 13087–13096. [Google Scholar] [CrossRef]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [CrossRef]
- Cortellino, S.; Wang, C.; Wang, B.; Bassi, M.R.; Caretti, E.; Champeval, D.; Calmont, A.; Jarnik, M.; Burch, J.; Zaret, K.S.; et al. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 2009, 325, 225–237. [Google Scholar] [CrossRef]
- Murcia, N.S.; Richards, W.G.; Yoder, B.K.; Mucenski, M.L.; Dunlap, J.R.; Woychik, R.P. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 2000, 127, 2347–2355. [Google Scholar] [PubMed]
- Stottmann, R.W.; Tran, P.V.; Turbe-Doan, A.; Beier, D.R. Ttc21b is required to restrict sonic hedgehog activity in the developing mouse forebrain. Dev. Biol. 2009, 335, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Gorivodsky, M.; Mukhopadhyay, M.; Wilsch-Braeuninger, M.; Phillips, M.; Teufel, A.; Kim, C.; Malik, N.; Huttner, W.; Westphal, H. Intraflagellar transport protein 172 is essential for primary cilia formation and plays a vital role in patterning the mammalian brain. Dev. Biol. 2009, 325, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Rix, S.; Calmont, A.; Scambler, P.J.; Beales, P.L. An Ift80 mouse model of short rib polydactyly syndromes shows defects in hedgehog signalling without loss or malformation of cilia. Hum. Mol. Genet. 2011, 20, 1306–1314. [Google Scholar] [CrossRef]
- Berbari, N.F.; Kin, N.W.; Sharma, N.; Michaud, E.J.; Kesterson, R.A.; Yoder, B.K. Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev. Biol. 2011, 360, 66–76. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Ko, H.W.; Liu, A.; Eggenschwiler, J.T. Analysis of hedgehog signaling in mouse intraflagellar transport mutants. Methods Cell Biol. 2009, 93, 347–369. [Google Scholar] [CrossRef]
- Gupta, P.R.; Pendse, N.; Greenwald, S.H.; Leon, M.; Liu, Q.; Pierce, E.A.; Bujakowska, K.M. Ift172 conditional knock-out mice exhibit rapid retinal degeneration and protein trafficking defects. Hum. Mol. Genet. 2018, 27, 2012–2024. [Google Scholar] [CrossRef]
- Keady, B.T.; Le, Y.Z.; Pazour, G.J. IFT20 is required for opsin trafficking and photoreceptor outer segment development. Mol. Biol. Cell 2011, 22, 921–930. [Google Scholar] [CrossRef]
- Resh, M.D. Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2006, 2, 584–590. [Google Scholar] [CrossRef]
- Schwarz, N.; Hardcastle, A.J.; Cheetham, M.E. Arl3 and RP2 mediated assembly and traffic of membrane associated cilia proteins. Vis. Res. 2012, 75, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Khan, N.; Hurd, T.; Ghosh, A.K.; Cheng, C.; Molday, R.; Heckenlively, J.R.; Swaroop, A.; Khanna, H. Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4503–4511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hanke-Gogokhia, C.; Jiang, L.; Li, X.; Wang, P.; Gerstner, C.D.; Frederick, J.M.; Yang, Z.; Baehr, W. Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. FASEB J. 2015, 29, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Wright, Z.C.; Singh, R.K.; Alpino, R.; Goldberg, A.F.; Sokolov, M.; Ramamurthy, V. ARL3 regulates trafficking of prenylated phototransduction proteins to the rod outer segment. Hum. Mol. Genet. 2016, 25, 2031–2044. [Google Scholar] [CrossRef]
- Schrick, J.J.; Vogel, P.; Abuin, A.; Hampton, B.; Rice, D.S. ADP-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am. J. Pathol. 2006, 168, 1288–1298. [Google Scholar] [CrossRef]
- Rao, K.N.; Zhang, W.; Li, L.; Anand, M.; Khanna, H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6delta and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum. Mol. Genet. 2016, 25, 4533–4545. [Google Scholar] [CrossRef]
- Xu, W.; Jin, M.; Hu, R.; Wang, H.; Zhang, F.; Yuan, S.; Cao, Y. The Joubert Syndrome Protein Inpp5e Controls Ciliogenesis by Regulating Phosphoinositides at the Apical Membrane. J. Am. Soc. Nephrol. 2017, 28, 118–129. [Google Scholar] [CrossRef]
- Gillespie, P.G.; Prusti, R.K.; Apel, E.D.; Beavo, J.A. A soluble form of bovine rod photoreceptor phosphodiesterase has a novel 15-kDa subunit. J. Biol. Chem. 1989, 264, 12187–12193. [Google Scholar]
- Thompson, D.A.; Khan, N.W.; Othman, M.I.; Chang, B.; Jia, L.; Grahek, G.; Wu, Z.; Hiriyanna, S.; Nellissery, J.; Li, T.; et al. Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15. PLoS ONE 2012, 7, e35865. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.S.; Shang, J.; Sandberg, M.A.; Berson, E.L.; Li, T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl. Acad. Sci. USA 2000, 97, 3649–3654. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.; Doan, T.; Rieke, F.; Detwiler, P.B.; Frederick, J.M.; Baehr, W. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 2007, 104, 8857–8862. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Niemi, G.A.; Reh, T.A.; Hurley, J.B. Leber congenital amaurosis linked to AIPL1: A mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13897–13902. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Artemyev, N.O. AIPL1: A specialized chaperone for the phototransduction effector. Cell Signal. 2017, 40, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Scheidecker, S.; Etard, C.; Pierce, N.W.; Geoffroy, V.; Schaefer, E.; Muller, J.; Chennen, K.; Flori, E.; Pelletier, V.; Poch, O.; et al. Exome sequencing of Bardet-Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J. Med. Genet. 2014, 51, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Eichers, E.R.; Abd-El-Barr, M.M.; Paylor, R.; Lewis, R.A.; Bi, W.; Lin, X.; Meehan, T.P.; Stockton, D.W.; Wu, S.M.; Lindsay, E.; et al. Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum. Genet. 2006, 120, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, K.; Mullins, R.F.; Andrews, M.; Chiang, A.P.; Swiderski, R.E.; Yang, B.; Braun, T.; Casavant, T.; Stone, E.M.; Sheffield, V.C. Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 8664–8669. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.W.; Dell’Orco, D. Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front. Mol. Neurosci. 2015, 8, 67. [Google Scholar] [CrossRef]
- Pinto, L.H.; Vitaterna, M.H.; Shimomura, K.; Siepka, S.M.; McDearmon, E.L.; Fenner, D.; Lumayag, S.L.; Omura, C.; Andrews, A.W.; Baker, M.; et al. Generation, characterization, and molecular cloning of the Noerg-1 mutation of rhodopsin in the mouse. Vis. Neurosci. 2005, 22, 619–629. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Xia, C.H.; Du, X.; Flannery, J.G.; Ridge, K.D.; Beutler, B.; Gong, X. Severe retinal degeneration caused by a novel rhodopsin mutation. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1059–1065. [Google Scholar] [CrossRef][Green Version]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J. Biol. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef]
- Chiang, W.C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, D.; Matthes, M.T.; Coppinger, J.A.; Palczewski, K.; LaVail, M.M.; Lin, J.H. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol. Neurobiol. 2015, 52, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Schiroli, D.; Montanari, M.; Marigo, V. Calpain Activation Is the Major Cause of Cell Death in Photoreceptors Expressing a Rhodopsin Misfolding Mutation. Mol. Neurobiol. 2020, 57, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bevilacqua, D.; Novoselov, S.S.; Parfitt, D.A.; Cheetham, M.E. The cell stress machinery and retinal degeneration. FEBS Lett. 2013, 587, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Kanuga, N.; Adamson, P.; Cheetham, M.E. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 4896–4905. [Google Scholar] [CrossRef]
- Zhang, N.; Kolesnikov, A.V.; Jastrzebska, B.; Mustafi, D.; Sawada, O.; Maeda, T.; Genoud, C.; Engel, A.; Kefalov, V.J.; Palczewski, K. Autosomal recessive retinitis pigmentosa E150K opsin mice exhibit photoreceptor disorganization. J. Clin. Investig. 2013, 123, 121–137. [Google Scholar] [CrossRef]
- Sakami, S.; Kolesnikov, A.V.; Kefalov, V.J.; Palczewski, K. P23H opsin knock-in mice reveal a novel step in retinal rod disc morphogenesis. Hum. Mol. Genet. 2014, 23, 1723–1741. [Google Scholar] [CrossRef]
- Hollingsworth, T.J.; Gross, A.K. The severe autosomal dominant retinitis pigmentosa rhodopsin mutant Ter349Glu mislocalizes and induces rapid rod cell death. J. Biol. Chem. 2013, 288, 29047–29055. [Google Scholar] [CrossRef]
- Humphries, M.M.; Rancourt, D.; Farrar, G.J.; Kenna, P.; Hazel, M.; Bush, R.A.; Sieving, P.A.; Sheils, D.M.; McNally, N.; Creighton, P.; et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat. Genet. 1997, 15, 216–219. [Google Scholar] [CrossRef]
- Lem, J.; Krasnoperova, N.V.; Calvert, P.D.; Kosaras, B.; Cameron, D.A.; Nicolo, M.; Makino, C.L.; Sidman, R.L. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 736–741. [Google Scholar] [CrossRef]
- Faber, S.; Roepman, R. Balancing the Photoreceptor Proteome: Proteostasis Network Therapeutics for Inherited Retinal Disease. Genes 2019, 10, 557. [Google Scholar] [CrossRef]
- Sancho-Pelluz, J.; Cui, X.; Lee, W.; Tsai, Y.T.; Wu, W.H.; Justus, S.; Washington, I.; Hsu, C.W.; Park, K.S.; Koch, S.; et al. Mechanisms of neurodegeneration in a preclinical autosomal dominant retinitis pigmentosa knock-in model with a Rho(D190N) mutation. Cell Mol. Life Sci. 2019, 76, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Park, P.S. Constitutively active rhodopsin and retinal disease. Adv. Pharmacol. 2014, 70, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Budzynski, E.; Gross, A.K.; McAlear, S.D.; Peachey, N.S.; Shukla, M.; He, F.; Edwards, M.; Won, J.; Hicks, W.L.; Wensel, T.G.; et al. Mutations of the opsin gene (Y102H and I307N) lead to light-induced degeneration of photoreceptors and constitutive activation of phototransduction in mice. J. Biol. Chem. 2010, 285, 14521–14533. [Google Scholar] [CrossRef] [PubMed]
- Daniele, L.L.; Insinna, C.; Chance, R.; Wang, J.; Nikonov, S.S.; Pugh, E.N., Jr. A mouse M-opsin monochromat: Retinal cone photoreceptors have increased M-opsin expression when S-opsin is knocked out. Vis. Res. 2011, 51, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, W.T.; Du, W.; Zhu, P.; Li, J.; Xu, F.; Sun, J.; Gerstner, C.D.; Baehr, W.; Boye, S.L.; et al. Gene-based Therapy in a Mouse Model of Blue Cone Monochromacy. Sci. Rep. 2017, 7, 6690. [Google Scholar] [CrossRef] [PubMed]
- Carter-Dawson, L.D.; LaVail, M.M.; Sidman, R.L. Differential effect of the rd mutation on rods and cones in the mouse retina. Investig. Ophthalmol. Vis. Sci. 1978, 17, 489–498. [Google Scholar] [PubMed]
- Calvert, P.D.; Krasnoperova, N.V.; Lyubarsky, A.L.; Isayama, T.; Nicolo, M.; Kosaras, B.; Wong, G.; Gannon, K.S.; Margolskee, R.F.; Sidman, R.L.; et al. Phototransduction in transgenic mice after targeted deletion of the rod transducin alpha -subunit. Proc. Natl. Acad. Sci. USA 2000, 97, 13913–13918. [Google Scholar] [CrossRef]
- Barber, A.C.; Hippert, C.; Duran, Y.; West, E.L.; Bainbridge, J.W.; Warre-Cornish, K.; Luhmann, U.F.; Lakowski, J.; Sowden, J.C.; Ali, R.R.; et al. Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad. Sci. USA 2013, 110, 354–359. [Google Scholar] [CrossRef]
- Miyamoto, M.; Aoki, M.; Sugimoto, S.; Kawasaki, K.; Imai, R. IRD1 and IRD2 mice, naturally occurring models of hereditary retinal dysfunction, show late-onset and progressive retinal degeneration. Curr. Eye Res. 2010, 35, 137–145. [Google Scholar] [CrossRef]
- Mejecase, C.; Laurent-Coriat, C.; Mayer, C.; Poch, O.; Mohand-Said, S.; Prevot, C.; Antonio, A.; Boyard, F.; Condroyer, C.; Michiels, C.; et al. Identification of a Novel Homozygous Nonsense Mutation Confirms the Implication of GNAT1 in Rod-Cone Dystrophy. PLoS ONE 2016, 11, e0168271. [Google Scholar] [CrossRef]
- Carrigan, M.; Duignan, E.; Humphries, P.; Palfi, A.; Kenna, P.F.; Farrar, G.J. A novel homozygous truncating GNAT1 mutation implicated in retinal degeneration. Br. J. Ophthalmol. 2016, 100, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Zenteno, J.C.; Garcia-Montano, L.A.; Cruz-Aguilar, M.; Ronquillo, J.; Rodas-Serrano, A.; Aguilar-Castul, L.; Matsui, R.; Vencedor-Meraz, C.I.; Arce-Gonzalez, R.; Graue-Wiechers, F.; et al. Extensive genic and allelic heterogeneity underlying inherited retinal dystrophies in Mexican patients molecularly analyzed by next-generation sequencing. Mol. Genet. Genomic Med. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.T.; Sakurai, K.; Liu, J.; Dinculescu, A.; Li, J.; Pang, J.; Min, S.H.; Chiodo, V.A.; Boye, S.L.; Chang, B.; et al. Functional interchangeability of rod and cone transducin alpha-subunits. Proc. Natl. Acad. Sci. USA 2009, 106, 17681–17686. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Wang, J.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. A New Mouse Model of Retinal Degeneration (rd17). Proceedings of ARVO Annual Meeting Abstract, Fort Lauderdale, FL, USA, 6–10 May 2007. [Google Scholar]
- Lobanova, E.S.; Finkelstein, S.; Herrmann, R.; Chen, Y.M.; Kessler, C.; Michaud, N.A.; Trieu, L.H.; Strissel, K.J.; Burns, M.E.; Arshavsky, V.Y. Transducin gamma-subunit sets expression levels of alpha- and beta-subunits and is crucial for rod viability. J. Neurosci. 2008, 28, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, A.V.; Rikimaru, L.; Hennig, A.K.; Lukasiewicz, P.D.; Fliesler, S.J.; Govardovskii, V.I.; Kefalov, V.J.; Kisselev, O.G. G-protein betagamma-complex is crucial for efficient signal amplification in vision. J. Neurosci. 2011, 31, 8067–8077. [Google Scholar] [CrossRef]
- Lobanova, E.S.; Finkelstein, S.; Skiba, N.P.; Arshavsky, V.Y. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9986–9991. [Google Scholar] [CrossRef]
- Jobling, A.I.; Vessey, K.A.; Waugh, M.; Mills, S.A.; Fletcher, E.L. A naturally occurring mouse model of achromatopsia: Characterization of the mutation in cone transducin and subsequent retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3350–3359. [Google Scholar] [CrossRef]
- Chang, B.; Dacey, M.S.; Hawes, N.L.; Hitchcock, P.F.; Milam, A.H.; Atmaca-Sonmez, P.; Nusinowitz, S.; Heckenlively, J.R. Cone photoreceptor function loss-3, a novel mouse model of achromatopsia due to a mutation in Gnat2. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5017–5021. [Google Scholar] [CrossRef]
- Ronning, K.E.; Allina, G.P.; Miller, E.B.; Zawadzki, R.J.; Pugh, E.N., Jr.; Herrmann, R.; Burns, M.E. Loss of cone function without degeneration in a novel Gnat2 knock-out mouse. Exp. Eye Res. 2018, 171, 111–118. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef]
- Michaelides, M.; Aligianis, I.A.; Holder, G.E.; Simunovic, M.; Mollon, J.D.; Maher, E.R.; Hunt, D.M.; Moore, A.T. Cone dystrophy phenotype associated with a frameshift mutation (M280fsX291) in the alpha-subunit of cone specific transducin (GNAT2). Br. J. Ophthalmol. 2003, 87, 1317–1320. [Google Scholar] [CrossRef]
- Du, J.; An, J.; Linton, J.D.; Wang, Y.; Hurley, J.B. How Excessive cGMP Impacts Metabolic Proteins in Retinas at the Onset of Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 289–295. [Google Scholar] [CrossRef]
- Tolone, A.; Belhadj, S.; Rentsch, A.; Schwede, F.; Paquet-Durand, F. The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Sothilingam, V.; Garcia Garrido, M.; Jiao, K.; Buena-Atienza, E.; Sahaboglu, A.; Trifunovic, D.; Balendran, S.; Koepfli, T.; Muhlfriedel, R.; Schon, C.; et al. Retinitis pigmentosa: Impact of different Pde6a point mutations on the disease phenotype. Hum. Mol. Genet. 2015, 24, 5486–5499. [Google Scholar] [CrossRef]
- Sakamoto, K.; McCluskey, M.; Wensel, T.G.; Naggert, J.K.; Nishina, P.M. New mouse models for recessive retinitis pigmentosa caused by mutations in the Pde6a gene. Hum. Mol. Genet. 2009, 18, 178–192. [Google Scholar] [CrossRef]
- Power, M.; Das, S.; Schutze, K.; Marigo, V.; Ekstrom, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Gouras, P.; Yamashita, C.K.; Kjeldbye, H.; Fisher, J.; Farber, D.B.; Goff, S.P. Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 1996, 272, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; Somervuo, V.; van den Born, L.I.; Roosing, S.; van Schooneveld, M.J.; Kuijpers, R.W.; van Moll-Ramirez, N.; Cremers, F.P.; Hoyng, C.B.; Klaver, C.C. Progressive loss of cones in achromatopsia: An imaging study using spectral-domain optical coherence tomography. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5952–5957. [Google Scholar] [CrossRef] [PubMed]
- Brennenstuhl, C.; Tanimoto, N.; Burkard, M.; Wagner, R.; Bolz, S.; Trifunovic, D.; Kabagema-Bilan, C.; Paquet-Durand, F.; Beck, S.C.; Huber, G.; et al. Targeted ablation of the Pde6h gene in mice reveals cross-species differences in cone and rod phototransduction protein isoform inventory. J. Biol. Chem. 2015, 290, 10242–10255. [Google Scholar] [CrossRef]
- Kohl, S.; Coppieters, F.; Meire, F.; Schaich, S.; Roosing, S.; Brennenstuhl, C.; Bolz, S.; van Genderen, M.M.; Riemslag, F.C.; European Retinal Disease, C.; et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am. J. Hum. Genet. 2012, 91, 527–532. [Google Scholar] [CrossRef]
- Pedurupillay, C.R.; Landsend, E.C.; Vigeland, M.D.; Ansar, M.; Frengen, E.; Misceo, D.; Stromme, P. Segregation of Incomplete Achromatopsia and Alopecia Due to PDE6H and LPAR6 Variants in a Consanguineous Family from Pakistan. Genes 2016, 7, 41. [Google Scholar] [CrossRef]
- Weitz, D.; Ficek, N.; Kremmer, E.; Bauer, P.J.; Kaupp, U.B. Subunit stoichiometry of the CNG channel of rod photoreceptors. Neuron 2002, 36, 881–889. [Google Scholar] [CrossRef]
- Zheng, J.; Trudeau, M.C.; Zagotta, W.N. Rod cyclic nucleotide-gated channels have a stoichiometry of three CNGA1 subunits and one CNGB1 subunit. Neuron 2002, 36, 891–896. [Google Scholar] [CrossRef]
- Huttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Vinberg, F.; Wang, T.; Molday, R.S.; Chen, J.; Kefalov, V.J. A new mouse model for stationary night blindness with mutant Slc24a1 explains the pathophysiology of the associated human disease. Hum. Mol. Genet. 2015, 24, 5915–5929. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.B.; Robinson, S.W.; Xiong, W.H.; Yau, K.W.; Birch, D.G.; Garbers, D.L. Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci. 1999, 19, 5889–5897. [Google Scholar] [CrossRef] [PubMed]
- Bouzia, Z.; Georgiou, M.; Hull, S.; Robson, A.G.; Fujinami, K.; Rotsos, T.; Pontikos, N.; Arno, G.; Webster, A.R.; Hardcastle, A.J.; et al. GUCY2D-Associated Leber Congenital Amaurosis: A Retrospective Natural History Study in Preparation for Trials of Novel Therapies. Am. J. Ophthalmol. 2020, 210, 59–70. [Google Scholar] [CrossRef]
- Baehr, W.; Karan, S.; Maeda, T.; Luo, D.G.; Li, S.; Bronson, J.D.; Watt, C.B.; Yau, K.W.; Frederick, J.M.; Palczewski, K. The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 2007, 282, 8837–8847. [Google Scholar] [CrossRef]
- Mendez, A.; Burns, M.E.; Sokal, I.; Dizhoor, A.M.; Baehr, W.; Palczewski, K.; Baylor, D.A.; Chen, J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. USA 2001, 98, 9948–9953. [Google Scholar] [CrossRef]
- Buch, P.K.; Mihelec, M.; Cottrill, P.; Wilkie, S.E.; Pearson, R.A.; Duran, Y.; West, E.L.; Michaelides, M.; Ali, R.R.; Hunt, D.M. Dominant cone-rod dystrophy: A mouse model generated by gene targeting of the GCAP1/Guca1a gene. PLoS ONE 2011, 6, e18089. [Google Scholar] [CrossRef][Green Version]
- Marino, V.; Dal Cortivo, G.; Oppici, E.; Maltese, P.E.; D’Esposito, F.; Manara, E.; Ziccardi, L.; Falsini, B.; Magli, A.; Bertelli, M.; et al. A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum. Mol. Genet. 2018, 27, 4204–4217. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Burns, M.E.; Spencer, M.; Niemi, G.A.; Chen, J.; Hurley, J.B.; Baylor, D.A.; Simon, M.I. Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase. Proc. Natl. Acad. Sci. USA 1999, 96, 3718–3722. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dodd, R.L.; Makino, C.L.; Simon, M.I.; Baylor, D.A.; Chen, J. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature 1997, 389, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Simon, M.I.; Matthes, M.T.; Yasumura, D.; LaVail, M.M. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Investig. Ophthalmol. Vis. Sci. 1999, 40, 2978–2982. [Google Scholar] [PubMed]
- Charette, J.R.; Samuels, I.S.; Yu, M.; Stone, L.; Hicks, W.; Shi, L.Y.; Krebs, M.P.; Naggert, J.K.; Nishina, P.M.; Peachey, N.S. A Chemical Mutagenesis Screen Identifies Mouse Models with ERG Defects. Adv. Exp. Med. Biol. 2016, 854, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Rajappa, M.; Goyal, A.; Kaur, J. Inherited metabolic disorders involving the eye: A clinico-biochemical perspective. Eye 2010, 24, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Poll-The, B.T.; Maillette de Buy Wenniger-Prick, C.J. The eye in metabolic diseases: Clues to diagnosis. Eur. J. Paediatr. Neurol. 2011, 15, 197–204. [Google Scholar] [CrossRef]
- Wright, A.F.; Chakarova, C.F.; Abd El-Aziz, M.M.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Anderson, R.E. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 1983, 22, 79–131. [Google Scholar] [CrossRef]
- Giusto, N.M.; Pasquare, S.J.; Salvador, G.A.; Ilincheta de Boschero, M.G. Lipid second messengers and related enzymes in vertebrate rod outer segments. J. Lipid Res. 2010, 51, 685–700. [Google Scholar] [CrossRef]
- Niu, S.L.; Mitchell, D.C.; Litman, B.J. Manipulation of cholesterol levels in rod disk membranes by methyl-beta-cyclodextrin: Effects on receptor activation. J. Biol. Chem. 2002, 277, 20139–20145. [Google Scholar] [CrossRef] [PubMed]
- Bretillon, L.; Thuret, G.; Gregoire, S.; Acar, N.; Joffre, C.; Bron, A.M.; Gain, P.; Creuzot-Garcher, C.P. Lipid and fatty acid profile of the retina, retinal pigment epithelium/choroid, and the lacrimal gland, and associations with adipose tissue fatty acids in human subjects. Exp. Eye Res. 2008, 87, 521–528. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Bretillon, L. The ins and outs of cholesterol in the vertebrate retina. J. Lipid Res. 2010, 51, 3399–3413. [Google Scholar] [CrossRef]
- German, O.L.; Agnolazza, D.L.; Politi, L.E.; Rotstein, N.P. Light, lipids and photoreceptor survival: Live or let die? Photochem. Photobiol. Sci. 2015, 14, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Koso, H.; Sasaki, J.; Nakanishi, H.; Sagara, H.; Nakagawa, K.M.; Takahashi, Y.; Hishikawa, D.; Iizuka-Hishikawa, Y.; Tokumasu, F.; et al. Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J. Biol. Chem. 2017, 292, 12054–12064. [Google Scholar] [CrossRef] [PubMed]
- Lobanova, E.S.; Schuhmann, K.; Finkelstein, S.; Lewis, T.R.; Cady, M.A.; Hao, Y.; Keuthan, C.; Ash, J.D.; Burns, M.E.; Shevchenko, A.; et al. Disrupted Blood-Retina Lysophosphatidylcholine Transport Impairs Photoreceptor Health But Not Visual Signal Transduction. J. Neurosci. 2019, 39, 9689–9701. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.L.; He, J.; Kakazu, A.H.; Jun, B.; Bazan, N.G.; Bazan, H.E.P. Defining a mechanistic link between pigment epithelium-derived factor, docosahexaenoic acid, and corneal nerve regeneration. J. Biol. Chem. 2017, 292, 18486–18499. [Google Scholar] [CrossRef]
- Comitato, A.; Subramanian, P.; Turchiano, G.; Montanari, M.; Becerra, S.P.; Marigo, V. Pigment epithelium-derived factor hinders photoreceptor cell death by reducing intracellular calcium in the degenerating retina. Cell Death Dis. 2018, 9, 560. [Google Scholar] [CrossRef]
- Bernstein, P.S.; Tammur, J.; Singh, N.; Hutchinson, A.; Dixon, M.; Pappas, C.M.; Zabriskie, N.A.; Zhang, K.; Petrukhin, K.; Leppert, M.; et al. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3331–3336. [Google Scholar]
- Vasireddy, V.; Jablonski, M.M.; Mandal, M.N.; Raz-Prag, D.; Wang, X.F.; Nizol, L.; Iannaccone, A.; Musch, D.C.; Bush, R.A.; Salem, N., Jr.; et al. Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4558–4568. [Google Scholar] [CrossRef][Green Version]
- Friedman, J.S.; Chang, B.; Krauth, D.S.; Lopez, I.; Waseem, N.H.; Hurd, R.E.; Feathers, K.L.; Branham, K.E.; Shaw, M.; Thomas, G.E.; et al. Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15523–15528. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, T.; Duh, D.; Peterlin, B.; Gregoric, J. The Str mouse as a model for incontinentia pigmenti. Pflugers Arch. 2000, 440, R53–R54. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Zhu, X.; Djajadi, H.R.; Molday, L.L.; Smith, R.S.; Libby, R.T.; John, S.W.; Molday, R.S. Phospholipid flippase ATP8A2 is required for normal visual and auditory function and photoreceptor and spiral ganglion cell survival. J. Cell Sci. 2014, 127, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Bryde, S.; Hennrich, H.; Verhulst, P.M.; Devaux, P.F.; Lenoir, G.; Holthuis, J.C. CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J. Biol. Chem. 2010, 285, 40562–40572. [Google Scholar] [CrossRef]
- van der Velden, L.M.; Wichers, C.G.; van Breevoort, A.E.; Coleman, J.A.; Molday, R.S.; Berger, R.; Klomp, L.W.; van de Graaf, S.F. Heteromeric interactions required for abundance and subcellular localization of human CDC50 proteins and class 1 P4-ATPases. J. Biol. Chem. 2010, 285, 40088–40096. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Y.; Li, S.; Zhang, S.; Zhu, X.; Tai, Z.; Yang, M.; Liu, Y.; Guo, X.; Chen, B.; et al. Loss of Tmem30a leads to photoreceptor degeneration. Sci. Rep. 2017, 7, 9296. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Energy metabolism of the visual system. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Rountree, A.; Cleghorn, W.M.; Contreras, L.; Lindsay, K.J.; Sadilek, M.; Gu, H.; Djukovic, D.; Raftery, D.; Satrustegui, J.; et al. Phototransduction Influences Metabolic Flux and Nucleotide Metabolism in Mouse Retina. J. Biol. Chem. 2016, 291, 4698–4710. [Google Scholar] [CrossRef]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, Z.; Zhao, P.; Huang, L.; Xu, M.; Yang, Y.; Chen, X.; Lu, F.; Zhang, X.; Wang, H.; et al. Whole-exome sequencing revealed HKDC1 as a candidate gene associated with autosomal-recessive retinitis pigmentosa. Hum. Mol. Genet. 2018, 27, 4157–4168. [Google Scholar] [CrossRef]
- Xia, C.H.; Lu, E.; Liu, H.; Du, X.; Beutler, B.; Gong, X. The role of Vldlr in intraretinal angiogenesis in mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6572–6579. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Jiang, A.; Liang, J.; Meng, H.; Chang, B.; Gao, H.; Qiao, X. Expression of VLDLR in the retina and evolution of subretinal neovascularization in the knockout mouse model’s retinal angiomatous proliferation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Moiseyev, G.; Zhou, K.K.; Chen, D.; Ma, J.X. Photoreceptor degeneration and retinal inflammation induced by very low-density lipoprotein receptor deficiency. Microvasc. Res. 2009, 78, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef]
- Greenwald, S.H.; Charette, J.R.; Staniszewska, M.; Shi, L.Y.; Brown, S.D.M.; Stone, L.; Liu, Q.; Hicks, W.L.; Collin, G.B.; Bowl, M.R.; et al. Mouse Models of NMNAT1-Leber Congenital Amaurosis (LCA9) Recapitulate Key Features of the Human Disease. Am. J. Pathol. 2016, 186, 1925–1938. [Google Scholar] [CrossRef]
- Bosl, M.R.; Stein, V.; Hubner, C.; Zdebik, A.A.; Jordt, S.E.; Mukhopadhyay, A.K.; Davidoff, M.S.; Holstein, A.F.; Jentsch, T.J. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(-) channel disruption. EMBO J. 2001, 20, 1289–1299. [Google Scholar] [CrossRef]
- Ng, L.; Lyubarsky, A.; Nikonov, S.S.; Ma, M.; Srinivas, M.; Kefas, B.; St Germain, D.L.; Hernandez, A.; Pugh, E.N., Jr.; Forrest, D. Type 3 deiodinase, a thyroid-hormone-inactivating enzyme, controls survival and maturation of cone photoreceptors. J. Neurosci. 2010, 30, 3347–3357. [Google Scholar] [CrossRef]
- Ng, L.; Hurley, J.B.; Dierks, B.; Srinivas, M.; Salto, C.; Vennstrom, B.; Reh, T.A.; Forrest, D. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat. Genet. 2001, 27, 94–98. [Google Scholar] [CrossRef]
- Gianesini, C.; Hiragaki, S.; Laurent, V.; Hicks, D.; Tosini, G. Cone Viability Is Affected by Disruption of Melatonin Receptors Signaling. Investig. Ophthalmol. Vis. Sci. 2016, 57, 94–104. [Google Scholar] [CrossRef]
- Baba, K.; Pozdeyev, N.; Mazzoni, F.; Contreras-Alcantara, S.; Liu, C.; Kasamatsu, M.; Martinez-Merlos, T.; Strettoi, E.; Iuvone, P.M.; Tosini, G. Melatonin modulates visual function and cell viability in the mouse retina via the MT1 melatonin receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 15043–15048. [Google Scholar] [CrossRef]
- Chen, Y.; Mehta, G.; Vasiliou, V. Antioxidant defenses in the ocular surface. Ocul. Surf. 2009, 7, 176–185. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lukas, T.J.; Du, N.; Suyeoka, G.; Neufeld, A.H. Dysfunction of the retinal pigment epithelium with age: Increased iron decreases phagocytosis and lysosomal activity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef]
- Frohns, A.; Frohns, F.; Naumann, S.C.; Layer, P.G.; Lobrich, M. Inefficient double-strand break repair in murine rod photoreceptors with inverted heterochromatin organization. Curr. Biol. 2014, 24, 1080–1090. [Google Scholar] [CrossRef][Green Version]
- Blasiak, J.; Petrovski, G.; Vereb, Z.; Facsko, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef]
- Biswal, M.R.; Ildefonso, C.J.; Mao, H.; Seo, S.J.; Wang, Z.; Li, H.; Le, Y.Z.; Lewin, A.S. Conditional Induction of Oxidative Stress in RPE: A Mouse Model of Progressive Retinal Degeneration. Adv. Exp. Med. Biol. 2016, 854, 31–37. [Google Scholar] [CrossRef]
- Cronin, T.; Raffelsberger, W.; Lee-Rivera, I.; Jaillard, C.; Niepon, M.L.; Kinzel, B.; Clerin, E.; Petrosian, A.; Picaud, S.; Poch, O.; et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010, 17, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, C.; Mouret, A.; Niepon, M.L.; Clerin, E.; Yang, Y.; Lee-Rivera, I.; Ait-Ali, N.; Millet-Puel, G.; Cronin, T.; Sedmak, T.; et al. Nxnl2 splicing results in dual functions in neuronal cell survival and maintenance of cell integrity. Hum. Mol. Genet. 2012, 21, 2298–2311. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Igarashi, K.; Uchihara, T.; Jishage, K.; Tomita, H.; Inaba, A.; Li, Y.; Arita, M.; Suzuki, H.; Mizusawa, H.; et al. Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 15185–15190. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Kolter, T.; Sandhoff, K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 2009, 1793, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Birch, D.G. Retinal degeneration in retinitis pigmentosa and neuronal ceroid lipofuscinosis: An overview. Mol. Genet. Metab. 1999, 66, 356–366. [Google Scholar] [CrossRef]
- Ostergaard, J.R. Juvenile neuronal ceroid lipofuscinosis (Batten disease): Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 73–83. [Google Scholar] [CrossRef]
- Leinonen, H.; Keksa-Goldsteine, V.; Ragauskas, S.; Kohlmann, P.; Singh, Y.; Savchenko, E.; Puranen, J.; Malm, T.; Kalesnykas, G.; Koistinaho, J.; et al. Retinal Degeneration In A Mouse Model Of CLN5 Disease Is Associated With Compromised Autophagy. Sci. Rep. 2017, 7, 1597. [Google Scholar] [CrossRef]
- Bartsch, U.; Galliciotti, G.; Jofre, G.F.; Jankowiak, W.; Hagel, C.; Braulke, T. Apoptotic photoreceptor loss and altered expression of lysosomal proteins in the nclf mouse model of neuronal ceroid lipofuscinosis. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6952–6959. [Google Scholar] [CrossRef]
- Jankowiak, W.; Brandenstein, L.; Dulz, S.; Hagel, C.; Storch, S.; Bartsch, U. Retinal Degeneration in Mice Deficient in the Lysosomal Membrane Protein CLN7. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4989–4998. [Google Scholar] [CrossRef]
- Chang, B.; Bronson, R.T.; Hawes, N.L.; Roderick, T.H.; Peng, C.; Hageman, G.S.; Heckenlively, J.R. Retinal degeneration in motor neuron degeneration: A mouse model of ceroid lipofuscinosis. Investig. Ophthalmol. Vis. Sci. 1994, 35, 1071–1076. [Google Scholar] [PubMed]
- Hafler, B.P.; Klein, Z.A.; Jimmy Zhou, Z.; Strittmatter, S.M. Progressive retinal degeneration and accumulation of autofluorescent lipopigments in Progranulin deficient mice. Brain Res. 2014, 1588, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Heldermon, C.D.; Hennig, A.K.; Ohlemiller, K.K.; Ogilvie, J.M.; Herzog, E.D.; Breidenbach, A.; Vogler, C.; Wozniak, D.F.; Sands, M.S. Development of sensory, motor and behavioral deficits in the murine model of Sanfilippo syndrome type B. PLoS ONE 2007, 2, e772. [Google Scholar] [CrossRef] [PubMed]
- Gelfman, C.M.; Vogel, P.; Issa, T.M.; Turner, C.A.; Lee, W.S.; Kornfeld, S.; Rice, D.S. Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5221–5228. [Google Scholar] [CrossRef] [PubMed]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.; Rothlin, C.V.; Burrola, P.; Burstyn-Cohen, T.; Lu, Q.; Garcia de Frutos, P.; Lemke, G. TAM receptor function in the retinal pigment epithelium. Mol. Cell Neurosci. 2006, 33, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; LaVail, M.M.; Yasumura, D.; Matthes, M.T.; Yang, H.; Trautmann, N.; Chappelow, A.V.; Feng, W.; Earp, H.S.; Matsushima, G.K.; et al. An RCS-like retinal dystrophy phenotype in mer knockout mice. Investig. Ophthalmol. Vis. Sci. 2003, 44, 826–838. [Google Scholar] [CrossRef]
- Houssier, M.; Raoul, W.; Lavalette, S.; Keller, N.; Guillonneau, X.; Baragatti, B.; Jonet, L.; Jeanny, J.C.; Behar-Cohen, F.; Coceani, F.; et al. CD36 deficiency leads to choroidal involution via COX2 down-regulation in rodents. PLoS Med. 2008, 5, e39. [Google Scholar] [CrossRef]
- Ying, G.; Boldt, K.; Ueffing, M.; Gerstner, C.D.; Frederick, J.M.; Baehr, W. The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J. Biol. Chem. 2018, 293, 17546–17558. [Google Scholar] [CrossRef]
- Lin, W.; Xu, G. Autophagy: A Role in the Apoptosis, Survival, Inflammation, and Development of the Retina. Ophthalmic Res. 2019, 61, 65–72. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Jansen, L.; JM, U.K.-I.; Siddiqui, A.; Lidov, H.G.; Bodi, I.; Smith, L.; Mein, R.; Cullup, T.; Dionisi-Vici, C.; et al. EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain 2016, 139, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Smucker, W.D.; Kontak, J.R. Adverse drug reactions causing hospital admission in an elderly population: Experience with a decision algorithm. J. Am. Board Fam. Pract. 1990, 3, 105–109. [Google Scholar] [PubMed]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Mohlin, C.; Taylor, L.; Ghosh, F.; Johansson, K. Autophagy and ER-stress contribute to photoreceptor degeneration in cultured adult porcine retina. Brain Res. 2014, 1585, 167–183. [Google Scholar] [CrossRef]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef]
- Falk, M.J. Neurodevelopmental manifestations of mitochondrial disease. J. Dev. Behav. Pediatr. 2010, 31, 610–621. [Google Scholar] [CrossRef]
- Davies, V.J.; Powell, K.A.; White, K.E.; Yip, W.; Hogan, V.; Hollins, A.J.; Davies, J.R.; Piechota, M.; Brownstein, D.G.; Moat, S.J.; et al. A missense mutation in the murine Opa3 gene models human Costeff syndrome. Brain 2008, 131, 368–380. [Google Scholar] [CrossRef][Green Version]
- Findlay, A.S.; Carter, R.N.; Starbuck, B.; McKie, L.; Novakova, K.; Budd, P.S.; Keighren, M.A.; Marsh, J.A.; Cross, S.H.; Simon, M.M.; et al. Mouse Idh3a mutations cause retinal degeneration and reduced mitochondrial function. Dis. Model. Mech. 2018, 11, 036426. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Caicci, F.; Bartolucci, M.; Calzia, D.; Santucci, L.; Manni, L.; Ramenghi, L.A.; Ghiggeri, G.; Traverso, C.E.; et al. Proteome of Bovine Mitochondria and Rod Outer Segment Disks: Commonalities and Differences. J. Proteome Res. 2018, 17, 918–925. [Google Scholar] [CrossRef]
- Calzia, D.; Barabino, S.; Bianchini, P.; Garbarino, G.; Oneto, M.; Caicci, F.; Diaspro, A.; Tacchetti, C.; Manni, L.; Candiani, S.; et al. New findings in ATP supply in rod outer segments: Insights for retinopathies. Biol. Cell 2013, 105, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Funk, R.H.; Schumann, U.; Engelmann, K.; Becker, K.A.; Roehlecke, C. Blue light induced retinal oxidative stress: Implications for macular degeneration. World J. Ophthalmol. 2014, 4, 29–34. [Google Scholar] [CrossRef]
- Calzia, D.; Garbarino, G.; Caicci, F.; Manni, L.; Candiani, S.; Ravera, S.; Morelli, A.; Traverso, C.E.; Panfoli, I. Functional expression of electron transport chain complexes in mouse rod outer segments. Biochimie 2014, 102, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef]
- Folz, S.J.; Trobe, J.D. The peroxisome and the eye. Surv. Ophthalmol. 1991, 35, 353–368. [Google Scholar] [CrossRef]
- Das, Y.; Roose, N.; De Groef, L.; Fransen, M.; Moons, L.; Van Veldhoven, P.P.; Baes, M. Differential distribution of peroxisomal proteins points to specific roles of peroxisomes in the murine retina. Mol. Cell Biochem. 2019, 456, 53–62. [Google Scholar] [CrossRef]
- Hiebler, S.; Masuda, T.; Hacia, J.G.; Moser, A.B.; Faust, P.L.; Liu, A.; Chowdhury, N.; Huang, N.; Lauer, A.; Bennett, J.; et al. The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder. Mol. Genet. Metab. 2014, 111, 522–532. [Google Scholar] [CrossRef]
- Pang, J.J.; Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Li, J.; Noorwez, S.M.; Malhotra, R.; McDowell, J.H.; Kaushal, S.; et al. Retinal degeneration 12 (rd12): A new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol. Vis. 2005, 11, 152–162. [Google Scholar]
- Wright, C.B.; Chrenek, M.A.; Feng, W.; Getz, S.E.; Duncan, T.; Pardue, M.T.; Feng, Y.; Redmond, T.M.; Boatright, J.H.; Nickerson, J.M. The Rpe65 rd12 allele exerts a semidominant negative effect on vision in mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2500–2515. [Google Scholar] [CrossRef]
- Redmond, T.M.; Yu, S.; Lee, E.; Bok, D.; Hamasaki, D.; Chen, N.; Goletz, P.; Ma, J.X.; Crouch, R.K.; Pfeifer, K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998, 20, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Tanabu, R.; Sato, K.; Monai, N.; Yamauchi, K.; Gonome, T.; Xie, Y.; Takahashi, S.; Ishiguro, S.I.; Nakazawa, M. The findings of optical coherence tomography of retinal degeneration in relation to the morphological and electroretinographic features in RPE65-/- mice. PLoS ONE 2019, 14, e0210439. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.L.; Wang, Z.; Chung, H.Y.; Redmond, T.M.; Fain, G.L.; Lem, J. Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat. Genet. 2003, 35, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Samardzija, M.; von Lintig, J.; Tanimoto, N.; Oberhauser, V.; Thiersch, M.; Reme, C.E.; Seeliger, M.; Grimm, C.; Wenzel, A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum. Mol. Genet. 2008, 17, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Suh, S.; Sander, C.L.; Hernandez, C.J.O.; Bulman, E.R.; Khadka, N.; Dong, Z.; Shi, W.; Palczewski, K.; Kiser, P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018, 27, 2225–2243. [Google Scholar] [CrossRef]
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3821–3829. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Molday, L.L.; Wahl, D.; Sarunic, M.V.; Molday, R.S. Localization and functional characterization of the p.Asn965Ser (N965S) ABCA4 variant in mice reveal pathogenic mechanisms underlying Stargardt macular degeneration. Hum. Mol. Genet. 2018, 27, 295–306. [Google Scholar] [CrossRef]
- Zhang, N.; Tsybovsky, Y.; Kolesnikov, A.V.; Rozanowska, M.; Swider, M.; Schwartz, S.B.; Stone, E.M.; Palczewska, G.; Maeda, A.; Kefalov, V.J.; et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Mol. Genet. 2015, 24, 3220–3237. [Google Scholar] [CrossRef]
- Wu, L.; Ueda, K.; Nagasaki, T.; Sparrow, J.R. Light damage in Abca4 and Rpe65rd12 mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1910–1918. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar] [CrossRef]
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem. 2012, 287, 5059–5069. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Maeda, A.; Chen, Y.; Chauhan, V.; Tang, J.; Palczewska, G.; Sakai, T.; Tsuneoka, H.; Palczewski, K.; Maeda, T. Retinal cone and rod photoreceptor cells exhibit differential susceptibility to light-induced damage. J. Neurochem. 2012, 121, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; Van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem. 2004, 279, 10422–10432. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Ghyselinck, N.B.; Mata, N.; Nusinowitz, S.; Lloyd, M.; Dennefeld, C.; Chambon, P.; Bok, D. Somatic ablation of the Lrat gene in the mouse retinal pigment epithelium drastically reduces its retinoid storage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5377–5387. [Google Scholar] [CrossRef]
- Liou, G.I.; Fei, Y.; Peachey, N.S.; Matragoon, S.; Wei, S.; Blaner, W.S.; Wang, Y.; Liu, C.; Gottesman, M.E.; Ripps, H. Early onset photoreceptor abnormalities induced by targeted disruption of the interphotoreceptor retinoid-binding protein gene. J. Neurosci. 1998, 18, 4511–4520. [Google Scholar] [CrossRef]
- Shen, J.; Shi, D.; Suzuki, T.; Xia, Z.; Zhang, H.; Araki, K.; Wakana, S.; Takeda, N.; Yamamura, K.; Jin, S.; et al. Severe ocular phenotypes in Rbp4-deficient mice in the C57BL/6 genetic background. Lab. Investig. 2016, 96, 680–691. [Google Scholar] [CrossRef]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar] [CrossRef]
- Amengual, J.; Zhang, N.; Kemerer, M.; Maeda, T.; Palczewski, K.; Von Lintig, J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum. Mol. Genet. 2014, 23, 5402–5417. [Google Scholar] [CrossRef]
- Wu, S.M. Synaptic transmission in the outer retina. Annu. Rev. Physiol. 1994, 56, 141–168. [Google Scholar] [CrossRef]
- Wu, S.M. Synaptic organization of the vertebrate retina: General principles and species-specific variations: The Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Mercer, A.J.; Thoreson, W.B. The dynamic architecture of photoreceptor ribbon synapses: Cytoskeletal, extracellular matrix, and intramembrane proteins. Vis. Neurosci. 2011, 28, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Ueno, A.; Omori, Y. Molecular mechanisms underlying selective synapse formation of vertebrate retinal photoreceptor cells. Cell Mol. Life Sci. 2019, 77, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.T.; Peachey, N.S. Mouse b-wave mutants. Doc. Ophthalmol. 2014, 128, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Pillers, D.A.; Weleber, R.G.; Woodward, W.R.; Green, D.G.; Chapman, V.M.; Ray, P.N. mdxCv3 mouse is a model for electroretinography of Duchenne/Becker muscular dystrophy. Investig. Ophthalmol. Vis. Sci. 1995, 36, 462–466. [Google Scholar]
- Satz, J.S.; Philp, A.R.; Nguyen, H.; Kusano, H.; Lee, J.; Turk, R.; Riker, M.J.; Hernandez, J.; Weiss, R.M.; Anderson, M.G.; et al. Visual impairment in the absence of dystroglycan. J. Neurosci. 2009, 29, 13136–13146. [Google Scholar] [CrossRef]
- Bytyqi, A.H.; Lockridge, O.; Duysen, E.; Wang, Y.; Wolfrum, U.; Layer, P.G. Impaired formation of the inner retina in an AChE knockout mouse results in degeneration of all photoreceptors. Eur. J. Neurosci. 2004, 20, 2953–2962. [Google Scholar] [CrossRef]
- Grisaru, D.; Sternfeld, M.; Eldor, A.; Glick, D.; Soreq, H. Structural roles of acetylcholinesterase variants in biology and pathology. Eur. J. Biochem. 1999, 264, 672–686. [Google Scholar] [CrossRef]
- Regus-Leidig, H.; Atorf, J.; Feigenspan, A.; Kremers, J.; Maw, M.A.; Brandstatter, J.H. Photoreceptor degeneration in two mouse models for congenital stationary night blindness type 2. PLoS ONE 2014, 9, e86769. [Google Scholar] [CrossRef]
- Kerov, V.; Laird, J.G.; Joiner, M.L.; Knecht, S.; Soh, D.; Hagen, J.; Gardner, S.H.; Gutierrez, W.; Yoshimatsu, T.; Bhattarai, S.; et al. alpha2delta-4 Is Required for the Molecular and Structural Organization of Rod and Cone Photoreceptor Synapses. J. Neurosci. 2018, 38, 6145–6160. [Google Scholar] [CrossRef]
- Ruether, K.; Grosse, J.; Matthiessen, E.; Hoffmann, K.; Hartmann, C. Abnormalities of the photoreceptor-bipolar cell synapse in a substrain of C57BL/10 mice. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4039–4047. [Google Scholar] [PubMed]
- Haeseleer, F.; Imanishi, Y.; Maeda, T.; Possin, D.E.; Maeda, A.; Lee, A.; Rieke, F.; Palczewski, K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat. Neurosci. 2004, 7, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Ishiba, Y.; Higashide, T.; Mori, N.; Kobayashi, A.; Kubota, S.; McLaren, M.J.; Satoh, H.; Wong, F.; Inana, G. Targeted inactivation of synaptic HRG4 (UNC119) causes dysfunction in the distal photoreceptor and slow retinal degeneration, revealing a new function. Exp. Eye Res. 2007, 84, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F. Interaction and colocalization of CaBP4 and Unc119 (MRG4) in photoreceptors. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2366–2375. [Google Scholar] [CrossRef]
- Giblin, J.P.; Comes, N.; Strauss, O.; Gasull, X. Ion Channels in the Eye: Involvement in Ocular Pathologies. Adv. Protein Chem. Struct. Biol. 2016, 104, 157–231. [Google Scholar] [CrossRef]
- Edwards, M.M.; Marin de Evsikova, C.; Collin, G.B.; Gifford, E.; Wu, J.; Hicks, W.L.; Whiting, C.; Varvel, N.H.; Maphis, N.; Lamb, B.T.; et al. Photoreceptor degeneration, azoospermia, leukoencephalopathy, and abnormal RPE cell function in mice expressing an early stop mutation in CLCN2. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3264–3272. [Google Scholar] [CrossRef]
- Stobrawa, S.M.; Breiderhoff, T.; Takamori, S.; Engel, D.; Schweizer, M.; Zdebik, A.A.; Bosl, M.R.; Ruether, K.; Jahn, H.; Draguhn, A.; et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001, 29, 185–196. [Google Scholar] [CrossRef]
- Rajan, I.; Read, R.; Small, D.L.; Perrard, J.; Vogel, P. An alternative splicing variant in Clcn7-/- mice prevents osteopetrosis but not neural and retinal degeneration. Vet. Pathol. 2011, 48, 663–675. [Google Scholar] [CrossRef]
- Dickerson, L.W.; Bonthius, D.J.; Schutte, B.C.; Yang, B.; Barna, T.J.; Bailey, M.C.; Nehrke, K.; Williamson, R.A.; Lamb, F.S. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002, 958, 227–250. [Google Scholar] [CrossRef]
- Kornak, U.; Kasper, D.; Bosl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef]
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poet, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Weinert, S.; Jabs, S.; Hohensee, S.; Chan, W.L.; Kornak, U.; Jentsch, T.J. Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 2014, 15, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Weber, P.; Bartsch, U.; Schachner, M.; Montag, D. Na,K-ATPase subunit beta1 knock-in prevents lethality of beta2 deficiency in mice. J. Neurosci. 1998, 18, 9192–9203. [Google Scholar] [CrossRef] [PubMed]
- Heller-Stilb, B.; van Roeyen, C.; Rascher, K.; Hartwig, H.G.; Huth, A.; Seeliger, M.W.; Warskulat, U.; Haussinger, D. Disruption of the taurine transporter gene (taut) leads to retinal degeneration in mice. FASEB J. 2002, 16, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Bok, D.; Galbraith, G.; Lopez, I.; Woodruff, M.; Nusinowitz, S.; BeltrandelRio, H.; Huang, W.; Zhao, S.; Geske, R.; Montgomery, C.; et al. Blindness and auditory impairment caused by loss of the sodium bicarbonate cotransporter NBC3. Nat. Genet. 2003, 34, 313–319. [Google Scholar] [CrossRef]
- Jin, Z.B.; Huang, X.F.; Lv, J.N.; Xiang, L.; Li, D.Q.; Chen, J.; Huang, C.; Wu, J.; Lu, F.; Qu, J. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat. Commun. 2014, 5, 3517. [Google Scholar] [CrossRef]
- Jadeja, S.; Barnard, A.R.; McKie, L.; Cross, S.H.; White, J.K.; Sanger Mouse Genetics, P.; Robertson, M.; Budd, P.S.; MacLaren, R.E.; Jackson, I.J. Mouse slc9a8 mutants exhibit retinal defects due to retinal pigmented epithelium dysfunction. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3015–3026. [Google Scholar] [CrossRef]
- Hori, K.; Katayama, N.; Kachi, S.; Kondo, M.; Kadomatsu, K.; Usukura, J.; Muramatsu, T.; Mori, S.; Miyake, Y. Retinal dysfunction in basigin deficiency. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3128–3133. [Google Scholar]
- Veleri, S.; Nellissery, J.; Mishra, B.; Manjunath, S.H.; Brooks, M.J.; Dong, L.; Nagashima, K.; Qian, H.; Gao, C.; Sergeev, Y.V.; et al. REEP6 mediates trafficking of a subset of Clathrin-coated vesicles and is critical for rod photoreceptor function and survival. Hum. Mol. Genet. 2017, 26, 2218–2230. [Google Scholar] [CrossRef]
- Ettaiche, M.; Deval, E.; Pagnotta, S.; Lazdunski, M.; Lingueglia, E. Acid-sensing ion channel 3 in retinal function and survival. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2417–2426. [Google Scholar] [CrossRef]
- Li, L.; Jiao, X.; D’Atri, I.; Ono, F.; Nelson, R.; Chan, C.C.; Nakaya, N.; Ma, Z.; Ma, Y.; Cai, X.; et al. Mutation in the intracellular chloride channel CLCC1 associated with autosomal recessive retinitis pigmentosa. PLoS Genet. 2018, 14, e1007504. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.H.; Chan, J.P.; Cazenave-Gassiot, A.; Poh, R.W.; Foo, J.C.; Galam, D.L.; Ghosh, S.; Nguyen, L.N.; Barathi, V.A.; Yeo, S.W.; et al. Mfsd2a Is a Transporter for the Essential omega-3 Fatty Acid Docosahexaenoic Acid (DHA) in Eye and Is Important for Photoreceptor Cell Development. J. Biol. Chem. 2016, 291, 10501–10514. [Google Scholar] [CrossRef] [PubMed]
- Mahimkar, R.M.; Visaya, O.; Pollock, A.S.; Lovett, D.H. The disintegrin domain of ADAM9: A ligand for multiple beta1 renal integrins. Biochem. J. 2005, 385, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.; Toomes, C.; Bida, L.; Danciger, M.; Towns, K.V.; McKibbin, M.; Jacobson, S.G.; Logan, C.V.; Ali, M.; Bond, J.; et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am. J. Hum. Genet. 2009, 84, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Vijayasarathy, C.; Ziccardi, L.; Sieving, P.A. Biology of retinoschisin. Adv. Exp. Med. Biol. 2012, 723, 513–518. [Google Scholar] [CrossRef]
- Jablonski, M.M.; Dalke, C.; Wang, X.; Lu, L.; Manly, K.F.; Pretsch, W.; Favor, J.; Pardue, M.T.; Rinchik, E.M.; Williams, R.W.; et al. An ENU-induced mutation in Rs1h causes disruption of retinal structure and function. Mol. Vis. 2005, 11, 569–581. [Google Scholar]
- Han, J.; Farmer, S.R.; Kirkland, J.L.; Corkey, B.E.; Yoon, R.; Pirtskhalava, T.; Ido, Y.; Guo, W. Octanoate attenuates adipogenesis in 3T3-L1 preadipocytes. J. Nutr. 2002, 132, 904–910. [Google Scholar] [CrossRef]
- Zeng, Y.; Takada, Y.; Kjellstrom, S.; Hiriyanna, K.; Tanikawa, A.; Wawrousek, E.; Smaoui, N.; Caruso, R.; Bush, R.A.; Sieving, P.A. RS-1 Gene Delivery to an Adult Rs1h Knockout Mouse Model Restores ERG b-Wave with Reversal of the Electronegative Waveform of X-Linked Retinoschisis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3279–3285. [Google Scholar] [CrossRef]
- Quinn, P.M.; Pellissier, L.P.; Wijnholds, J. The CRB1 Complex: Following the Trail of Crumbs to a Feasible Gene Therapy Strategy. Front. Neurosci. 2017, 11, 175. [Google Scholar] [CrossRef]
- Mehalow, A.K.; Kameya, S.; Smith, R.S.; Hawes, N.L.; Denegre, J.M.; Young, J.A.; Bechtold, L.; Haider, N.B.; Tepass, U.; Heckenlively, J.R.; et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 2003, 12, 2179–2189. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Kantardzhieva, A.; Malysheva, A.; Meuleman, J.; Versteeg, I.; Levelt, C.; Klooster, J.; Geiger, S.; Seeliger, M.W.; Rashbass, P.; et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J. Cell Sci. 2004, 117, 4169–4177. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.J.; Wijnholds, J. Retinogenesis of the Human Fetal Retina: An Apical Polarity Perspective. Genes 2019, 10, 987. [Google Scholar] [CrossRef]
- Moore, B.A.; Leonard, B.C.; Sebbag, L.; Edwards, S.G.; Cooper, A.; Imai, D.M.; Straiton, E.; Santos, L.; Reilly, C.; Griffey, S.M.; et al. Identification of genes required for eye development by high-throughput screening of mouse knockouts. Commun. Biol. 2018, 1, 236. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.R.; Owen, D.; Mott, H.R. Cdc42 in actin dynamics: An ordered pathway governed by complex equilibria and directional effector handover. Small GTPases 2017, 8, 237–244. [Google Scholar] [CrossRef]
- Yokokura, S.; Wada, Y.; Nakai, S.; Sato, H.; Yao, R.; Yamanaka, H.; Ito, S.; Sagara, Y.; Takahashi, M.; Nakamura, Y.; et al. Targeted disruption of FSCN2 gene induces retinopathy in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2905–2915. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, X.; Zhao, M.; Xie, Y.; Li, P.; Wang, O.; Zhou, B.; Yang, L.; Nie, Y.; Cheng, L.; Song, X.; et al. Null Mutation of the Fascin2 Gene by TALEN Leading to Progressive Hearing Loss and Retinal Degeneration in C57BL/6J Mice. G3 2018, 8, 3221–3230. [Google Scholar] [CrossRef]
- Saksens, N.T.; Krebs, M.P.; Schoenmaker-Koller, F.E.; Hicks, W.; Yu, M.; Shi, L.; Rowe, L.; Collin, G.B.; Charette, J.R.; Letteboer, S.J.; et al. Mutations in CTNNA1 cause butterfly-shaped pigment dystrophy and perturbed retinal pigment epithelium integrity. Nat. Genet. 2016, 48, 144–151. [Google Scholar] [CrossRef]
- Maddox, D.M.; Collin, G.B.; Ikeda, A.; Pratt, C.H.; Ikeda, S.; Johnson, B.A.; Hurd, R.E.; Shopland, L.S.; Naggert, J.K.; Chang, B.; et al. A Mutation in Syne2 Causes Early Retinal Defects in Photoreceptors, Secondary Neurons, and Muller Glia. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3776–3787. [Google Scholar] [CrossRef]
- Yu, J.; Lei, K.; Zhou, M.; Craft, C.M.; Xu, G.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. KASH protein Syne-2/Nesprin-2 and SUN proteins SUN1/2 mediate nuclear migration during mammalian retinal development. Hum. Mol. Genet. 2011, 20, 1061–1073. [Google Scholar] [CrossRef]
- Gordon, M.D.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef]
- Liu, C.; Nathans, J. An essential role for frizzled 5 in mammalian ocular development. Development 2008, 135, 3567–3576. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, C.H.; Sun, Y.; Gong, Y.; Favazza, T.L.; Morss, P.C.; Saba, N.J.; Fredrick, T.W.; He, X.; Akula, J.D.; et al. Pharmacologic Activation of Wnt Signaling by Lithium Normalizes Retinal Vasculature in a Murine Model of Familial Exudative Vitreoretinopathy. Am. J. Pathol. 2016, 186, 2588–2600. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; van de Pol, D.; Bachner, D.; Oerlemans, F.; Winkens, H.; Hameister, H.; Wieringa, B.; Hendriks, W.; Ropers, H.H. An animal model for Norrie disease (ND): Gene targeting of the mouse ND gene. Hum. Mol. Genet. 1996, 5, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Hackam, A.S. The Wnt signaling pathway in retinal degenerations. IUBMB Life 2005, 57, 381–388. [Google Scholar] [CrossRef]
- Hawes, N.L.; Chang, B.; Hageman, G.S.; Nusinowitz, S.; Nishina, P.M.; Schneider, B.S.; Smith, R.S.; Roderick, T.H.; Davisson, M.T.; Heckenlively, J.R. Retinal degeneration 6 (rd6): A new mouse model for human retinitis punctata albescens. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3149–3157. [Google Scholar]
- Hawkes, W.G. Bibliography: Nurses and smoking. J. N. Y. State Nurses Assoc. 1990, 21, 14. [Google Scholar]
- Chavali, V.R.; Khan, N.W.; Cukras, C.A.; Bartsch, D.U.; Jablonski, M.M.; Ayyagari, R. A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum. Mol. Genet. 2011, 20, 2000–2014. [Google Scholar] [CrossRef]
- Cai, Z.; Simons, D.L.; Fu, X.Y.; Feng, G.S.; Wu, S.M.; Zhang, X. Loss of Shp2-mediated mitogen-activated protein kinase signaling in Muller glial cells results in retinal degeneration. Mol. Cell Biol. 2011, 31, 2973–2983. [Google Scholar] [CrossRef][Green Version]
- Chavez-Solano, M.; Ibarra-Sanchez, A.; Trevino, M.; Gonzalez-Espinosa, C.; Lamas, M. Fyn kinase genetic ablation causes structural abnormalities in mature retina and defective Muller cell function. Mol. Cell Neurosci. 2016, 72, 91–100. [Google Scholar] [CrossRef]
- Ji, X.; Liu, Y.; Hurd, R.; Wang, J.; Fitzmaurice, B.; Nishina, P.M.; Chang, B. Retinal Pigment Epithelium Atrophy 1 (rpea1): A New Mouse Model With Retinal Detachment Caused by a Disruption of Protein Kinase C, theta. Investig. Ophthalmol. Vis. Sci. 2016, 57, 877–888. [Google Scholar] [CrossRef][Green Version]
- Ivanovic, I.; Anderson, R.E.; Le, Y.Z.; Fliesler, S.J.; Sherry, D.M.; Rajala, R.V. Deletion of the p85alpha regulatory subunit of phosphoinositide 3-kinase in cone photoreceptor cells results in cone photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3775–3783. [Google Scholar] [CrossRef] [PubMed]
- Rajala, R.V.; Ranjo-Bishop, M.; Wang, Y.; Rajala, A.; Anderson, R.E. The p110alpha isoform of phosphoinositide 3-kinase is essential for cone photoreceptor survival. Biochimie 2015, 112, 35–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yi, X.; Schubert, M.; Peachey, N.S.; Suzuma, K.; Burks, D.J.; Kushner, J.A.; Suzuma, I.; Cahill, C.; Flint, C.L.; Dow, M.A.; et al. Insulin receptor substrate 2 is essential for maturation and survival of photoreceptor cells. J. Neurosci. 2005, 25, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, H.; Kajikawa, S.; Ishii, Y.; Yamamoto, S.; Hamashima, T.; Azuma, E.; Sato, H.; Matsushima, T.; Shibuya, M.; Shimada, Y.; et al. The Novel Pathogenesis of Retinopathy Mediated by Multiple RTK Signals is Uncovered in Newly Developed Mouse Model. EBioMedicine 2018, 31, 190–201. [Google Scholar] [CrossRef]
- Mongan, M.; Wang, J.; Liu, H.; Fan, Y.; Jin, C.; Kao, W.Y.; Xia, Y. Loss of MAP3K1 enhances proliferation and apoptosis during retinal development. Development 2011, 138, 4001–4012. [Google Scholar] [CrossRef]
- Toyofuku, T.; Nojima, S.; Ishikawa, T.; Takamatsu, H.; Tsujimura, T.; Uemura, A.; Matsuda, J.; Seki, T.; Kumanogoh, A. Endosomal sorting by Semaphorin 4A in retinal pigment epithelium supports photoreceptor survival. Genes Dev. 2012, 26, 816–829. [Google Scholar] [CrossRef]
- Carter-Dawson, L.D.; LaVail, M.M. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J. Comp. Neurol. 1979, 188, 263–272. [Google Scholar] [CrossRef]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef]
- Brzezinski, J.A.; Reh, T.A. Photoreceptor cell fate specification in vertebrates. Development 2015, 142, 3263–3273. [Google Scholar] [CrossRef]
- Khidr, L.; Chen, P.L. RB, the conductor that orchestrates life, death and differentiation. Oncogene 2006, 25, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Roger, J.E.; Hiriyanna, A.; Gotoh, N.; Hao, H.; Cheng, D.F.; Ratnapriya, R.; Kautzmann, M.A.; Chang, B.; Swaroop, A. OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. J. Clin. Investig. 2014, 124, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.M.; Zhang, A.; Zhang, X.; Huecker, J.B.; Hennig, A.K.; Chen, S. Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS Genet. 2014, 10, e1004111. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Kitamura, T.; Yoshida, S.; Kuwahara, R.; Chaya, T.; Irie, S.; Furukawa, T. Mef2d is essential for the maturation and integrity of retinal photoreceptor and bipolar cells. Genes Cells 2015, 20, 408–426. [Google Scholar] [CrossRef]
- Kiyama, T.; Chen, C.K.; Wang, S.W.; Pan, P.; Ju, Z.; Wang, J.; Takada, S.; Klein, W.H.; Mao, C.A. Essential roles of mitochondrial biogenesis regulator Nrf1 in retinal development and homeostasis. Mol. Neurodegener. 2018, 13, 56. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, R.; Umino, Y.; Kaczynski, T.J.; Sapkota, D.; Li, S.; Xiang, M.; Fliesler, S.J.; Sherry, D.M.; Gannon, M.; et al. Onecut1 is essential for horizontal cell genesis and retinal integrity. J. Neurosci. 2013, 33, 13053–13065. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef]
- Zelinger, L.; Swaroop, A. RNA Biology in Retinal Development and Disease. Trends Genet. 2018, 34, 341–351. [Google Scholar] [CrossRef]
- Barabino, A.; Plamondon, V.; Abdouh, M.; Chatoo, W.; Flamier, A.; Hanna, R.; Zhou, S.; Motoyama, N.; Hebert, M.; Lavoie, J.; et al. Loss of Bmi1 causes anomalies in retinal development and degeneration of cone photoreceptors. Development 2016, 143, 1571–1584. [Google Scholar] [CrossRef]
- Susaki, K.; Kaneko, J.; Yamano, Y.; Nakamura, K.; Inami, W.; Yoshikawa, T.; Ozawa, Y.; Shibata, S.; Matsuzaki, O.; Okano, H.; et al. Musashi-1, an RNA-binding protein, is indispensable for survival of photoreceptors. Exp. Eye Res. 2009, 88, 347–355. [Google Scholar] [CrossRef]
- McKinnon, P.J. Maintaining genome stability in the nervous system. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, L.H.; Svejstrup, J.Q. The Cellular Response to Transcription-Blocking DNA Damage. Trends Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; De Vivo, A.; Uprety, N.; Kim, J.; Stevens, S.M., Jr.; Kee, Y. BMI1-UBR5 axis regulates transcriptional repression at damaged chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, 11243–11248. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Dhillon, K.K.; Tucker, N.; Chi, Y.; Huang, J.W.; Kawasumi, M.; Nghiem, P.; Wang, Y.; Clurman, B.E.; Jacquemont, C.; et al. DGCR8 Mediates Repair of UV-Induced DNA Damage Independently of RNA Processing. Cell Rep. 2017, 19, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Burger, K.; Schlackow, M.; Potts, M.; Hester, S.; Mohammed, S.; Gullerova, M. Nuclear phosphorylated Dicer processes double-stranded RNA in response to DNA damage. J. Cell Biol. 2017, 216, 2373–2389. [Google Scholar] [CrossRef]
- Montaldo, N.P.; Bordin, D.L.; Brambilla, A.; Rosinger, M.; Fordyce Martin, S.L.; Bjoras, K.O.; Bradamante, S.; Aas, P.A.; Furrer, A.; Olsen, L.C.; et al. Alkyladenine DNA glycosylase associates with transcription elongation to coordinate DNA repair with gene expression. Nat. Commun. 2019, 10, 5460. [Google Scholar] [CrossRef]
- Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. [Google Scholar] [CrossRef]
- de Araujo, P.R.; Gorthi, A.; da Silva, A.E.; Tonapi, S.S.; Vo, D.T.; Burns, S.C.; Qiao, M.; Uren, P.J.; Yuan, Z.M.; Bishop, A.J.; et al. Musashi1 Impacts Radio-Resistance in Glioblastoma by Controlling DNA-Protein Kinase Catalytic Subunit. Am. J. Pathol. 2016, 186, 2271–2278. [Google Scholar] [CrossRef]
- Onn, L.; Portillo, M.; Ilic, S.; Cleitman, G.; Stein, D.; Kaluski, S.; Shirat, I.; Slobodnik, Z.; Einav, M.; Erdel, F.; et al. SIRT6 is a DNA double-strand break sensor. Elife 2020, 9. [Google Scholar] [CrossRef]
- Calderwood, S.K. A critical role for topoisomerase IIb and DNA double strand breaks in transcription. Transcription 2016, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046.e1045–1061.e1045. [Google Scholar] [CrossRef]
- Nishi, R.; Wijnhoven, P.W.G.; Kimura, Y.; Matsui, M.; Konietzny, R.; Wu, Q.; Nakamura, K.; Blundell, T.L.; Kessler, B.M. The deubiquitylating enzyme UCHL3 regulates Ku80 retention at sites of DNA damage. Sci. Rep. 2018, 8, 17891. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lin, Z.; Li, F.; Diao, W.; Dong, C.; Zhou, H.; Xie, X.; Wang, Z.; Shen, Y.; Long, J. Dimerization of elongator protein 1 is essential for Elongator complex assembly. Proc. Natl. Acad. Sci. USA 2015, 112, 10697–10702. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hao, H.; Swerdel, M.R.; Cho, H.Y.; Lee, K.B.; Hart, R.P.; Lyu, Y.L.; Cai, L. Top2b is involved in the formation of outer segment and synapse during late-stage photoreceptor differentiation by controlling key genes of photoreceptor transcriptional regulatory network. J. Neurosci. Res. 2017, 95, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Sanchez, L.; De la Cerda, B.; Diaz-Corrales, F.J.; Massalini, S.; Chakarova, C.F.; Wright, A.F.; Bhattacharya, S.S. ATR localizes to the photoreceptor connecting cilium and deficiency leads to severe photoreceptor degeneration in mice. Hum. Mol. Genet. 2013, 22, 1507–1515. [Google Scholar] [CrossRef]
- Bian, C.; Zhang, C.; Luo, T.; Vyas, A.; Chen, S.H.; Liu, C.; Kassab, M.A.; Yang, Y.; Kong, M.; Yu, X. NADP(+) is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat. Commun. 2019, 10, 693. [Google Scholar] [CrossRef]
- Sundermeier, T.R.; Sakami, S.; Sahu, B.; Howell, S.J.; Gao, S.; Dong, Z.; Golczak, M.; Maeda, A.; Palczewski, K. MicroRNA-processing Enzymes Are Essential for Survival and Function of Mature Retinal Pigmented Epithelial Cells in Mice. J. Biol. Chem. 2017, 292, 3366–3378. [Google Scholar] [CrossRef]
- Damiani, D.; Alexander, J.J.; O’Rourke, J.R.; McManus, M.; Jadhav, A.P.; Cepko, C.L.; Hauswirth, W.W.; Harfe, B.D.; Strettoi, E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J. Neurosci. 2008, 28, 4878–4887. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Ramirez, G.; Salcedo, E.; Stabio, M.E.; Lefcort, F. Loss of Ikbkap Causes Slow, Progressive Retinal Degeneration in a Mouse Model of Familial Dysautonomia. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Spoor, M.; Nagtegaal, A.P.; Ridwan, Y.; Borgesius, N.Z.; van Alphen, B.; van der Pluijm, I.; Hoeijmakers, J.H.; Frens, M.A.; Borst, J.G. Accelerated loss of hearing and vision in the DNA-repair deficient Ercc1(delta/-) mouse. Mech. Ageing Dev. 2012, 133, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Gorgels, T.G.; van der Pluijm, I.; Brandt, R.M.; Garinis, G.A.; van Steeg, H.; van den Aardweg, G.; Jansen, G.H.; Ruijter, J.M.; Bergen, A.A.; van Norren, D.; et al. Retinal degeneration and ionizing radiation hypersensitivity in a mouse model for Cockayne syndrome. Mol. Cell Biol. 2007, 27, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Eblimit, A.; Zaneveld, S.A.; Liu, W.; Thomas, K.; Wang, K.; Li, Y.; Mardon, G.; Chen, R. NMNAT1 E257K variant, associated with Leber Congenital Amaurosis (LCA9), causes a mild retinal degeneration phenotype. Exp. Eye Res. 2018, 173, 32–43. [Google Scholar] [CrossRef]
- Peshti, V.; Obolensky, A.; Nahum, L.; Kanfi, Y.; Rathaus, M.; Avraham, M.; Tinman, S.; Alt, F.W.; Banin, E.; Cohen, H.Y. Characterization of physiological defects in adult SIRT6-/- mice. PLoS ONE 2017, 12, e0176371. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Park, C.W.; Ryu, K.Y.; Chung, H. Disruption of the polyubiquitin gene Ubb causes retinal degeneration in mice. Biochem. Biophys. Res. Commun. 2019, 513, 35–40. [Google Scholar] [CrossRef]
- Semenova, E.; Wang, X.; Jablonski, M.M.; Levorse, J.; Tilghman, S.M. An engineered 800 kilobase deletion of Uchl3 and Lmo7 on mouse chromosome 14 causes defects in viability, postnatal growth and degeneration of muscle and retina. Hum. Mol. Genet. 2003, 12, 1301–1312. [Google Scholar] [CrossRef]
- Pinelli, M.; Carissimo, A.; Cutillo, L.; Lai, C.H.; Mutarelli, M.; Moretti, M.N.; Singh, M.V.; Karali, M.; Carrella, D.; Pizzo, M.; et al. An atlas of gene expression and gene co-regulation in the human retina. Nucleic Acids Res. 2016, 44, 5773–5784. [Google Scholar] [CrossRef]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.R.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763.e764–779.e764. [Google Scholar] [CrossRef]
- Graziotto, J.J.; Farkas, M.H.; Bujakowska, K.; Deramaudt, B.M.; Zhang, Q.; Nandrot, E.F.; Inglehearn, C.F.; Bhattacharya, S.S.; Pierce, E.A. Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 190–198. [Google Scholar] [CrossRef]
- Xu, M.; Xie, Y.A.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef]
- Chen, B.J.; Lam, T.C.; Liu, L.Q.; To, C.H. Post-translational modifications and their applications in eye research (Review). Mol. Med. Rep. 2017, 15, 3923–3935. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.R.; Kolandaivelu, S.; Bergo, M.O.; Ramamurthy, V. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc. Natl. Acad. Sci. USA 2011, 108, 8862–8866. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.R.; Pendse, N.D.; Kolandaivelu, S.; Bergo, M.O.; Young, S.G.; Ramamurthy, V. Deficiency of Isoprenylcysteine Carboxyl Methyltransferase (ICMT) Leads to Progressive Loss of Photoreceptor Function. J. Neurosci. 2016, 36, 5107–5114. [Google Scholar] [CrossRef][Green Version]
- Bosch Grau, M.; Masson, C.; Gadadhar, S.; Rocha, C.; Tort, O.; Marques Sousa, P.; Vacher, S.; Bieche, I.; Janke, C. Alterations in the balance of tubulin glycylation and glutamylation in photoreceptors leads to retinal degeneration. J. Cell Sci. 2017, 130, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Park, J.H.; Gumerson, J.; Wu, Z.; Swaroop, A.; Qian, H.; Roll-Mecak, A.; Li, T. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E2925–E2934. [Google Scholar] [CrossRef] [PubMed]
- Blanks, J.C.; Spee, C. Retinal degeneration in the pcd/pcd mutant mouse: Accumulation of spherules in the interphotoreceptor space. Exp. Eye Res. 1992, 54, 637–644. [Google Scholar] [CrossRef]
- Chen, J.; Qian, H.; Horai, R.; Chan, C.C.; Falick, Y.; Caspi, R.R. Comparative analysis of induced vs. spontaneous models of autoimmune uveitis targeting the interphotoreceptor retinoid binding protein. PLoS ONE 2013, 8, e72161. [Google Scholar] [CrossRef]
- Yu, M.; Zou, W.; Peachey, N.S.; McIntyre, T.M.; Liu, J. A novel role of complement in retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7684–7692. [Google Scholar] [CrossRef]
- Lyzogubov, V.V.; Bora, P.S.; Wu, X.; Horn, L.E.; de Roque, R.; Rudolf, X.V.; Atkinson, J.P.; Bora, N.S. The Complement Regulatory Protein CD46 Deficient Mouse Spontaneously Develops Dry-Type Age-Related Macular Degeneration-Like Phenotype. Am. J. Pathol. 2016, 186, 2088–2104. [Google Scholar] [CrossRef]
- Jobling, A.I.; Waugh, M.; Vessey, K.A.; Phipps, J.A.; Trogrlic, L.; Greferath, U.; Mills, S.A.; Tan, Z.L.; Ward, M.M.; Fletcher, E.L. The Role of the Microglial Cx3cr1 Pathway in the Postnatal Maturation of Retinal Photoreceptors. J. Neurosci. 2018, 38, 4708–4723. [Google Scholar] [CrossRef]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Y.; Wang, L.; Li, W. Age-related macular degeneration phenotypes are associated with increased tumor necrosis-alpha and subretinal immune cells in aged Cxcr5 knockout mice. PLoS ONE 2017, 12, e0173716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, J.; Chen, X.; Jie-Qiong, Z.; Li, X.; Luo, L.; Huang, H.; Liu, W.; Zhou, X.; Yan, J.; et al. Interferon Regulatory Factor 3 Deficiency Induces Age-Related Alterations of the Retina in Young and Old Mice. Front. Cell Neurosci. 2019, 13, 272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, Y.; Miller, M.J.; Peng, B.; Liu, L.; Soderland, C.; Tang, J.; Kern, T.S.; Pintar, J.; Steinle, J.J. IGFBP-3 and TNF-alpha regulate retinal endothelial cell apoptosis. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5376–5384. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ambati, J.; Anand, A.; Fernandez, S.; Sakurai, E.; Lynn, B.C.; Kuziel, W.A.; Rollins, B.J.; Ambati, B.K. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Coffey, P.J.; Gias, C.; McDermott, C.J.; Lundh, P.; Pickering, M.C.; Sethi, C.; Bird, A.; Fitzke, F.W.; Maass, A.; Chen, L.L.; et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. USA 2007, 104, 16651–16656. [Google Scholar] [CrossRef]
- Ueda, Y.; Mohammed, I.; Song, D.; Gullipalli, D.; Zhou, L.; Sato, S.; Wang, Y.; Gupta, S.; Cheng, Z.; Wang, H.; et al. Murine systemic thrombophilia and hemolytic uremic syndrome from a factor H point mutation. Blood 2017, 129, 1184–1196. [Google Scholar] [CrossRef]
- Ma, W.; Silverman, S.M.; Zhao, L.; Villasmil, R.; Campos, M.M.; Amaral, J.; Wong, W.T. Absence of TGFbeta signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. Elife 2019, 8. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Huang, L.; Yang, Y.; Zhang, L.; Yang, M.; Liu, W.; Ramasamy, K.; Jiang, Z.; Sundaresan, P.; et al. A splicing mutation in aryl hydrocarbon receptor associated with retinitis pigmentosa. Hum. Mol. Genet. 2018, 27, 2563–2572. [Google Scholar] [CrossRef]
- Swiderski, R.E.; Nakano, Y.; Mullins, R.F.; Seo, S.; Banfi, B. A mutation in the mouse ttc26 gene leads to impaired hedgehog signaling. PLoS Genet. 2014, 10, e1004689. [Google Scholar] [CrossRef]
- Omori, Y.; Kubo, S.; Kon, T.; Furuhashi, M.; Narita, H.; Kominami, T.; Ueno, A.; Tsutsumi, R.; Chaya, T.; Yamamoto, H.; et al. Samd7 is a cell type-specific PRC1 component essential for establishing retinal rod photoreceptor identity. Proc. Natl. Acad. Sci. USA 2017, 114, E8264–E8273. [Google Scholar] [CrossRef] [PubMed]
- Behnen, P.; Felline, A.; Comitato, A.; Di Salvo, M.T.; Raimondi, F.; Gulati, S.; Kahremany, S.; Palczewski, K.; Marigo, V.; Fanelli, F. A Small Chaperone Improves Folding and Routing of Rhodopsin Mutants Linked to Inherited Blindness. iScience 2018, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Chiang, W.C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef]
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef]
- Zhao, Y.; Hong, D.H.; Pawlyk, B.; Yue, G.; Adamian, M.; Grynberg, M.; Godzik, A.; Li, T. The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: Subserving RPGR function and participating in disk morphogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3965–3970. [Google Scholar] [CrossRef]
- Won, J.; Gifford, E.; Smith, R.S.; Yi, H.; Ferreira, P.A.; Hicks, W.L.; Li, T.; Naggert, J.K.; Nishina, P.M. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum. Mol. Genet. 2009, 18, 4329–4339. [Google Scholar] [CrossRef]
- Zhang, C.; Quan, R.; Wang, J. Development and application of CRISPR/Cas9 technologies in genomic editing. Hum. Mol. Genet. 2018, 27, R79–R88. [Google Scholar] [CrossRef]
- Comitato, A.; Schiroli, D.; La Marca, C.; Marigo, V. Differential Contribution of Calcium-Activated Proteases and ER-Stress in Three Mouse Models of Retinitis Pigmentosa Expressing P23H Mutant RHO. Adv. Exp. Med. Biol. 2019, 1185, 311–316. [Google Scholar] [CrossRef]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef]
- Kumar, M.V.; Nagineni, C.N.; Chin, M.S.; Hooks, J.J.; Detrick, B. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J. Neuroimmunol. 2004, 153, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Shiose, S.; Chen, Y.; Okano, K.; Roy, S.; Kohno, H.; Tang, J.; Pearlman, E.; Maeda, T.; Palczewski, K.; Maeda, A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J. Biol. Chem. 2011, 286, 15543–15555. [Google Scholar] [CrossRef] [PubMed]
- Vollrath, D.; Yasumura, D.; Benchorin, G.; Matthes, M.T.; Feng, W.; Nguyen, N.M.; Sedano, C.D.; Calton, M.A.; LaVail, M.M. Tyro3 Modulates Mertk-Associated Retinal Degeneration. PLoS Genet. 2015, 11, e1005723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Seo, S.; Bhattarai, S.; Bugge, K.; Searby, C.C.; Zhang, Q.; Drack, A.V.; Stone, E.M.; Sheffield, V.C. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum. Mol. Genet. 2014, 23, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Rachel, R.A.; May-Simera, H.L.; Veleri, S.; Gotoh, N.; Choi, B.Y.; Murga-Zamalloa, C.; McIntyre, J.C.; Marek, J.; Lopez, I.; Hackett, A.N.; et al. Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis. J. Clin. Investig. 2012, 122, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.N.; Zhang, W.; Li, L.; Ronquillo, C.; Baehr, W.; Khanna, H. Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum. Mol. Genet. 2016, 25, 2005–2012. [Google Scholar] [CrossRef]
- Schon, C.; Asteriti, S.; Koch, S.; Sothilingam, V.; Garcia Garrido, M.; Tanimoto, N.; Herms, J.; Seeliger, M.W.; Cangiano, L.; Biel, M.; et al. Loss of HCN1 enhances disease progression in mouse models of CNG channel-linked retinitis pigmentosa and achromatopsia. Hum. Mol. Genet. 2016, 25, 1165–1175. [Google Scholar] [CrossRef][Green Version]
- Quinn, P.M.; Mulder, A.A.; Henrique Alves, C.; Desrosiers, M.; de Vries, S.I.; Klooster, J.; Dalkara, D.; Koster, A.J.; Jost, C.R.; Wijnholds, J. Loss of CRB2 in Muller glial cells modifies a CRB1-associated retinitis pigmentosa phenotype into a Leber congenital amaurosis phenotype. Hum. Mol. Genet. 2019, 28, 105–123. [Google Scholar] [CrossRef]
- Ng, L.; Liu, H.; St Germain, D.L.; Hernandez, A.; Forrest, D. Deletion of the Thyroid Hormone-Activating Type 2 Deiodinase Rescues Cone Photoreceptor Degeneration but Not Deafness in Mice Lacking Type 3 Deiodinase. Endocrinology 2017, 158, 1999–2010. [Google Scholar] [CrossRef]
- van der Pluijm, I.; Garinis, G.A.; Brandt, R.M.; Gorgels, T.G.; Wijnhoven, S.W.; Diderich, K.E.; de Wit, J.; Mitchell, J.R.; van Oostrom, C.; Beems, R.; et al. Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007, 5, e2. [Google Scholar] [CrossRef]
- Chang, B.; Heckenlively, J.R.; Bayley, P.R.; Brecha, N.C.; Davisson, M.T.; Hawes, N.L.; Hirano, A.A.; Hurd, R.E.; Ikeda, A.; Johnson, B.A.; et al. The nob2 mouse, a null mutation in Cacna1f: Anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis. Neurosci. 2006, 23, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.M.; Kiang, S.; McNally, N.; Donovan, M.A.; Sieving, P.A.; Bush, R.A.; Machida, S.; Cotter, T.; Hobson, A.; Farrar, J.; et al. Comparative structural and functional analysis of photoreceptor neurons of Rho-/- mice reveal increased survival on C57BL/6J in comparison to 129Sv genetic background. Vis. Neurosci. 2001, 18, 437–443. [Google Scholar] [CrossRef]
- Liu, Q.; Saveliev, A.; Pierce, E.A. The severity of retinal degeneration in Rp1h gene-targeted mice is dependent on genetic background. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1566–1574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Haider, N.B.; Zhang, W.; Hurd, R.; Ikeda, A.; Nystuen, A.M.; Naggert, J.K.; Nishina, P.M. Mapping of genetic modifiers of Nr2e3 rd7/rd7 that suppress retinal degeneration and restore blue cone cells to normal quantity. Mamm. Genome 2008, 19, 145–154. [Google Scholar] [CrossRef]
- Threadgill, D.W.; Miller, D.R.; Churchill, G.A.; de Villena, F.P. The collaborative cross: A recombinant inbred mouse population for the systems genetic era. ILAR J. 2011, 52, 24–31. [Google Scholar] [CrossRef]
- Churchill, G.A.; Gatti, D.M.; Munger, S.C.; Svenson, K.L. The Diversity Outbred mouse population. Mamm. Genome 2012, 23, 713–718. [Google Scholar] [CrossRef]
- Cruz, N.M.; Yuan, Y.; Leehy, B.D.; Baid, R.; Kompella, U.; DeAngelis, M.M.; Escher, P.; Haider, N.B. Modifier genes as therapeutics: The nuclear hormone receptor Rev Erb alpha (Nr1d1) rescues Nr2e3 associated retinal disease. PLoS ONE 2014, 9, e87942. [Google Scholar] [CrossRef]
- Danciger, M.; Ogando, D.; Yang, H.; Matthes, M.T.; Yu, N.; Ahern, K.; Yasumura, D.; Williams, R.W.; Lavail, M.M. Genetic modifiers of retinal degeneration in the rd3 mouse. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2863–2869. [Google Scholar] [CrossRef]
- van Wyk, M.; Schneider, S.; Kleinlogel, S. Variable phenotypic expressivity in inbred retinal degeneration mouse lines: A comparative study of C3H/HeOu and FVB/N rd1 mice. Mol. Vis. 2015, 21, 811–827. [Google Scholar]
- Luhmann, U.F.; Carvalho, L.S.; Holthaus, S.M.; Cowing, J.A.; Greenaway, S.; Chu, C.J.; Herrmann, P.; Smith, A.J.; Munro, P.M.; Potter, P.; et al. The severity of retinal pathology in homozygous Crb1rd8/rd8 mice is dependent on additional genetic factors. Hum. Mol. Genet. 2015, 24, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Charette, J.R.; Philip, V.M.; Stearns, T.M.; Zhang, W.; Naggert, J.K.; Krebs, M.P.; Nishina, P.M. Genetic modifier loci of mouse Mfrp(rd6) identified by quantitative trait locus analysis. Exp. Eye Res. 2014, 118, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Maddox, D.M.; Ikeda, S.; Ikeda, A.; Zhang, W.; Krebs, M.P.; Nishina, P.M.; Naggert, J.K. An allele of microtubule-associated protein 1A (Mtap1a) reduces photoreceptor degeneration in Tulp1 and Tub Mutant Mice. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1663–1669. [Google Scholar] [CrossRef]
- Kong, Y.; Zhao, L.; Charette, J.R.; Hicks, W.L.; Stone, L.; Nishina, P.M.; Naggert, J.K. An FRMD4B variant suppresses dysplastic photoreceptor lesions in models of enhanced S-cone syndrome and of Nrl deficiency. Hum. Mol. Genet. 2018, 27, 3340–3352. [Google Scholar] [CrossRef] [PubMed]
- Paskowitz, D.M.; LaVail, M.M.; Duncan, J.L. Light and inherited retinal degeneration. Br. J. Ophthalmol. 2006, 90, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Dellett, M.; Sasai, N.; Nishide, K.; Becker, S.; Papadaki, V.; Limb, G.A.; Moore, A.T.; Kondo, T.; Ohnuma, S. Genetic background and light-dependent progression of photoreceptor cell degeneration in Prominin-1 knockout mice. Investig. Ophthalmol. Vis. Sci. 2014, 56, 164–176. [Google Scholar] [CrossRef]
- Naash, M.L.; Peachey, N.S.; Li, Z.Y.; Gryczan, C.C.; Goto, Y.; Blanks, J.; Milam, A.H.; Ripps, H. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Investig. Ophthalmol. Vis. Sci. 1996, 37, 775–782. [Google Scholar]
- Rascher, K.; Servos, G.; Berthold, G.; Hartwig, H.G.; Warskulat, U.; Heller-Stilb, B.; Haussinger, D. Light deprivation slows but does not prevent the loss of photoreceptors in taurine transporter knockout mice. Vis. Res. 2004, 44, 2091–2100. [Google Scholar] [CrossRef]
- Smith, S.B.; Cope, B.K.; McCoy, J.R. Effects of dark-rearing on the retinal degeneration of the C57BL/6-mivit/mivit mouse. Exp. Eye Res. 1994, 58, 77–84. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Sun, W.; Jastrzebska, B.; Hatala, D.A.; Winkens, H.J.; Hofmann, K.P.; Janssen, J.J.; Baehr, W.; et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J. Biol. Chem. 2006, 281, 37697–37704. [Google Scholar] [CrossRef]
- Ettaiche, M.; Guy, N.; Hofman, P.; Lazdunski, M.; Waldmann, R. Acid-sensing ion channel 2 is important for retinal function and protects against light-induced retinal degeneration. J. Neurosci. 2004, 24, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.W.; Zallocchi, M.; Wang, W.M.; Delimont, D.; Cosgrove, D. Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6421–6427. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Wang, W.; Delimont, D.; Cheung, L.; Zallocchi, M.; Cosgrove, D.; Peng, Y.W. Photoreceptors in whirler mice show defective transducin translocation and are susceptible to short-term light/dark changes-induced degeneration. Exp. Eye Res. 2014, 118, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Anderson, R.E.; Tomita, H.; Adler, R.; Liu, X.; Zack, D.J.; Rajala, R.V. Nonredundant role of Akt2 for neuroprotection of rod photoreceptor cells from light-induced cell death. J. Neurosci. 2007, 27, 203–211. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Yates, J.R.; Bradley, M.; Moore, A.T.; Bird, A.C.; Genetic Factors in AMD Study. Smoking and age related macular degeneration: The number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 2006, 90, 75–80. [Google Scholar] [CrossRef]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef]
- Fujihara, M.; Nagai, N.; Sussan, T.E.; Biswal, S.; Handa, J.T. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE 2008, 3, e3119. [Google Scholar] [CrossRef]
- Ebrahimi, K.B.; Cano, M.; Rhee, J.; Datta, S.; Wang, L.; Handa, J.T. Oxidative Stress Induces an Interactive Decline in Wnt and Nrf2 Signaling in Degenerating Retinal Pigment Epithelium. Antioxid. Redox Signal. 2018, 29, 389–407. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Veniant, M.M.; Cham, C.M.; Flynn, L.M.; Pierotti, V.; Loring, J.F.; Traber, M.; Ruland, S.; Stokowski, R.S.; Huszar, D.; et al. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc. Natl. Acad. Sci. USA 1996, 93, 6393–6398. [Google Scholar] [CrossRef]
- Schmidt-Erfurth, U.; Rudolf, M.; Funk, M.; Hofmann-Rummelt, C.; Franz-Haas, N.S.; Aherrahrou, Z.; Schlotzer-Schrehardt, U. Ultrastructural changes in a murine model of graded Bruch membrane lipoidal degeneration and corresponding VEGF164 detection. Investig. Ophthalmol. Vis. Sci. 2008, 49, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Malek, G.; Johnson, L.V.; Mace, B.E.; Saloupis, P.; Schmechel, D.E.; Rickman, D.W.; Toth, C.A.; Sullivan, P.M.; Bowes Rickman, C. Apolipoprotein E allele-dependent pathogenesis: A model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 11900–11905. [Google Scholar] [CrossRef] [PubMed]
- Perusek, L.; Maeda, T. Vitamin A derivatives as treatment options for retinal degenerative diseases. Nutrients 2013, 5, 2646–2666. [Google Scholar] [CrossRef] [PubMed]
- Perusek, L.; Maeda, A.; Maeda, T. Supplementation with vitamin a derivatives to rescue vision in animal models of degenerative retinal diseases. Methods Mol. Biol. 2015, 1271, 345–362. [Google Scholar] [CrossRef]
- Guadagni, V.; Novelli, E.; Piano, I.; Gargini, C.; Strettoi, E. Pharmacological approaches to retinitis pigmentosa: A laboratory perspective. Prog. Retin. Eye Res. 2015, 48, 62–81. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; McGinnis, J.F. Oxidative stress: The achilles’ heel of neurodegenerative diseases of the retina. Front. Biosci. Landmark. Ed. 2012, 17, 1976–1995. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef]
- Ebert, S.; Weigelt, K.; Walczak, Y.; Drobnik, W.; Mauerer, R.; Hume, D.A.; Weber, B.H.; Langmann, T. Docosahexaenoic acid attenuates microglial activation and delays early retinal degeneration. J. Neurochem. 2009, 110, 1863–1875. [Google Scholar] [CrossRef]
- Yu, M.; Yan, W.; Beight, C. Lutein and Zeaxanthin Isomers Reduce Photoreceptor Degeneration in the Pde6b (rd10) Mouse Model of Retinitis Pigmentosa. Biomed. Res. Int. 2018, 2018, 4374087. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef]
- Barone, I.; Novelli, E.; Piano, I.; Gargini, C.; Strettoi, E. Environmental enrichment extends photoreceptor survival and visual function in a mouse model of retinitis pigmentosa. PLoS ONE 2012, 7, e50726. [Google Scholar] [CrossRef]
- Sundin, O.H.; Leppert, G.S.; Silva, E.D.; Yang, J.M.; Dharmaraj, S.; Maumenee, I.H.; Santos, L.C.; Parsa, C.F.; Traboulsi, E.I.; Broman, K.W.; et al. Extreme hyperopia is the result of null mutations in MFRP, which encodes a Frizzled-related protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9553–9558. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H. The mouse’s eye and Mfrp: Not quite human. Ophthalmic Genet. 2005, 26, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Kameya, S.; Hawes, N.L.; Chang, B.; Heckenlively, J.R.; Naggert, J.K.; Nishina, P.M. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum. Mol. Genet. 2002, 11, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Almoallem, B.; Arno, G.; De Zaeytijd, J.; Verdin, H.; Balikova, I.; Casteels, I.; de Ravel, T.; Hull, S.; Suzani, M.; Destree, A.; et al. The majority of autosomal recessive nanophthalmos and posterior microphthalmia can be attributed to biallelic sequence and structural variants in MFRP and PRSS56. Sci. Rep. 2020, 10, 1289. [Google Scholar] [CrossRef] [PubMed]
- Chekuri, A.; Sahu, B.; Chavali, V.R.M.; Voronchikhina, M.; Soto-Hermida, A.; Suk, J.J.; Alapati, A.N.; Bartsch, D.U.; Ayala-Ramirez, R.; Zenteno, J.C.; et al. Long-Term Effects of Gene Therapy in a Novel Mouse Model of Human MFRP-Associated Retinopathy. Hum. Gene Ther. 2019, 30, 632–650. [Google Scholar] [CrossRef] [PubMed]
- Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Brown, K.A.; Antonio, M.; Beisel, K.W.; Steel, K.P.; Brown, S.D. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 1995, 374, 62–64. [Google Scholar] [CrossRef]
- Lentz, J.J.; Gordon, W.C.; Farris, H.E.; MacDonald, G.H.; Cunningham, D.E.; Robbins, C.A.; Tempel, B.L.; Bazan, N.G.; Rubel, E.W.; Oesterle, E.C.; et al. Deafness and retinal degeneration in a novel USH1C knock-in mouse model. Dev. Neurobiol. 2010, 70, 253–267. [Google Scholar] [CrossRef]
- Karan, G.; Lillo, C.; Yang, Z.; Cameron, D.J.; Locke, K.G.; Zhao, Y.; Thirumalaichary, S.; Li, C.; Birch, D.G.; Vollmer-Snarr, H.R.; et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: A model for macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 4164–4169. [Google Scholar] [CrossRef]
- Grishchuk, Y.; Stember, K.G.; Matsunaga, A.; Olivares, A.M.; Cruz, N.M.; King, V.E.; Humphrey, D.M.; Wang, S.L.; Muzikansky, A.; Betensky, R.A.; et al. Retinal Dystrophy and Optic Nerve Pathology in the Mouse Model of Mucolipidosis IV. Am. J. Pathol. 2016, 186, 199–209. [Google Scholar] [CrossRef]
- Amir, N.; Zlotogora, J.; Bach, G. Mucolipidosis type IV: Clinical spectrum and natural history. Pediatrics 1987, 79, 953–959. [Google Scholar] [PubMed]
- Park, H.; Qazi, Y.; Tan, C.; Jabbar, S.B.; Cao, Y.; Schmid, G.; Pardue, M.T. Assessment of axial length measurements in mouse eyes. Optom. Vis. Sci. 2012, 89, 296–303. [Google Scholar] [CrossRef]
- Ruppersburg, C.C.; Hartzell, H.C. The Ca2+-activated Cl- channel ANO1/TMEM16A regulates primary ciliogenesis. Mol. Biol. Cell 2014, 25, 1793–1807. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ye, W.; Wang, W.J.; Sison, E.S.; Jan, Y.N.; Jan, L.Y. Cytoplasmic Cl(-) couples membrane remodeling to epithelial morphogenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E11161–E11169. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Fabian, L.; Brill, J.A. Phosphatidylinositol 4,5-bisphosphate regulates cilium transition zone maturation in Drosophila melanogaster. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Bhatia, V.; Valdes-Sanchez, L.; Rodriguez-Martinez, D.; Bhattacharya, S.S. Formation of 53BP1 foci and ATM activation under oxidative stress is facilitated by RNA:DNA hybrids and loss of ATM-53BP1 expression promotes photoreceptor cell survival in mice. F1000Res 2018, 7, 1233. [Google Scholar] [CrossRef]
- Muller, B.; Ellinwood, N.M.; Lorenz, B.; Stieger, K. Detection of DNA Double Strand Breaks by gammaH2AX Does Not Result in 53bp1 Recruitment in Mouse Retinal Tissues. Front. Neurosci. 2018, 12, 286. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collin, G.B.; Gogna, N.; Chang, B.; Damkham, N.; Pinkney, J.; Hyde, L.F.; Stone, L.; Naggert, J.K.; Nishina, P.M.; Krebs, M.P. Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells 2020, 9, 931. https://doi.org/10.3390/cells9040931
Collin GB, Gogna N, Chang B, Damkham N, Pinkney J, Hyde LF, Stone L, Naggert JK, Nishina PM, Krebs MP. Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells. 2020; 9(4):931. https://doi.org/10.3390/cells9040931
Chicago/Turabian StyleCollin, Gayle B., Navdeep Gogna, Bo Chang, Nattaya Damkham, Jai Pinkney, Lillian F. Hyde, Lisa Stone, Jürgen K. Naggert, Patsy M. Nishina, and Mark P. Krebs. 2020. "Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss" Cells 9, no. 4: 931. https://doi.org/10.3390/cells9040931
APA StyleCollin, G. B., Gogna, N., Chang, B., Damkham, N., Pinkney, J., Hyde, L. F., Stone, L., Naggert, J. K., Nishina, P. M., & Krebs, M. P. (2020). Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells, 9(4), 931. https://doi.org/10.3390/cells9040931