Tumor-Intrinsic or Drug-Induced Immunogenicity Dictates the Therapeutic Success of the PD1/PDL Axis Blockade

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell lines and Reagents
2.3. Primary Cells
2.4. In Vitro Treatments
2.5. Therapeutic Protocol
2.6. IFNγ ELISpot
2.7. Flow Cytometry
2.8. Intracellular Staining
2.9. mRNA Extraction and Real-Time PCR
2.10. Morphometric Analysis of MCA205 Tumor Cells
2.11. Statistical Analysis
3. Results
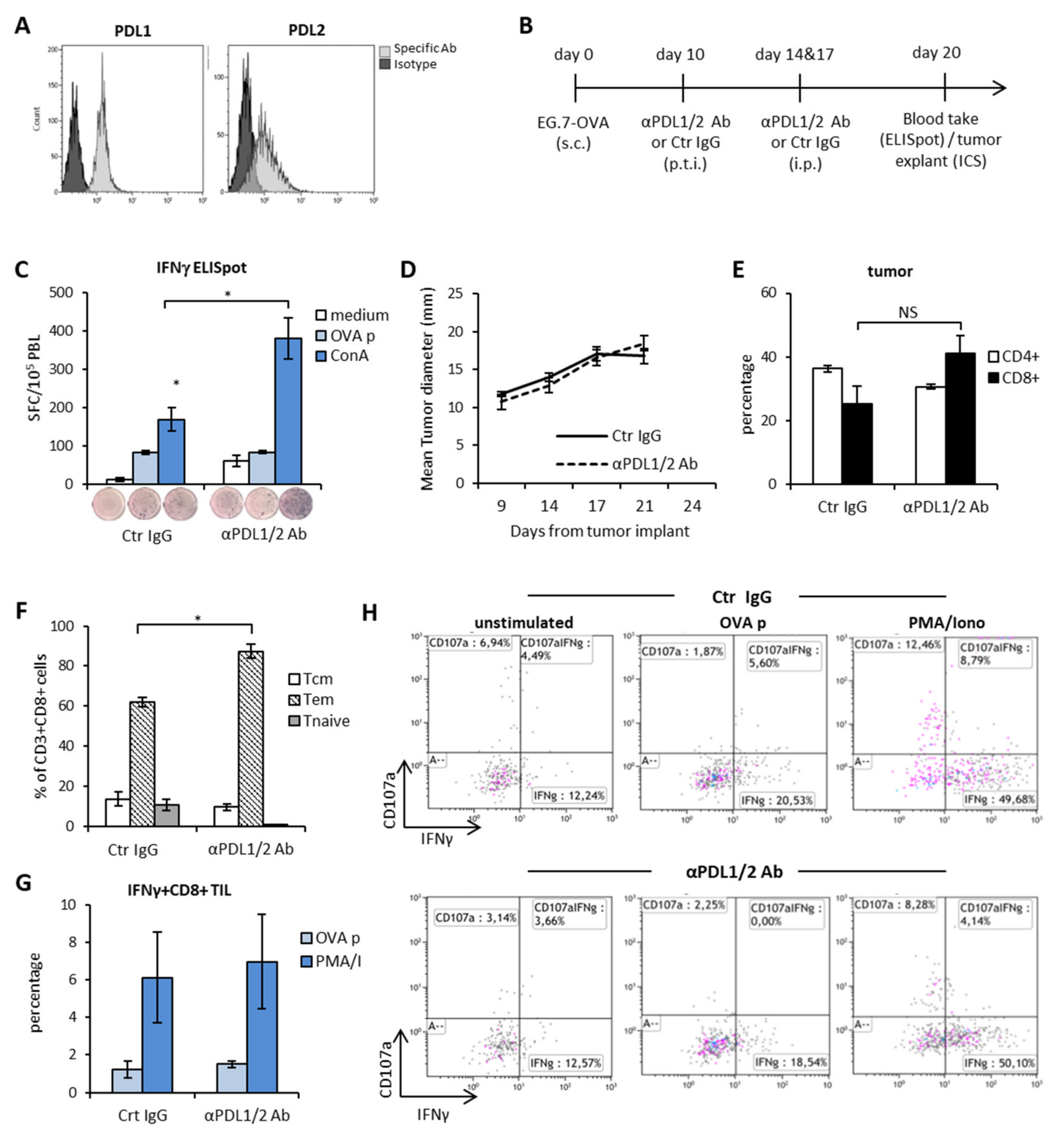
3.1. PD1-PDL Axis Blockade Induces Systemic Polyclonal Immune Activation but no Tumor Specific Immune Responses or CD8+ T Cells and Mice Survival
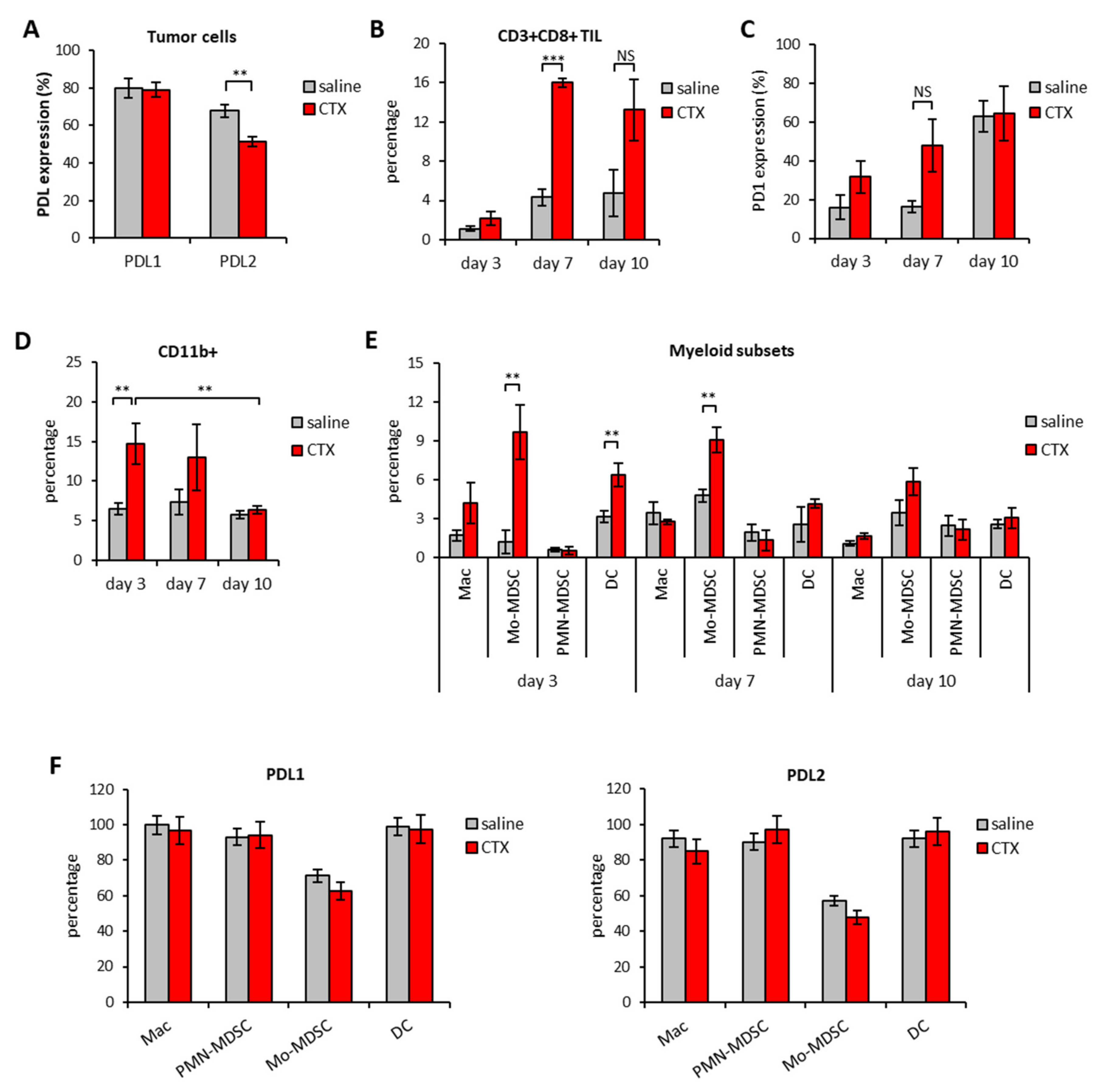
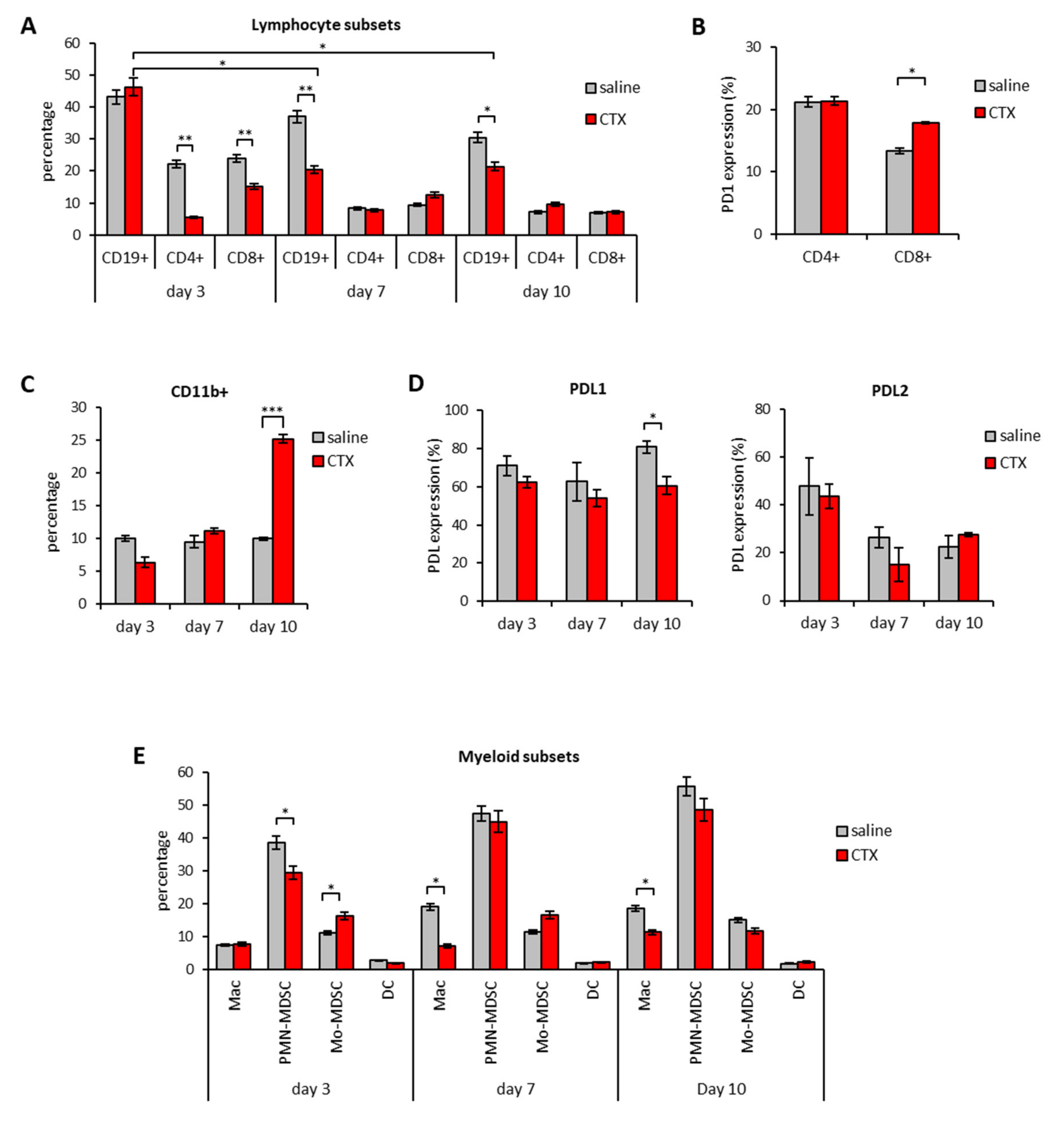
3.2. Modulation of PD1/PDL Molecules in Tumor and Spleen of Mice Treated with CTX
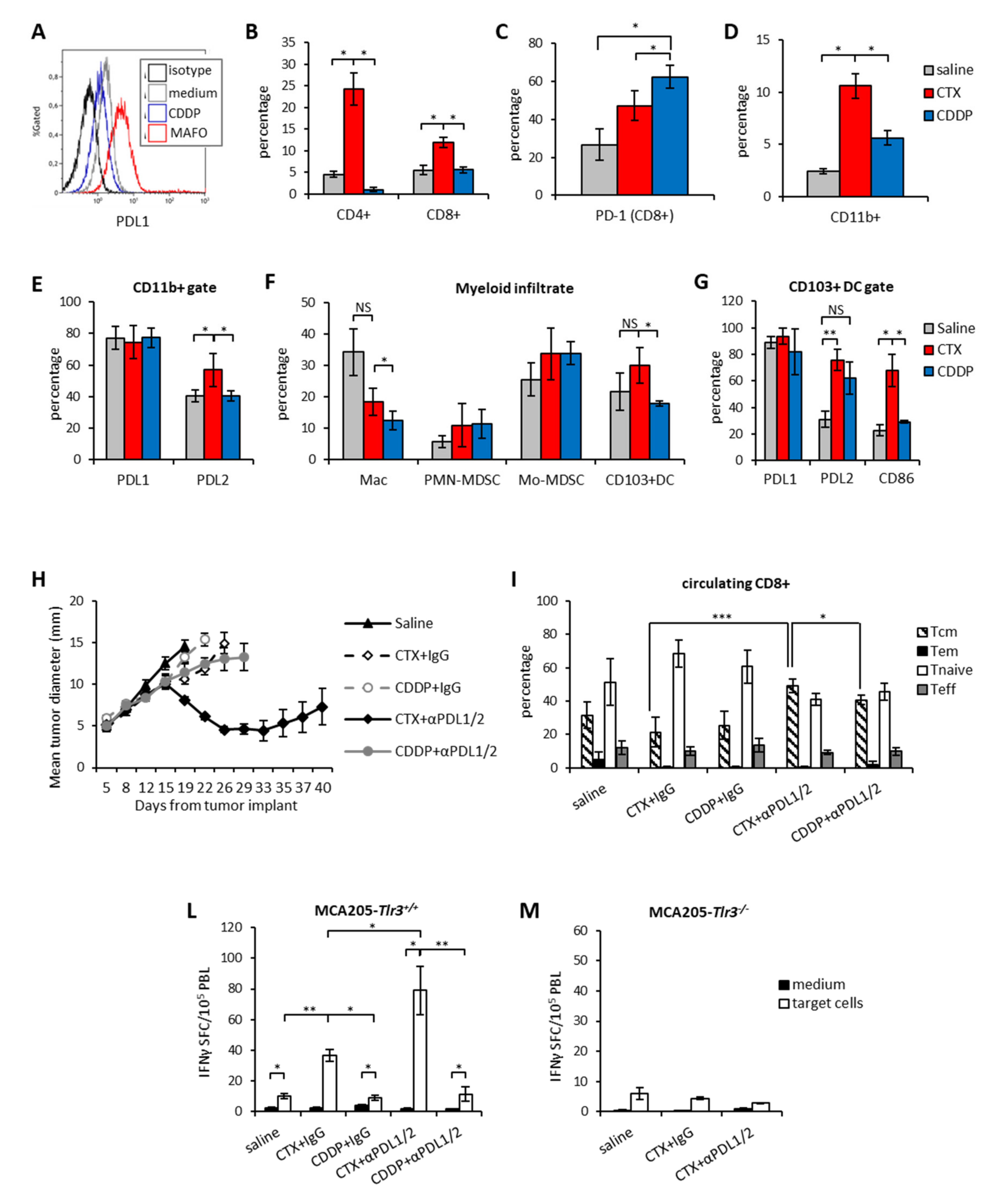
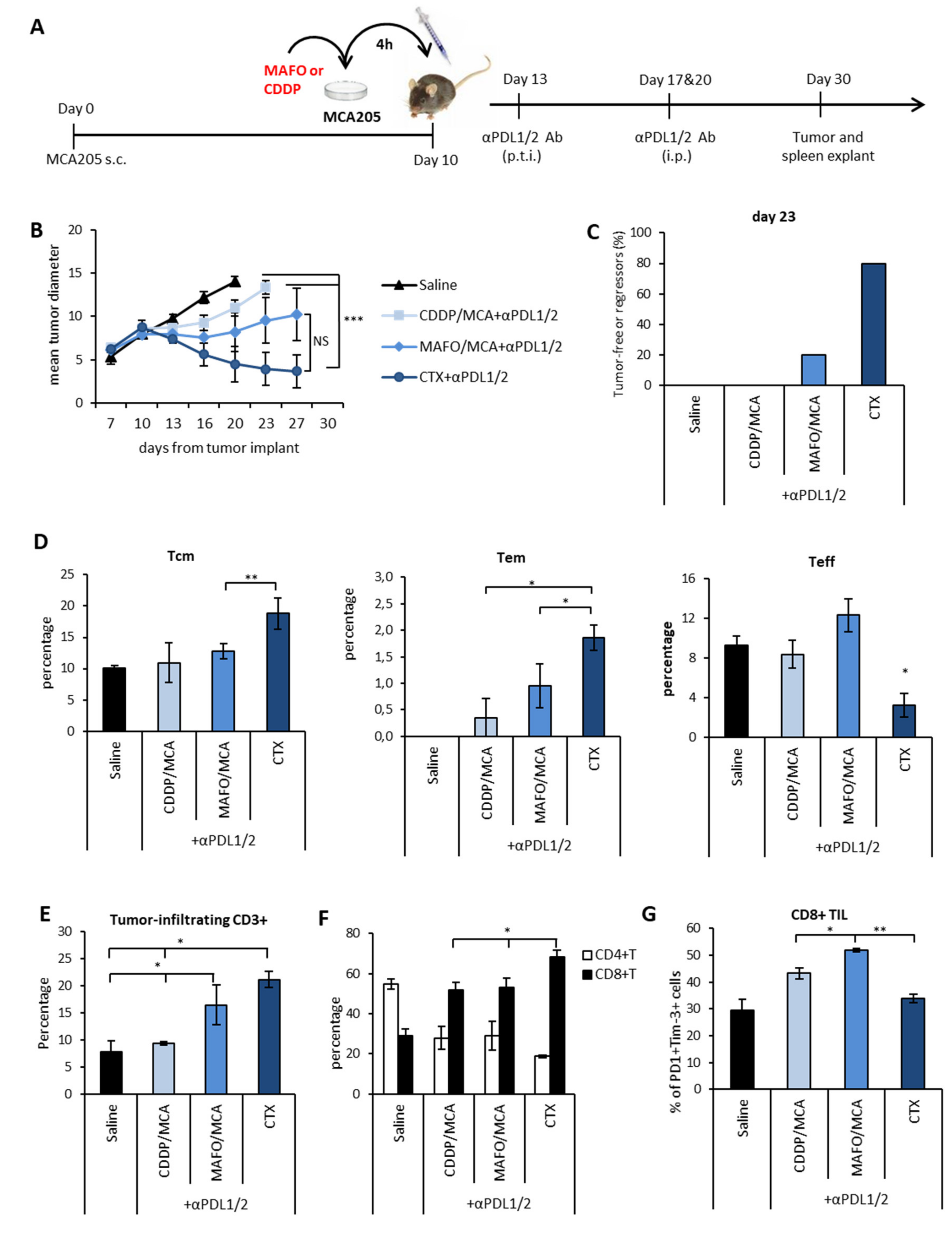
3.3. Therapeutic Synergism between CTX and PDL1/2 Blockade and Immune Correlates of Therapeutic Efficacy
3.4. Immunogenic Chemotherapy Augments the Therapeutic Efficacy of PDL Blockade in Poorly Immunogenic Tumors
3.5. Chemotherapy-Induced Vaccination only Partially Explains the Therapeutic Synergism between Immunogenic Chemotherapy and PDL Blockade
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Larkins, E.; Blumenthal, G.M.; Yuan, W.; He, K.; Sridhara, R.; Subramaniam, S.; Zhao, H.; Liu, C.; Yu, J.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma with Disease Progression on or After Platinum-Containing Chemotherapy. Oncol. 2017, 22, 873–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front. Immunol. 2018, 9, 1739. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.J.; Kendall, S.M.; Makarov, V.; Walsh, L.; Desrichard, A.; Weinhold, N.; Chan, T.A. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016, 48, 1327–1329. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Sci. 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signaling prevents anti-tumour immunity. Nat. 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2015, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, E.M.; Topalian, S.L.; et al. Colocalization of Inflammatory Response with B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive Resistance Mechanism of Immune Escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Jacquelot, N.; Yamazaki, T.; Roberti, M.P.; Duong, C.P.M.; Andrews, M.C.; Verlingue, L.; Ferrere, G.; Becharef, S.; Vétizou, M.; Daillère, R.; et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019, 29, 846–861. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2013, 21, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavoni, G.; Sistigu, A.; Valentíni, M.; Mattei, F.; Sestili, P.; Spadaro, F.; Sánchez, M.; Lorenzi, S.; D’Urso, M.T.; Belardelli, F.; et al. Cyclophosphamide Synergizes with Type I Interferons through Systemic Dendritic Cell Reactivation and Induction of Immunogenic Tumor Apoptosis. Cancer Res. 2010, 71, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2006, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Negri, D.; Sestili, P.; Borghi, M.; Ciccolella, M.; Bracci, L. Enzyme-linked immunospot assay to monitor antigen-specific cellular immune responses in mouse tumor models. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 632, pp. 457–477. [Google Scholar]
- Montoya, M.; Schiavoni, G.; Mattei, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’Urso, M.T.; et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. OncoImmunology 2017, 6, e1317420. [Google Scholar] [CrossRef] [Green Version]
- The R Foundation. The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 April 2020).
- Raivo, K. pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-projectproject.org/web/packages/pheatmap/ (accessed on 10 April 2020).
- Research Services Branch. Available online: https://imagej.nih (accessed on 10 April 2020).
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic microenvironment plays a key role in human melanoma progression through a sustained exosome mediated transfer of clinically relevant metastatic molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.-S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 2019, 8, 26. [Google Scholar] [CrossRef]
- Sistigu, A.; Viaud, S.; Chaput, N.; Bracci, L.; Proietti, E.; Zitvogel, L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 2011, 33, 369–383. [Google Scholar] [CrossRef]
- Michaud, M.; Sukkurwala, A.Q.; Di Sano, F.; Zitvogel, L.; Kepp, O.; Kroemer, G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. OncoImmunology 2014, 3, e28276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boes, M.; Meyer-Wentrup, F. TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett. 2015, 361, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; E Baracco, E.; Remédios, C.; et al. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Takeda, Y.; Yoshida, S.; Takashima, K.; Ishii-Mugikura, N.; Shime, H.; Seya, T.; Matsumoto, M. Vaccine immunotherapy with ARNAX induces tumor-specific memory T cells and durable anti-tumor immunity in mouse models. Cancer Sci. 2018, 109, 2119–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, E.; Greco, G.; Garrone, B.; Baccarini, S.; Mauri, C.; Venditti, M.; Carlei, D.; Belardelli, F. Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. J. Clin. Investig. 1998, 101, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Moschella, F.; Sestili, P.; La Sorsa, V.; Valentini, M.; Canini, I.; Baccarini, S.; Maccari, S.; Ramoni, C.; Belardelli, F.; et al. Cyclophosphamide Enhances the Antitumor Efficacy of Adoptively Transferred Immune Cells through the Induction of Cytokine Expression, B-Cell and T-Cell Homeostatic Proliferation, and Specific Tumor Infiltration. Clin. Cancer Res. 2007, 13, 644–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsiak, M.E.C.; Semnani, R.T.; De Pascalis, R.; Kashmiri, S.V.S.; Schlom, J.; Sabzevari, H. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef]
- Moschella, F.; Valentini, M.; Macchia, I.; Sestili, P.; D’Urso, M.T.; Alessandri, C.; Belardelli, F.; Proietti, E.; Aricò, E. Unraveling Cancer Chemoimmunotherapy Mechanisms by Gene and Protein Expression Profiling of Responses to Cyclophosphamide. Cancer Res. 2011, 71, 3528–3539. [Google Scholar] [CrossRef] [Green Version]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Kataoka, K.; Yamagishi, J.; Ogawa, S.; Seya, T.; Matsumoto, M. A TLR3-Specific Adjuvant Relieves Innate Resistance to PD-L1 Blockade without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep. 2017, 19, 1874–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Huang, X.; Huang, G.; Chen, Y.; Chen, L.; Song, H. Preconditioning Chemotherapy with Cisplatin Enhances the Antitumor Activity of Cytokine-Induced Killer Cells in a Murine Melanoma Model. Cancer Biotherapy Radiopharm. 2012, 27, 210–220. [Google Scholar] [CrossRef]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immun. 2018, 48, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 938. [Google Scholar] [CrossRef]
- Matsuo, K.; Kitahata, K.; Kawabata, F.; Kamei, M.; Hara, Y.; Takamura, S.; Oiso, N.; Kawada, A.; Yoshie, O.; Nakayama, T. A Highly Active Form of XCL1/Lymphotactin Functions as an Effective Adjuvant to Recruit Cross-Presenting Dendritic Cells for Induction of Effector and Memory CD8+ T Cells. Front. Immunol. 2018, 9, 2775. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef] [Green Version]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef]
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar] [CrossRef]
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Andreone, S.; Spadaro, F.; Buccione, C.; Mancini, J.; Tinari, A.; Sestili, P.; Gambardella, A.; Lucarini, V.; Ziccheddu, G.; Parolini, I.; et al. IL-33 Promotes CD11b/CD18-Mediated Adhesion of Eosinophils to Cancer Cells and Synapse-Polarized Degranulation Leading to Tumor Cell Killing. Cancers 2019, 11, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, S.C.S.; Hu, X.; Panten, J.; Grees, M.; Renders, S.; Thomas, D.; Weber, R.; Schulze, T.J.; Utikal, J.; Umansky, V. Eosinophil accumulation predicts response to melanoma treatment with immune checkpoint inhibitors. OncoImmunology 2020, 9, 1727116. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward and Reverse Primers (5′–3′) | NCBI Accession Number | Amplicon Size (Base Pairs) |
|---|---|---|---|
| IFN-γ | TCAAGTGGCATAGATGTGGAAGAA TGGCTCTGCAGGATTTTCATG | NM_008337.4 | 92 bp |
| IL-2 | CCTGAGCAGGATGGAGAATTACA TCCAGAACATGCCGCAGAG | NM_008366.3 | 141 bp |
| IL-4 | ACAGGAGAAGGGACGCCAT GAAGCCCTACAGACGAGCTCA | NM_021283.2 | 95 bp |
| IL-10 | GGTTGCCAAGCCTTATCGGA ACCTGCTCCACTGCCTTGCT | NM_010548.2 | 191 bp |
| IL-12 | GGAAGCACGGCAGCAGAATA AACTTGAGGGAGAAGTAGGAATGG | NM_001303244.1 | 180 bp |
| IL-13 | AGACCAGACTCCCCTGTGCA TGGGTCCTGTAGATGGCATTG | NM_008355.3 | 123 bp |
| IL-17 | GCTCCAGAAGGCCCTCAGA AGCTTTCCCTCCGCATTGA | NM_010552.3 | 142 bp |
| IL-33 | GGGCTCACTGCAGGA GGACCAGGGCTTCGC | NM_ 001164724.2 | 147 bp |
| TGF-β1 | TGACGTCACTGGAGTTGTACGG GGTTCATGTCATGGATGGTGC | NM_ 011577.2 | 170 bp |
| Prf1 | GTGTCGCATGTACAG TGTGGTAAGCATGCT | NM_ 011073.3 | 116 bp |
| Gzmb | GATCGGGAGTGTGAGTCCTAC GAAAGCACGTGGAGGTGAAC | NM_013542.2 | 183 bp |
| TLR-3 | TTGCGTTGCGAAGTGAAGAA CAGTTGGGCGTTGTTCAAGA | NM_126166.5 | 149 bp |
| HPRT | CTGGTGAAAAGGACCTCTCG TGAAGTACTCATTATAGTCAAGGGCA | NM_013556.2 | 109 bp |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; Lucarini, V.; Macchia, I.; Sestili, P.; Buccione, C.; Donati, S.; Ciccolella, M.; Sistigu, A.; D’Urso, M.T.; Pacca, A.M.; et al. Tumor-Intrinsic or Drug-Induced Immunogenicity Dictates the Therapeutic Success of the PD1/PDL Axis Blockade. Cells 2020, 9, 940. https://doi.org/10.3390/cells9040940
Rossi A, Lucarini V, Macchia I, Sestili P, Buccione C, Donati S, Ciccolella M, Sistigu A, D’Urso MT, Pacca AM, et al. Tumor-Intrinsic or Drug-Induced Immunogenicity Dictates the Therapeutic Success of the PD1/PDL Axis Blockade. Cells. 2020; 9(4):940. https://doi.org/10.3390/cells9040940
Chicago/Turabian StyleRossi, Alessandra, Valeria Lucarini, Iole Macchia, Paola Sestili, Carla Buccione, Simona Donati, Maria Ciccolella, Antonella Sistigu, Maria Teresa D’Urso, Anna Maria Pacca, and et al. 2020. "Tumor-Intrinsic or Drug-Induced Immunogenicity Dictates the Therapeutic Success of the PD1/PDL Axis Blockade" Cells 9, no. 4: 940. https://doi.org/10.3390/cells9040940