Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny


Abstract
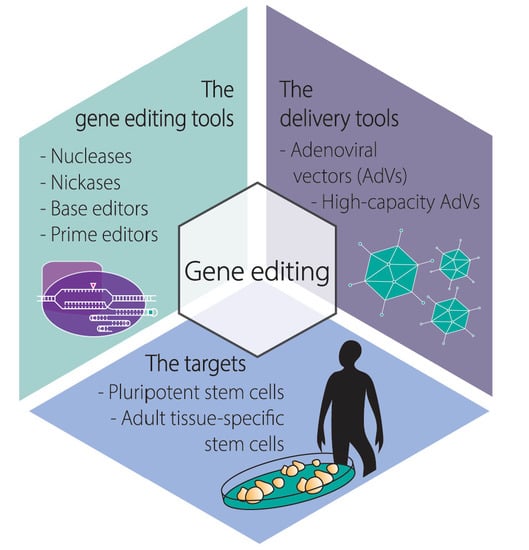
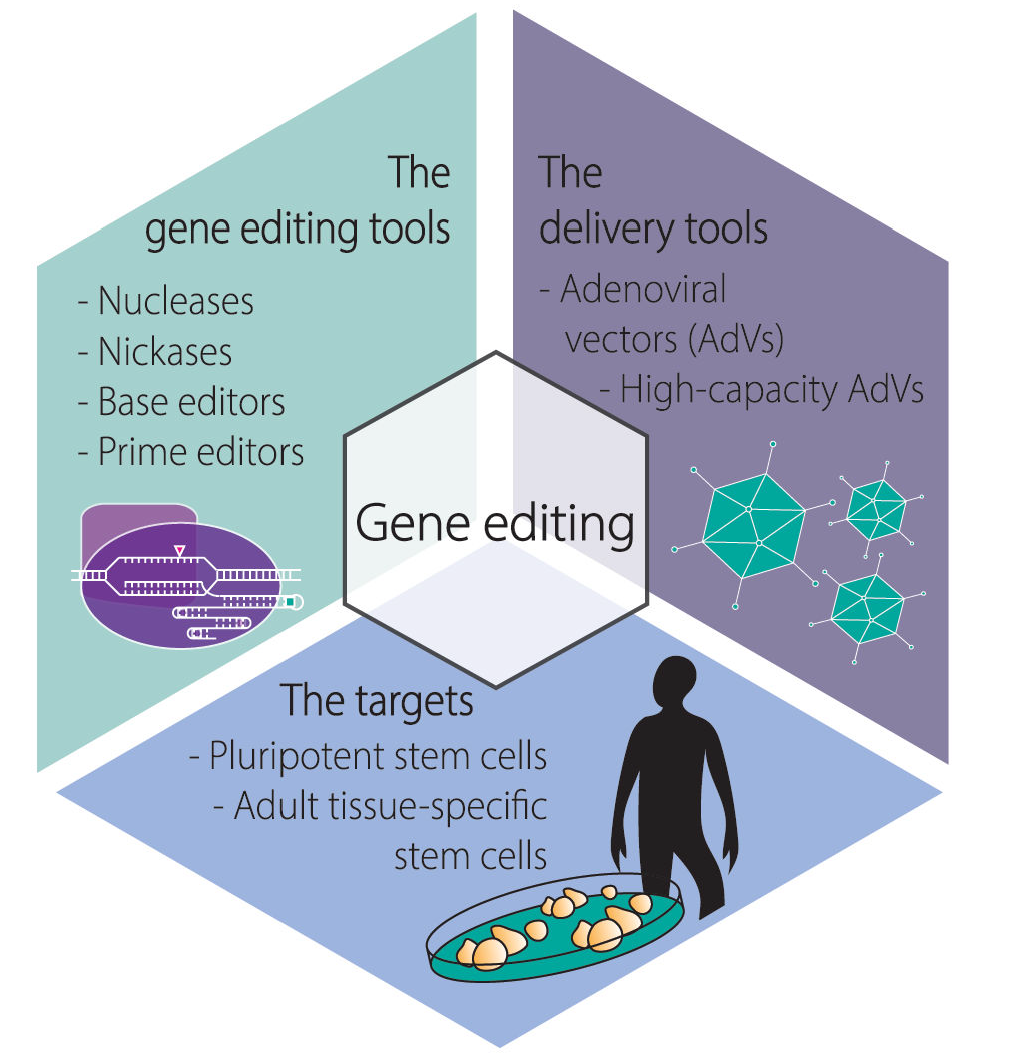
:
{kind=link}
{kind=link}
{kind=link}
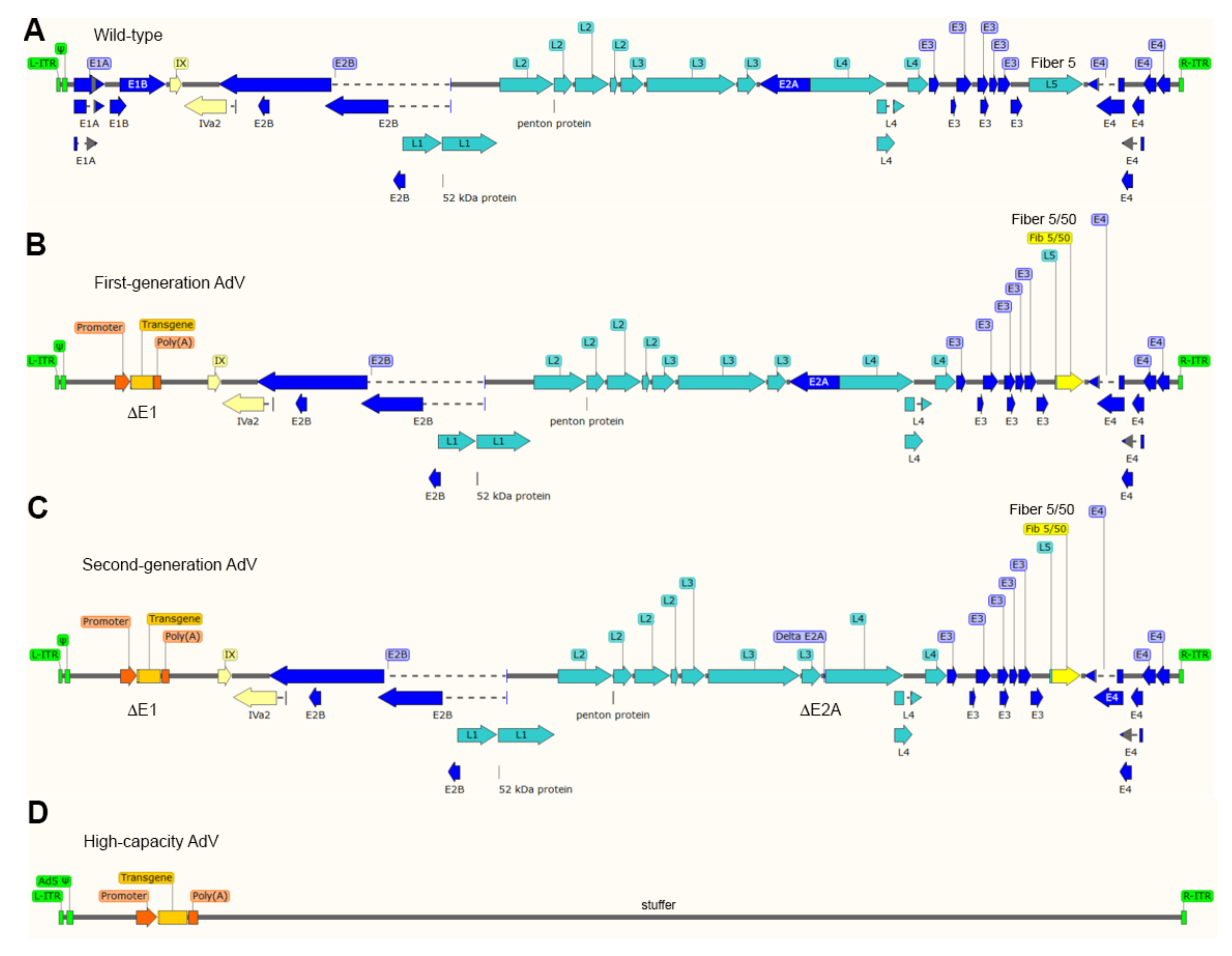{kind=link}
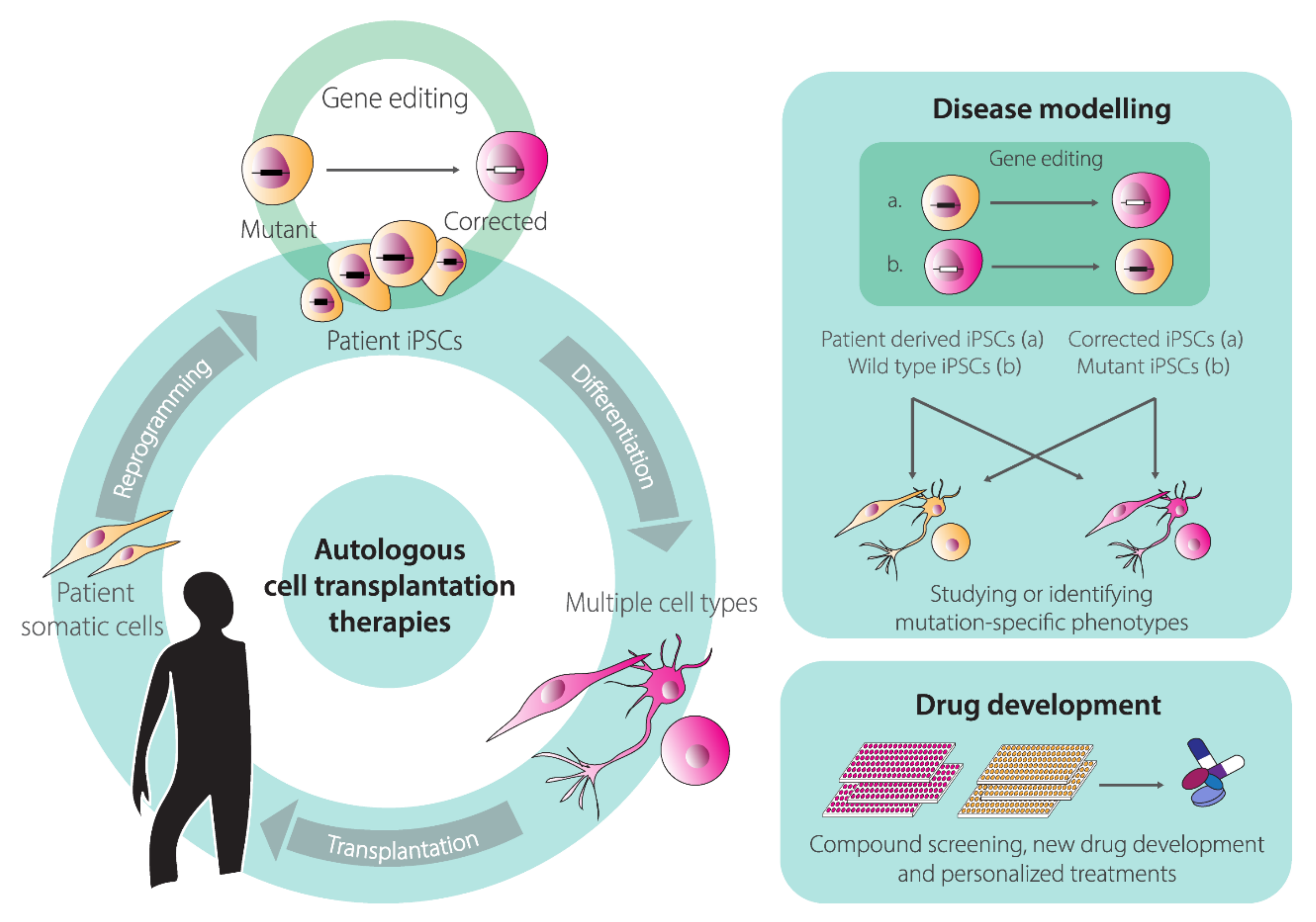{kind=link}
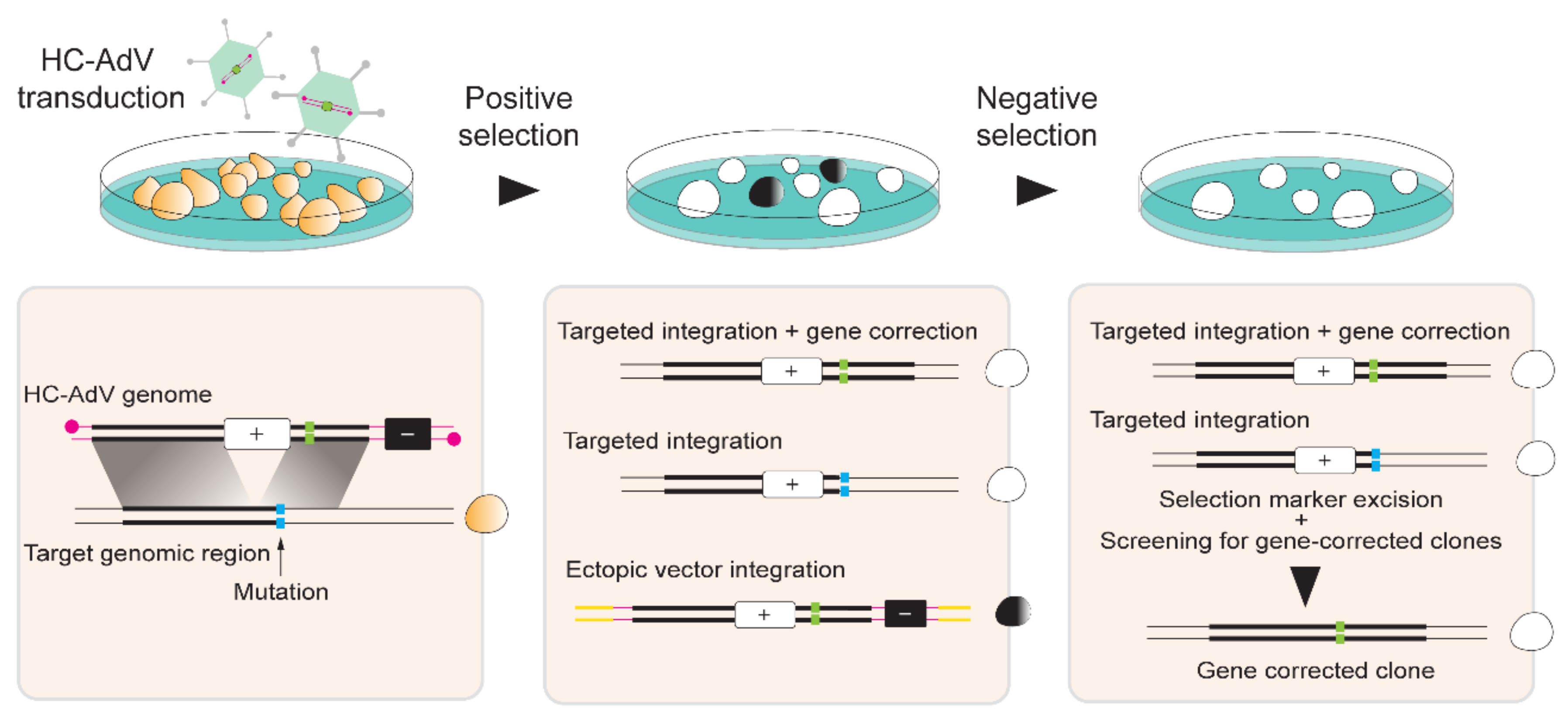{kind=link}
1. Introduction
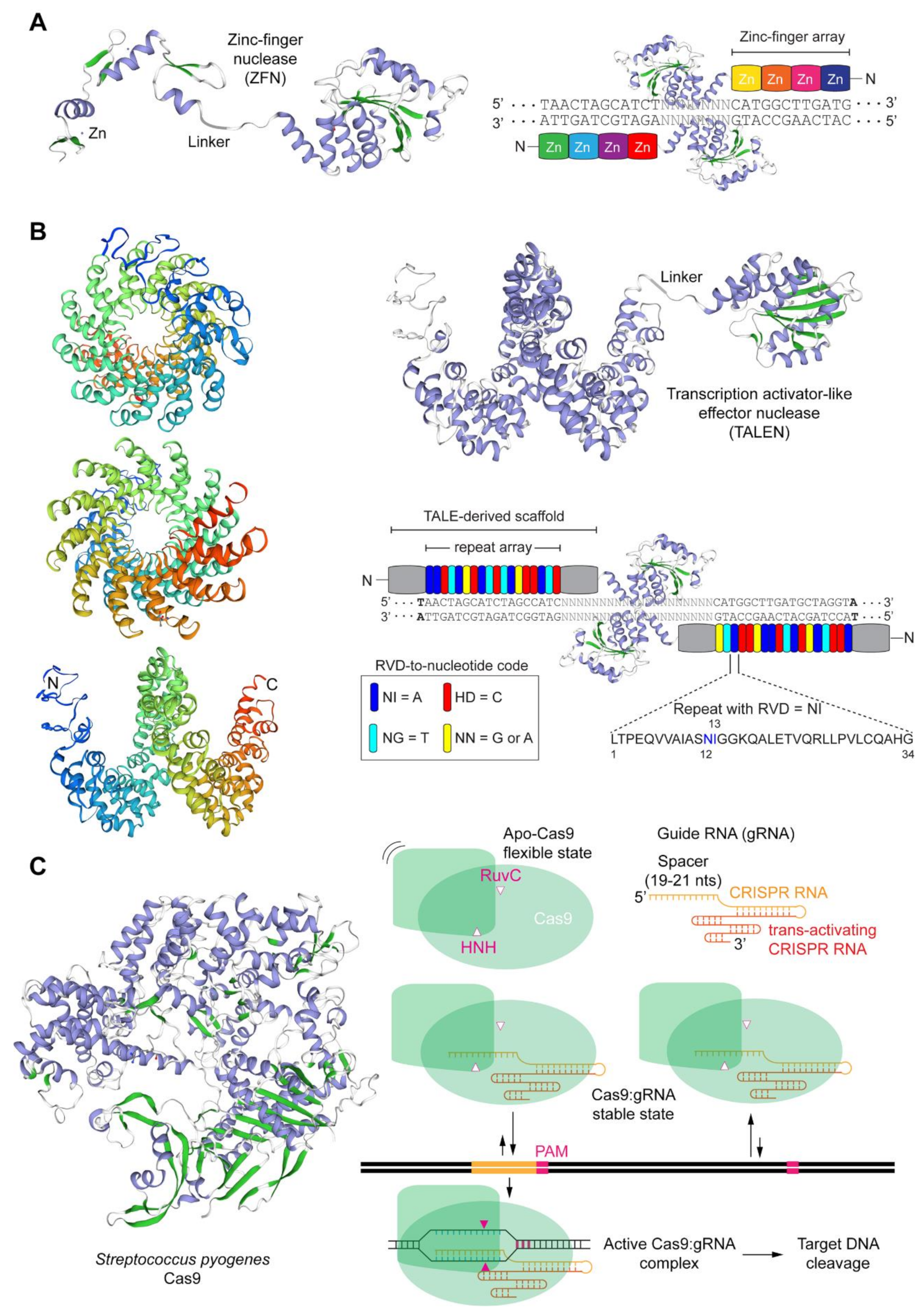
1.1. The Main Gene Editing Principles Based on Programmable Nucleases and Their Key Pros and Cons
1.2. The Main Programmable Nuclease Platforms and Their Key Pros and Cons
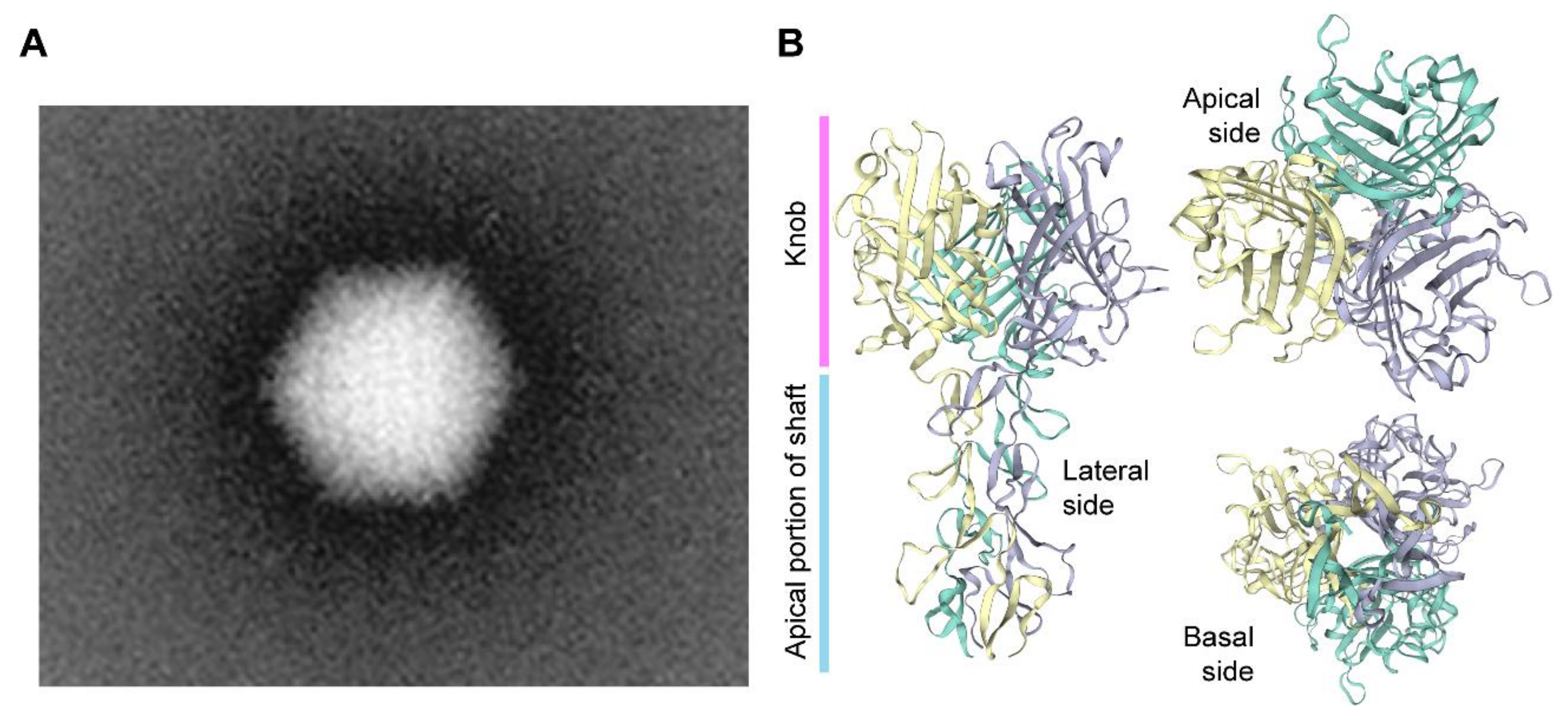
1.3. A Brief Overview on the Biology of Adenoviruses and Their Recombinant Types
2. Adenoviral Vector-Based Gene Editing in Human Adult Stem Cells and Their Progeny
2.1. Targeted Gene Disruption
2.2. Targeted Gene Integration
3. Human Embryonic Stem Cells (hESCs) and Human Induced Pluripotent Stem Cells (hiPSCs) Genome Editing
3.1. High-Capacity Adenoviral Vector (HC-AdV)-Based Gene Editing in hESCs and hiPSCs
3.2. HC-AdV-Based Gene Editing for Targeted Gene Correction in Human Pluripotent Stem Cells (hPSCs)
4. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell. Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kweon, J.; Kim, J.S. TALENs and ZFNs are associated with different mutation signatures. Nat. Methods 2013, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef]
- Ata, H.; Ekstrom, T.L.; Martínez-Gálvez, G.; Mann, C.M.; Dvornikov, A.V.; Schaefbauer, K.J.; Ma, A.C.; Dobbs, D.; Clark, K.J.; Ekker, S.C. Robust activation of microhomology-mediated end joining for precision gene editing applications. PLoS Genet. 2018, 14, e1007652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri-Ghahfarokhi, A.; Taylor, B.J.M.; Nitsch, R.; Lundin, A.; Cavallo, A.L.; Madeyski-Bengtson, K.; Karlsson, F.; Clausen, M.; Hicks, R.; Mayr, L.M.; et al. Decoding non-random mutational signatures at Cas9 targeted sites. Nucleic Acids Res. 2018, 46, 8417–8434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Van Overbeek, M.; Capurso, D.; Carter, M.M.; Thompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gonçalves, M.A. Engineered Viruses as Genome Editing Devices. Mol. Ther. 2016, 24, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Chandrasegaran, S.; Carroll, D. Origins of Programmable Nucleases for Genome Engineering. J. Mol. Biol. 2016, 428, 963–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhu, P.; Shi, H.; Guo, L.; Zhang, Q.; Chen, Y.; Chen, S.; Zhang, Z.; Peng, J.; Chen, J. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 2019, 568, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Tuladhar, R.; Yeu, Y.; Piazza, J.T.; Tan, Z.; Clemenceau, J.R.; Wu, X.; Barrett, Q.; Herbert, J.; Mathews, D.H.; Kim, J.; et al. CRISPR–Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nat. Commun. 2019, 10, 4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söllü, C.; Pars, K.; Cornu, T.I.; Thibodeau-Beganny, S.; Maeder, M.L.; Joung, J.K.; Heilbronn, R.; Cathomen, T. Autonomous zinc-finger nuclease pairs for targeted chromosomal deletion. Nucleic Acids Res. 2010, 38, 8269–8276. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Gonçalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef] [Green Version]
- Choi, P.S.; Meyerson, M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat. Commun. 2014, 5, 3728. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Gerbi, S.A. Making ends meet: Targeted integration of DNA fragments by genome editing. Chromosoma 2018, 127, 405–420. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell. Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Deng, C.; Capecchi, M.R. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol. Cell. Biol. 1992, 12, 3365–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, A.C.; Itzhaki, J.E. Gene targeting in human somatic cells. Complete inactivation of an interferon-inducible gene. Eur. J. Biochem. 1993, 218, 273–281. [Google Scholar] [CrossRef]
- Ganguly, A.; Smelt, S.; Mewar, R.; Fertala, A.; Sieron, A.L.; Overhauser, J.; Prockop, D.J. Targeted insertions of two exogenous collagen genes into both alleles of their endogenous loci in cultures human cells: The insertions are directed by relatively short fragments containing the promoters and the 5′ ends of the genes. Proc. Natl. Acad. Sci. USA 1994, 91, 7365–7369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thyagarajan, B.; Johnson, B.L.; Campbell, C. The effect of target site transcription on gene targeting in human cells in vitro. Nucleic Acids Res. 1995, 23, 2784–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacsovich, T.; Yang, D.; Waldman, A.S. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 1994, 22, 5649–5657. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 1994, 91, 6064–6068. [Google Scholar] [CrossRef] [Green Version]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [Green Version]
- Maggio, I.; Gonçalves, M.A. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol. 2015, 33, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Huang, S.; Zhao, X.; Wright, D.A.; Carpenter, S.; Spalding, M.H.; Weeks, D.P.; Yang, B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011, 39, 6315–6325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfouz, M.M.; Li, L.; Shamimuzzaman, M.; Wibowo, A.; Fang, X.; Zhu, J.K. De novo-engineered transcription activator-like effector (TALE) hybrid nuclease with novel DNA binding specificity creates double-strand breaks. Proc. Natl. Acad. Sci. USA 2011, 108, 2623–2628. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lütge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; McLachlan, A.D.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [Green Version]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [Green Version]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef]
- Cathomen, T.; Joung, K.J. Zinc-finger Nucleases: The next generation emerges. Mol. Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef]
- Mussolino, C.; Cathomen, T. TALE nucleases: Tailored genome engineering made easy. Curr. Opin. Biotechnol. 2012, 23, 644–650. [Google Scholar] [CrossRef]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [Green Version]
- Valton, J.; Dupuy, A.; Daboussi, F.; Thomas, S.; Maréchal, A.; Macmaster, R.; Melliand, K.; Juillerat, A.; Duchateau, P. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J. Biol. Chem. 2012, 287, 38427–38432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Rinsma, M.; Janssen, J.M.; Liu, J.; Maggio, I.; Gonçalves, M.A. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016, 44, 6482–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Nishimasu, H.; Kodera, N.; Hirano, S.; Ando, T.; Uchihashi, T.; Nureki, O. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat. Commun. 2017, 8, 1430. [Google Scholar] [CrossRef] [Green Version]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [Green Version]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef]
- Chen, X.; Tasca, F.; Wang, Q.; Liu, J.; Janssen, J.M.; Brescia, M.D.; Bellin, M.; Szuhai, K.; Kenrick, J.; Frock, R.L.; et al. Expanding the editable genome and CRISPR-Cas9 versatility using DNA cutting-free gene targeting based on in trans paired nicking. Nucleic Acids Res. 2020, 48, 974–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, J.; Janssen, J.M.; Gonçalves, M.A. The chromatin structure differentially impacts high-specificity CRISPR-Cas9 nuclease strategies. Mol. Ther. Nucleic Acids 2017, 8, 558–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The impact of chromatin dynamics on Cas9-mediated genome editing in human cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.T.; Fløe, L.; Petersen, T.S.; Huang, J.; Xu, F.; Bolund, L.; Luo, Y.; Lin, L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017, 591, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gonçalves, M.A. DNA, RNA, and Protein Tools for Editing the Genetic Information in Human Cells. iScience 2018, 6, 247–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.K.; Gaj, T. Innovations in CRISPR technology. Curr. Opin. Biotechnol. 2018, 52, 95–101. [Google Scholar] [CrossRef]
- Hajizadeh Dastjerdi, A.; Newman, A.; Burgio, G. The Expanding Class 2 CRISPR Toolbox: Diversity, Applicability, and Targeting Drawbacks. BioDrugs 2019, 33, 503–513. [Google Scholar] [CrossRef]
- Wold, W.S.M.; Horwitz, M.S. Adenoviruses. In Fields Virology, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Gonçalves, M.A.; de Vries, A.A. Adenovirus: From foe to friend. Rev. Med. Virol. 2006, 16, 167–186. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, J.; van der Heijdt, S.; Hoeben, R.C. The adenovirus capsid: Major progress in minor proteins. J. Gen. Virol. 2005, 86, 1581–1588. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef]
- Nilsson, E.C.; Storm, R.J.; Bauer, J.; Johansson, S.M.; Lookene, A.; Ångström, J.; Hedenström, M.; Eriksson, T.L.; Frängsmyr, L.; Rinaldi, S.; et al. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nat. Med. 2011, 17, 105–109. [Google Scholar] [CrossRef]
- Arnberg, N.; Edlund, K.; Kidd, A.H.; Wadell, G. Adenovirus type 37 uses sialic acid as a cellular receptor. J. Virol. 2000, 74, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Padilla, J.; Papp, T.; Kaján, G.L.; Benkő, M.; Havenga, M.; Lemckert, A.; Harrach, B.; Baker, A.H. Development of Novel Adenoviral Vectors to Overcome Challenges Observed With HAdV-5-based Constructs. Mol. Ther. 2016, 24, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Barry, M.A.; Rubin, J.D.; Lu, S.C. Retargeting adenoviruses for therapeutic applications and vaccines. FEBS Lett. 2020. [Google Scholar] [CrossRef] [Green Version]
- Shayakhmetov, D.M.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Lieber, A. Efficient gene transfer into human CD34+ cells by a retargeted adenovirus vector. J. Virol. 2000, 74, 2567–2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knaän-Shanzer, S.; Van Der Velde, I.; Havenga, M.J.E.; Lemckert, A.A.C.; De Vries, A.A.F.; Valerio, D. Highly efficient targeted transduction of undifferentiated human hematopoietic cells by adenoviral vectors displaying fiber knobs of subgroup B. Hum. Gene Ther. 2001, 12, 1989–2005. [Google Scholar]
- Gonçalves, M.A.; de Vries, A.A.; Holkers, M.; van de Watering, M.J.M.; van der Velde, I.; van Nierop, G.P.; Valerio, D.; Knaän-Shanzer, S. Human mesenchymal stem cells ectopically expressing full-length dystrophin can complement Duchenne muscular dystrophy myotubes by cell fusion. Hum. Mol. Genet. 2005, 15, 213–221. [Google Scholar]
- Knaän-Shanzer, S.; van de Watering, M.J.M.; van der Velde, I.; Gonçalves, M.A.F.V.; Valerio, D.; de Vries, A.A.F. Endowing human adenovirus serotype 5 vectors with fiber domains of species B greatly enhances gene transfer into human mesenchymal stem cells. Stem Cells 2005, 23, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.A.; alves, M.A.; Holkers, M.; Cudré-Mauroux, C.; Van Nierop, G.P.; Knaän-Shanzer, S.; Van Der Velde, I.; Valerio, D.; De Vries, A.A. Transduction of myogenic cells by retargeted dual high-capacity hybrid viral vectors: Robust dystrophin synthesis in duchenne muscular dystrophy muscle cells. Mol. Ther. 2006, 13, 976–986. [Google Scholar]
- Meier, O.; Greber, U.F. Adenovirus endocytosis. J. Gene Med. 2004, 6, S152–S163. [Google Scholar] [CrossRef]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Shukarev, G.; Callendret, B.; Luhn, K.; Douoguih, M. A two-dose heterologous prime-boost vaccine regimen eliciting sustained immune responses to Ebola Zaire could support a preventive strategy for future outbreaks. Hum. Vaccin. Immunother. 2017, 13, 266–270. [Google Scholar] [CrossRef]
- Barouch, D.H.; Tomaka, F.L.; Wegmann, F.; Stieh, D.J.; Alter, G.; Robb, M.L.; Michael, N.L.; Peter, L.; Nkolola, J.P.; Borducchi, E.N.; et al. Evaluation of a mosaic HIV-1 vaccine in a multicentre, randomised, double-blind, placebo-controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13-19). Lancet 2018, 392, 232–243. [Google Scholar] [CrossRef]
- Havenga, M.J.; Holterman, L.; Melis, I.; Smits, S.; Kaspers, J.; Heemskerk, E.; van der Vlugt, R.; Koldijk, M.; Schouten, G.J.; Hateboer, G.; et al. Serum-free transient protein production system based on adenoviral vector and PER.C6 technology: High yield and preserved bioactivity. Biotechnol. Bioeng. 2008, 100, 273–283. [Google Scholar] [CrossRef]
- Parks, R.J.; Graham, F.L. A helper-dependent system for adenovirus vector production helps define a lower limit for efficient DNA packaging. J. Virol. 1997, 71, 3293–3298. [Google Scholar] [CrossRef] [Green Version]
- Jager, L.; Hausl, M.A.; Rauschhuber, C.; Wolf, N.M.; Kay, M.A.; Ehrhardt, A. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat. Protoc. 2009, 4, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Krymskaya, L.; Wang, J.; Henley, J.; Rao, A.; Cao, L.-F.; Tran, C.-A.; Torres-Coronado, M.; Gardner, A.; Gonzalez, N.; et al. Genomic Editing of the HIV-1 Coreceptor CCR5 in Adult Hematopoietic Stem and Progenitor Cells Using Zinc Finger Nucleases. Mol. Ther. 2013, 21, 1259–1269. [Google Scholar] [CrossRef]
- Maier, D.A.; Brennan, A.L.; Jiang, S.; Binder-Scholl, G.K.; Lee, G.; Plesa, G.; Zheng, Z.; Cotte, J.; Carpenito, C.; Wood, T.; et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum. Gene Ther. 2013, 24, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saydaminova, K.; Ye, X.; Wang, H.; Richter, M.; Ho, M.; Chen, H.; Xu, N.; Kim, J.S.; Papapetrou, E.; Holmes, M.C.; et al. Efficient genome editing in hematopoietic stem cells with helper-dependent Ad5/35 vectors expressing site-specific endonucleases under microRNA regulation. Mol. Ther. Methods Clin. Dev. 2015, 1, 14057. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Yuan, J.; Wang, J.; Crain, K.; Fearns, C.; Kim, K.A.; Hua, K.L.; Gregory, P.D.; Holmes, M.C.; Torbett, B.E. Zinc-finger Nuclease Editing of Human cxcr4 Promotes HIV-1 CD4+ T Cell Resistance and Enrichment. Mol. Ther. 2012, 20, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holkers, M.; Maggio, I.; Liu, J.; Janssen, J.M.; Miselli, F.; Mussolino, C.; Recchia, A.; Cathomen, T.; Gonçalves, M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013, 41, e63. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Stroukov, W.; Cathomen, T.; Mussolino, C. Chimerization Enables Gene Synthesis and Lentiviral Delivery of Customizable TALE-Based Effectors. Int. J. Mol. Sci. 2020, 21, 795. [Google Scholar] [CrossRef] [Green Version]
- Pelascini, L.P.; Maggio, I.; Liu, J.; Holkers, M.; Cathomen, T.; Gonçalves, M.A. Histone deacetylase inhibition rescues gene knockout levels achieved with integrase-defective lentiviral vectors encoding zinc-finger nucleases. Hum. Gene Ther. Methods 2013, 24, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelascini, L.P.; Janssen, J.M.; Gonçalves, M.A. Histone deacetylase inhibition activates transgene expression from integration-defective lentiviral vectors in dividing and non-dividing cells. Hum. Gene Ther. 2013, 24, 78–96. [Google Scholar] [CrossRef]
- Maggio, I.; Holkers, M.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vector delivery of RNA-guided CRISPR/Cas9 nuclease complexes induces targeted mutagenesis in a diverse array of human cells. Sci. Rep. 2014, 4, 5105. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Liu, M.; Klomp, J.; Merrill, B.J.; Rehman, J.; Malik, A.B. Method for Dual Viral Vector Mediated CRISPR-Cas9 Gene Disruption in Primary Human Endothelial Cells. Sci. Rep. 2017, 7, 42127. [Google Scholar] [CrossRef] [Green Version]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [Green Version]
- Brescia, M.; Janssen, J.M.; Liu, J.; Gonçalves, M.A. High-capacity adenoviral vectors permit robust and versatile testing of DMD gene repair tools and strategies in human cells. Cells 2020, 9, 869. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Psatha, N.; Sova, P.; Gil, S.; Wang, H.; Kim, J.; Kulkarni, C.; Valensisi, C.; Hawkins, R.D.; Stamatoyannopoulos, G.; et al. Reactivation of gamma-globin in adult beta-YAC mice after ex vivo and in vivo hematopoietic stem cell genome editing. Blood 2018, 131, 2915–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35(++) Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Conti, A.; Ferrari, S.; Della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coluccio, A.; Miselli, F.; Lombardo, A.; Marconi, A.; Malagoli Tagliazucchi, G.; Gonçalves, M.A.; Pincelli, C.; Maruggi, G.; Del Rio, M.; Naldini, L.; et al. Targeted gene addition in human epithelial stem cells by zinc-finger nuclease-mediated homologous recombination. Mol. Ther. 2013, 21, 1695–1704. [Google Scholar] [CrossRef] [Green Version]
- Holkers, M.; Maggio, I.; Henriques, S.F.; Janssen, J.M.; Cathomen, T.; Gonçalves, M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef]
- Li, C.; Mishra, A.S.; Gil, S.; Wang, M.; Georgakopoulou, A.; Papayannopoulou, T.; Hawkins, R.D.; Lieber, A. Targeted Integration and High-Level Transgene Expression in AAVS1 Transgenic Mice after In Vivo HSC Transduction with HDAd5/35++ Vectors. Mol. Ther. 2019, 27, 2195–2212. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- de Miguel-Beriain, I. The ethics of stem cells revisited. Adv. Drug Deliv. Rev. 2015, 82-83, 176–180. [Google Scholar] [CrossRef]
- Hu, J.; Wang, J. From embryonic stem cells to induced pluripotent stem cells-Ready for clinical therapy? Clin. Transplant 2019, 33, e13573. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbrel, E.A.; Lanza, R. Pluripotent stem cells: The last 10 years. Regen. Med. 2016, 11, 831–847. [Google Scholar] [CrossRef] [Green Version]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.H.; Izpisua Belmonte, J.C. A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J. Biol. Chem. 2014, 289, 4594–4599. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Janssen, J.M.; Liu, J.; Maggio, I.; t Jong, A.E.J.; Mikkers, H.M.M.; Gonçalves, M. In trans paired nicking triggers seamless genome editing without double-stranded DNA cutting. Nat. Commun. 2017, 8, 657. [Google Scholar] [CrossRef]
- Nakajima, K.; Zhou, Y.; Tomita, A.; Hirade, Y.; Gurumurthy, C.B.; Nakada, S. Precise and efficient nucleotide substitution near genomic nick via noncanonical homology-directed repair. Genome Res. 2018, 28, 223–230. [Google Scholar] [CrossRef]
- Hyodo, T.; Rahman, M.L.; Karnan, S.; Ito, T.; Toyoda, A.; Ota, A.; Wahiduzzaman, M.; Tsuzuki, S.; Okada, Y.; Hosokawa, Y.; et al. Tandem Paired Nicking Promotes Precise Genome Editing with Scarce Interference by p53. Cell Rep. 2020, 30, 1195–1207. [Google Scholar] [CrossRef] [Green Version]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.A.; van Nierop, G.P.; Holkers, M.; de Vries, A.A. Concerted nicking of donor and chromosomal acceptor DNA promotes homology-directed gene targeting in human cells. Nucleic Acids Res. 2012, 40, 3443–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.; Wilson, D.R.; Green, J.J. Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol. 2019, 37, 281–293. [Google Scholar] [CrossRef]
- Ohbayashi, F.; Balamotis, M.A.; Kishimoto, A.; Aizawa, E.; Diaz, A.; Hasty, P.; Graham, F.L.; Caskey, C.T.; Mitani, K. Correction of chromosomal mutation and random integration in embryonic stem cells with helper-dependent adenoviral vectors. Proc. Natl. Acad. Sci. USA 2005, 102, 13628–13633. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Mitsui, K.; Aizawa, E.; Hasegawa, K.; Kawase, E.; Yamagishi, T.; Shimizu, Y.; Suemori, H.; Nakatsuji, N.; Mitani, K. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc. Natl. Acad. Sci. USA 2008, 105, 13781–13786. [Google Scholar] [CrossRef] [Green Version]
- Aizawa, E.; Hirabayashi, Y.; Iwanaga, Y.; Suzuki, K.; Sakurai, K.; Shimoji, M.; Aiba, K.; Wada, T.; Tooi, N.; Kawase, E.; et al. Efficient and accurate homologous recombination in hESCs and hiPSCs using helper-dependent adenoviral vectors. Mol. Ther. 2012, 20, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Umeda, K.; Suzuki, K.; Yamazoe, T.; Shiraki, N.; Higuchi, Y.; Tokieda, K.; Kume, K.; Mitani, K.; Kume, S. Albumin gene targeting in human embryonic stem cells and induced pluripotent stem cells with helper-dependent adenoviral vector to monitor hepatic differentiation. Stem Cell Res. 2013, 10, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Sone, T.; Shin, M.; Ouchi, T.; Sasanuma, H.; Miyamoto, A.; Ohte, S.; Tsukamoto, S.; Nakanishi, M.; Okano, H.; Katagiri, T.; et al. Dual usage of a stage-specific fluorescent reporter system based on a helper-dependent adenoviral vector to visualize osteogenic differentiation. Sci. Rep. 2019, 9, 9705. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Suzuki, K.; Qu, J.; Sancho-Martinez, I.; Yi, F.; Li, M.; Kumar, S.; Nivet, E.; Kim, J.; Soligalla, R.D.; et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 2011, 8, 688–694. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Suzuki, K.; Qu, J.; Saini, P.; Dubova, I.; Yi, F.; Lee, J.; Sancho-Martinez, I.; Liu, G.H.; Izpisua Belmonte, J.C. Efficient correction of hemoglobinopathy-causing mutations by homologous recombination in integration-free patient iPSCs. Cell Res. 2011, 21, 1740–1744. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.J.; Grove, N.C.; Ing, J.; Crane, A.M.; Venken, K.; Davis, B.R.; Ng, P. Homology Requirements for Efficient, Footprintless Gene Editing at the CFTR Locus in Human iPSCs with Helper-dependent Adenoviral Vectors. Mol. Ther. Nucleic Acids 2016, 5, e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.J.; Grove, N.C.; Turner, D.L.; Ng, P. Gene Editing with Helper-Dependent Adenovirus Can Efficiently Introduce Multiple Changes Simultaneously over a Large Genomic Region. Mol. Ther. Nucleic Acids 2017, 8, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Suzuki, K.; Li, M.; Qu, J.; Montserrat, N.; Tarantino, C.; Gu, Y.; Yi, F.; Xu, X.; Zhang, W.; et al. Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat. Commun. 2014, 5, 4330. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ozawa, Y.; Suzuki, K.; Yuki, K.; Ohyama, M.; Akamatsu, W.; Matsuzaki, Y.; Shimmura, S.; Mitani, K.; Tsubota, K.; et al. The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol. Brain 2014, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, J.; Suzuki, K.; Qu, J.; Wang, P.; Zhou, J.; Liu, X.; Ren, R.; Xu, X.; Ocampo, A.; et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 2015, 348, 1160–1163. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Yu, C.; Qu, J.; Li, M.; Yao, X.; Yuan, T.; Goebl, A.; Tang, S.; Ren, R.; Aizawa, E.; et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 2014, 15, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.G.; Petek, L.M.; Russell, D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004, 36, 767. [Google Scholar] [CrossRef] [Green Version]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Park, S.; Beal, P.A. Off-Target Editing by CRISPR-Guided DNA Base Editors. Biochemistry 2019, 58, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
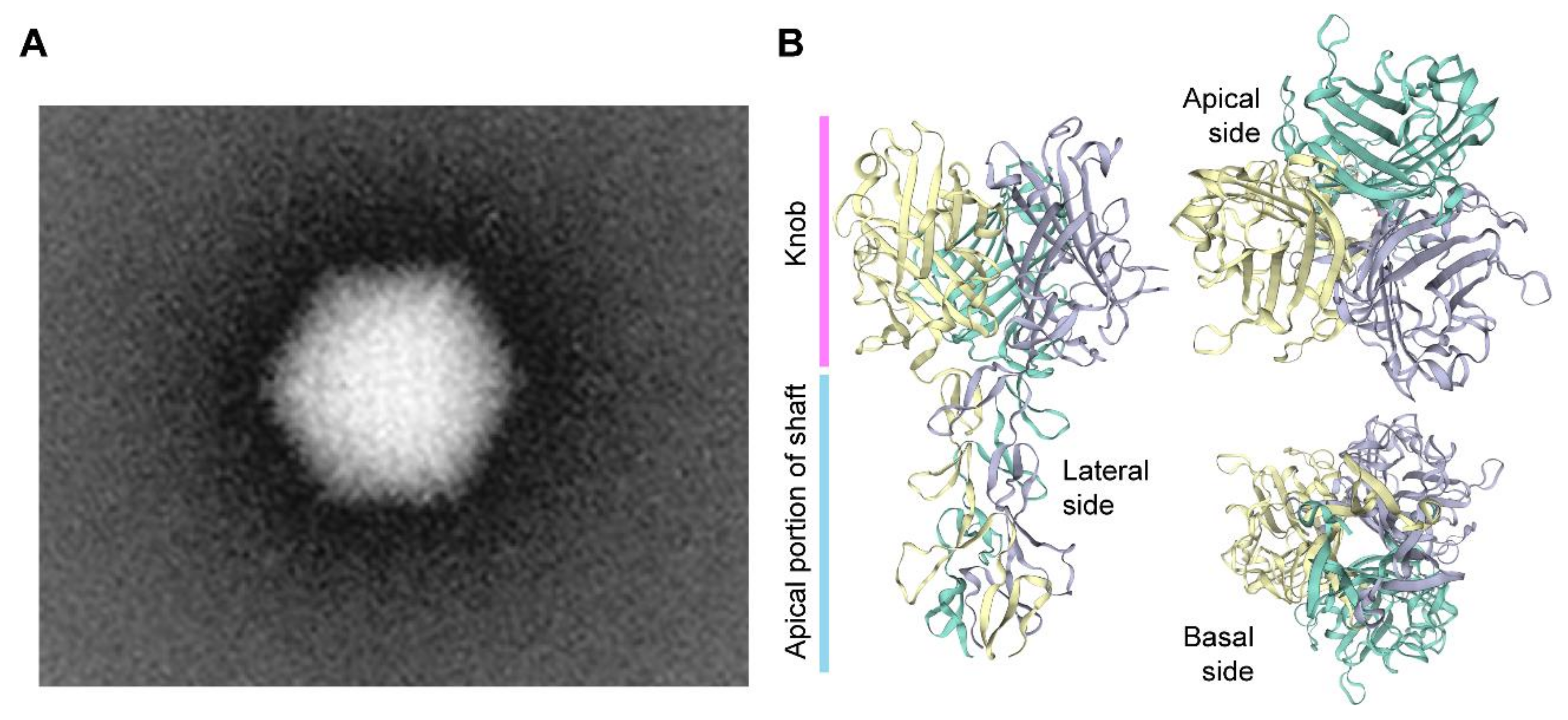
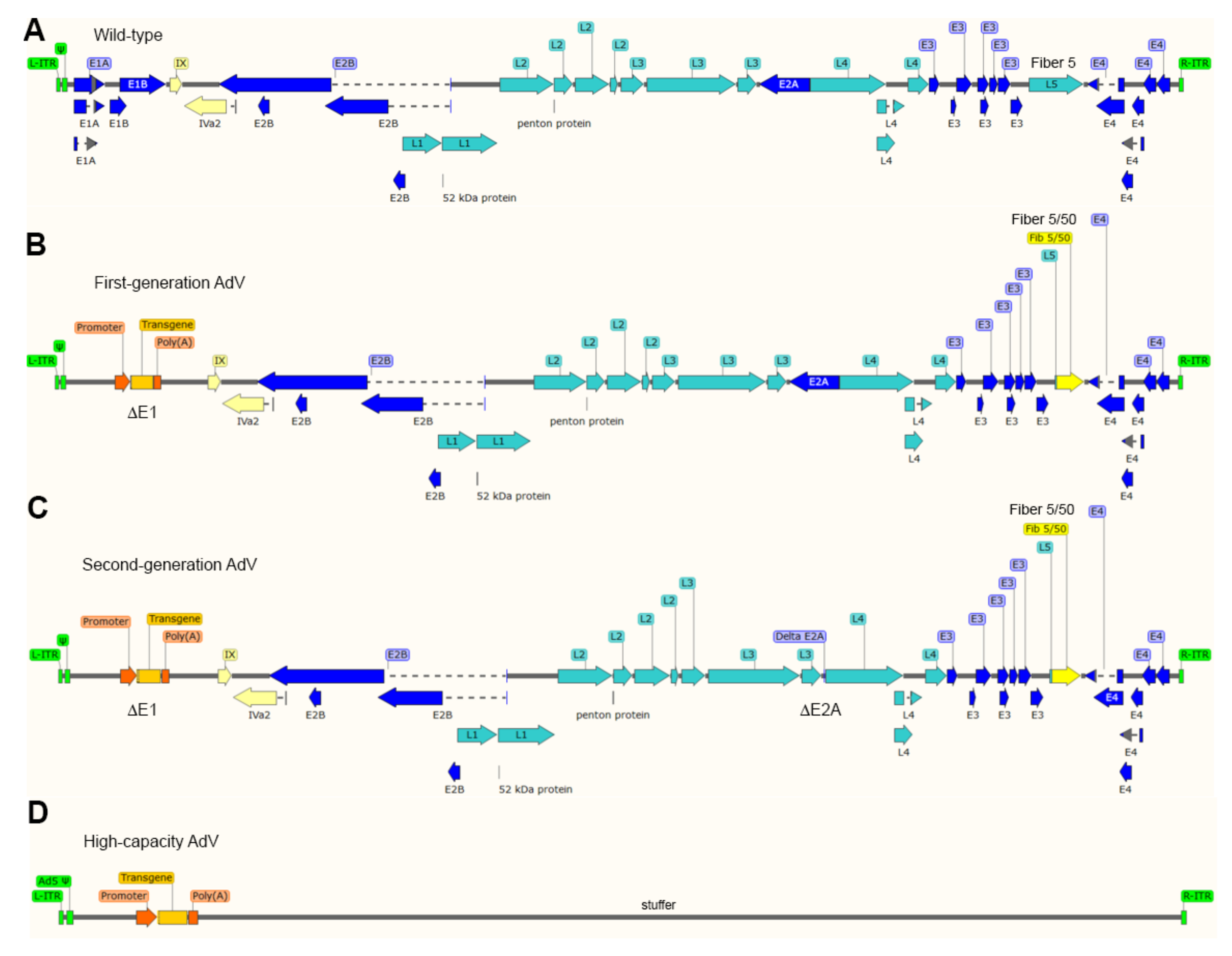
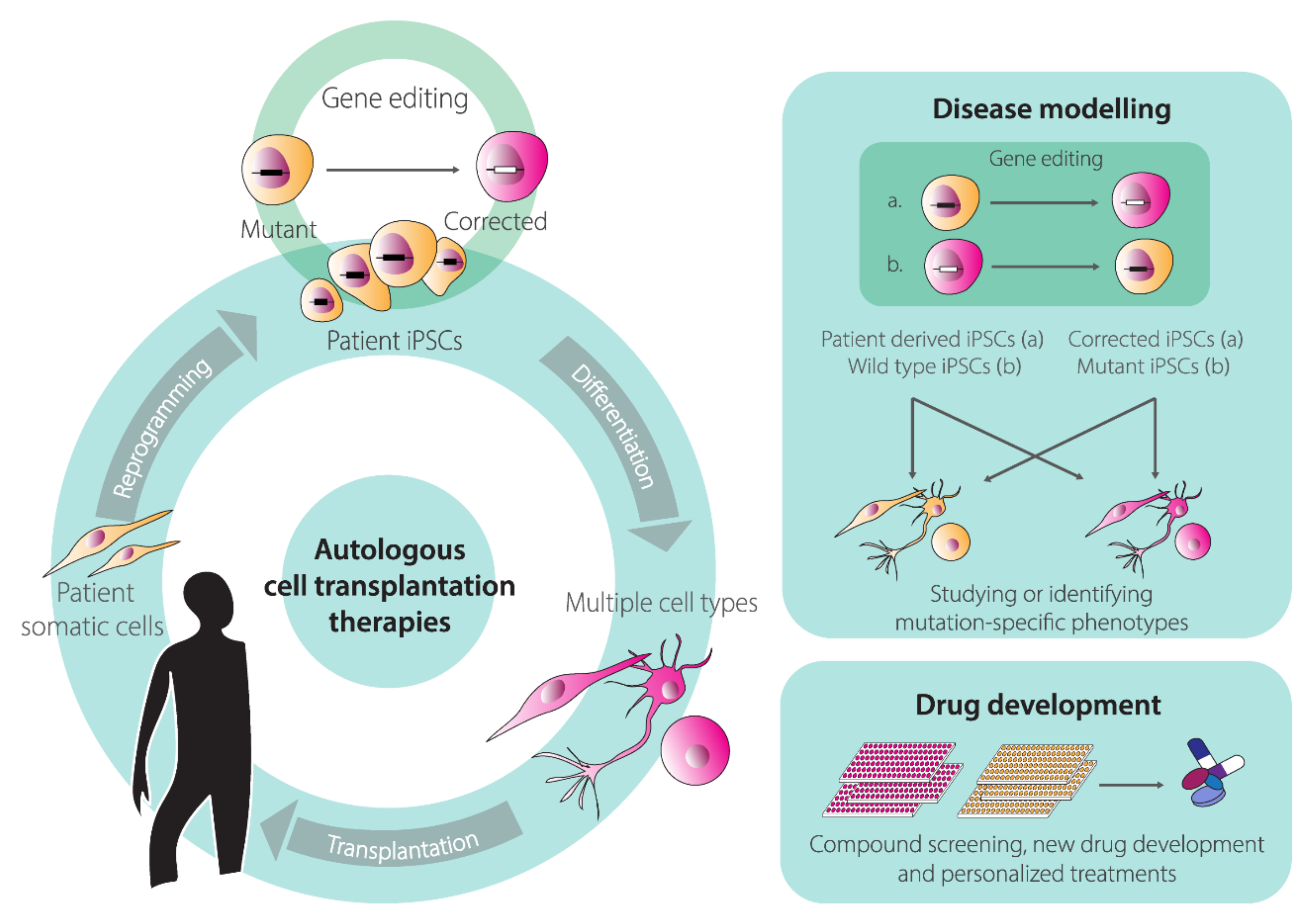
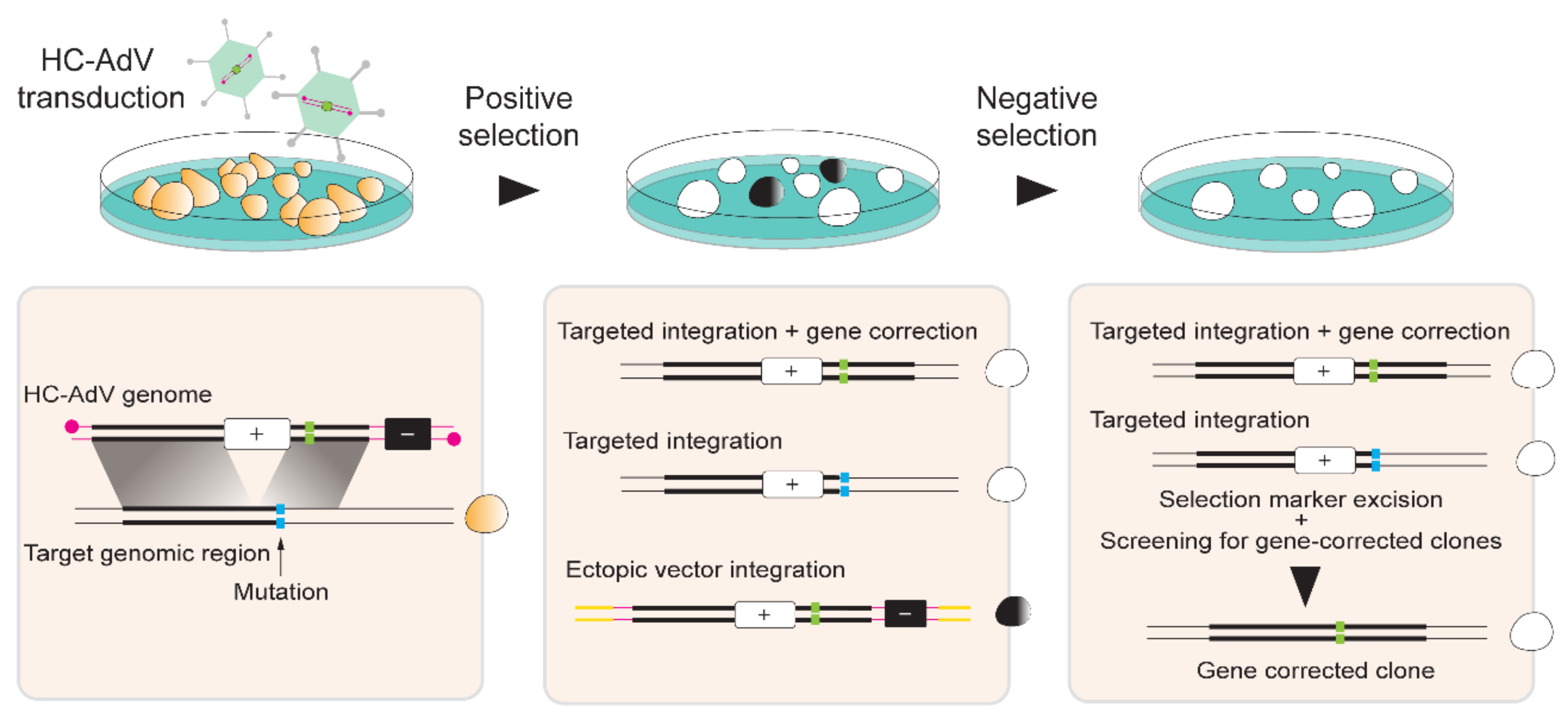
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasca, F.; Wang, Q.; Gonçalves, M.A.F.V. Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells 2020, 9, 953. https://doi.org/10.3390/cells9040953
Tasca F, Wang Q, Gonçalves MAFV. Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells. 2020; 9(4):953. https://doi.org/10.3390/cells9040953
Chicago/Turabian StyleTasca, Francesca, Qian Wang, and Manuel A.F.V. Gonçalves. 2020. "Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny" Cells 9, no. 4: 953. https://doi.org/10.3390/cells9040953
APA StyleTasca, F., Wang, Q., & Gonçalves, M. A. F. V. (2020). Adenoviral Vectors Meet Gene Editing: A Rising Partnership for the Genomic Engineering of Human Stem Cells and Their Progeny. Cells, 9(4), 953. https://doi.org/10.3390/cells9040953