The Catalytic-Dependent and -Independent Roles of Lsd1 and Lsd2 Lysine Demethylases in Heterochromatin Formation in Schizosaccharomyces pombe

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Cell Culture
2.2. Dilution Assay
2.3. RNA-Seq and ChIP-seq
2.4. qRT-PCR
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. Fluorescence Microscopy
2.7. Heterochromatin Formation and Maintenance Assay Using an ade6+ Reporter
2.8. Growth Curve
3. Results
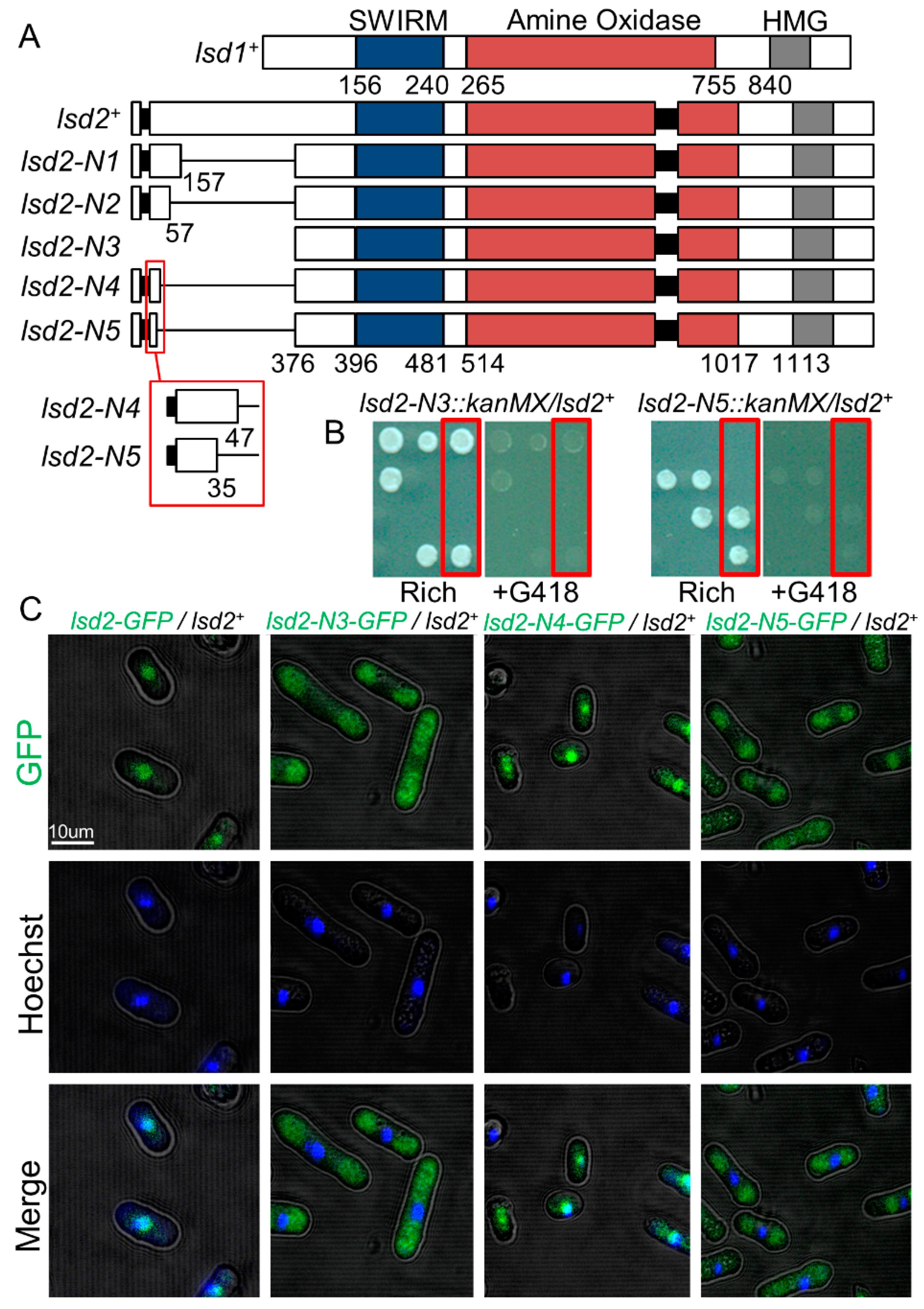
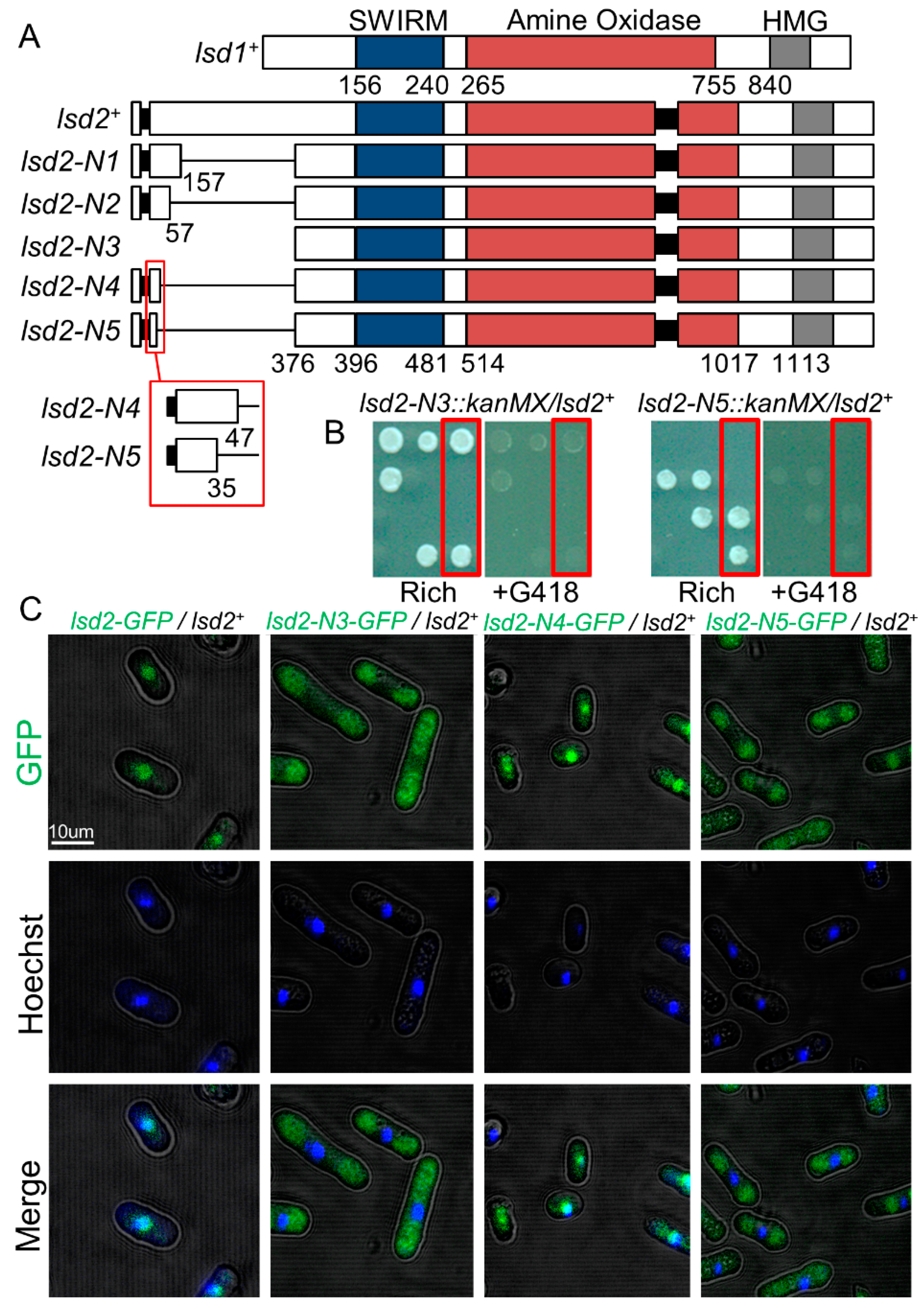
3.1. A N-Terminal Region of Lsd2 Is Required for Nuclear Localization and Viability
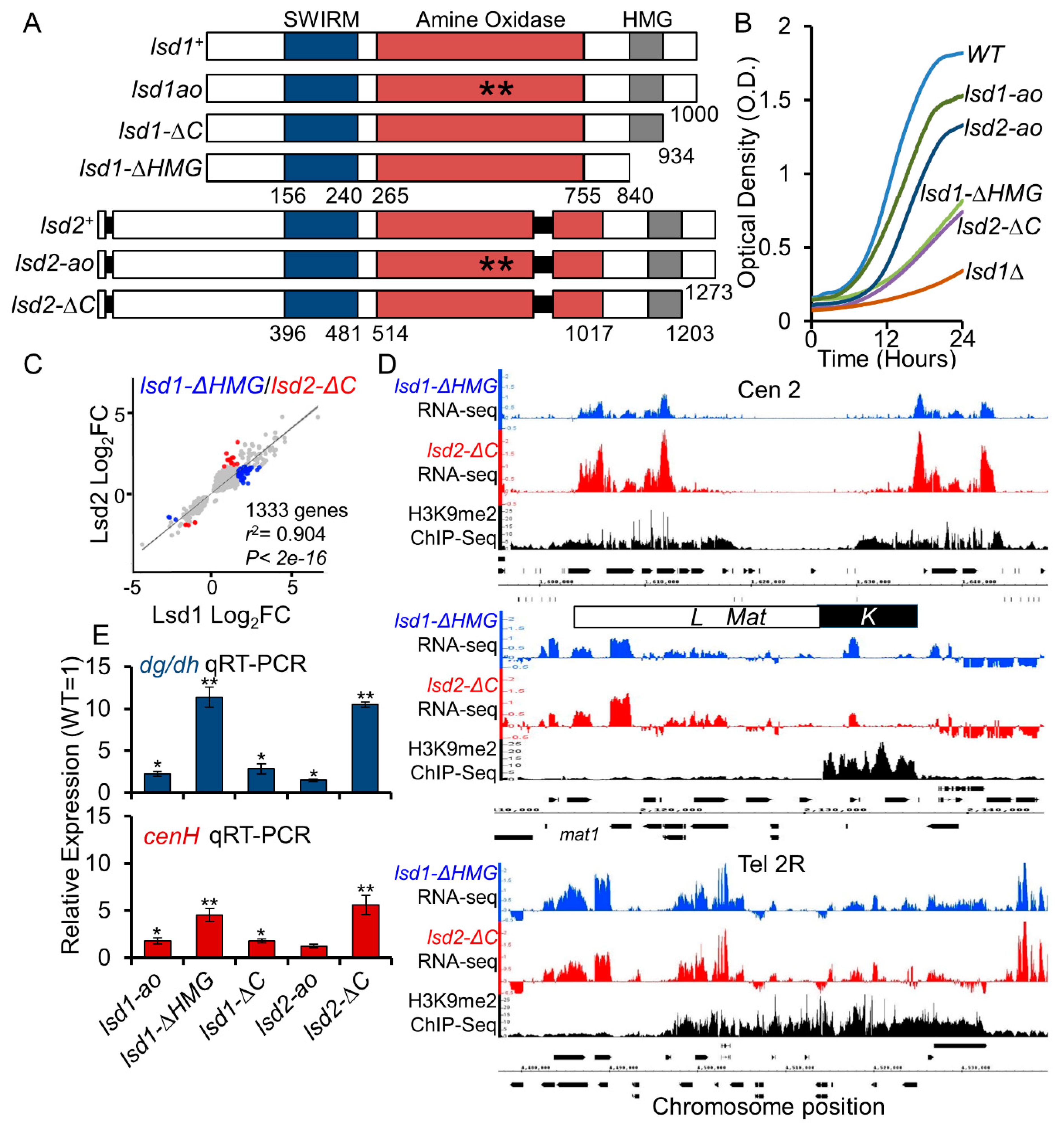
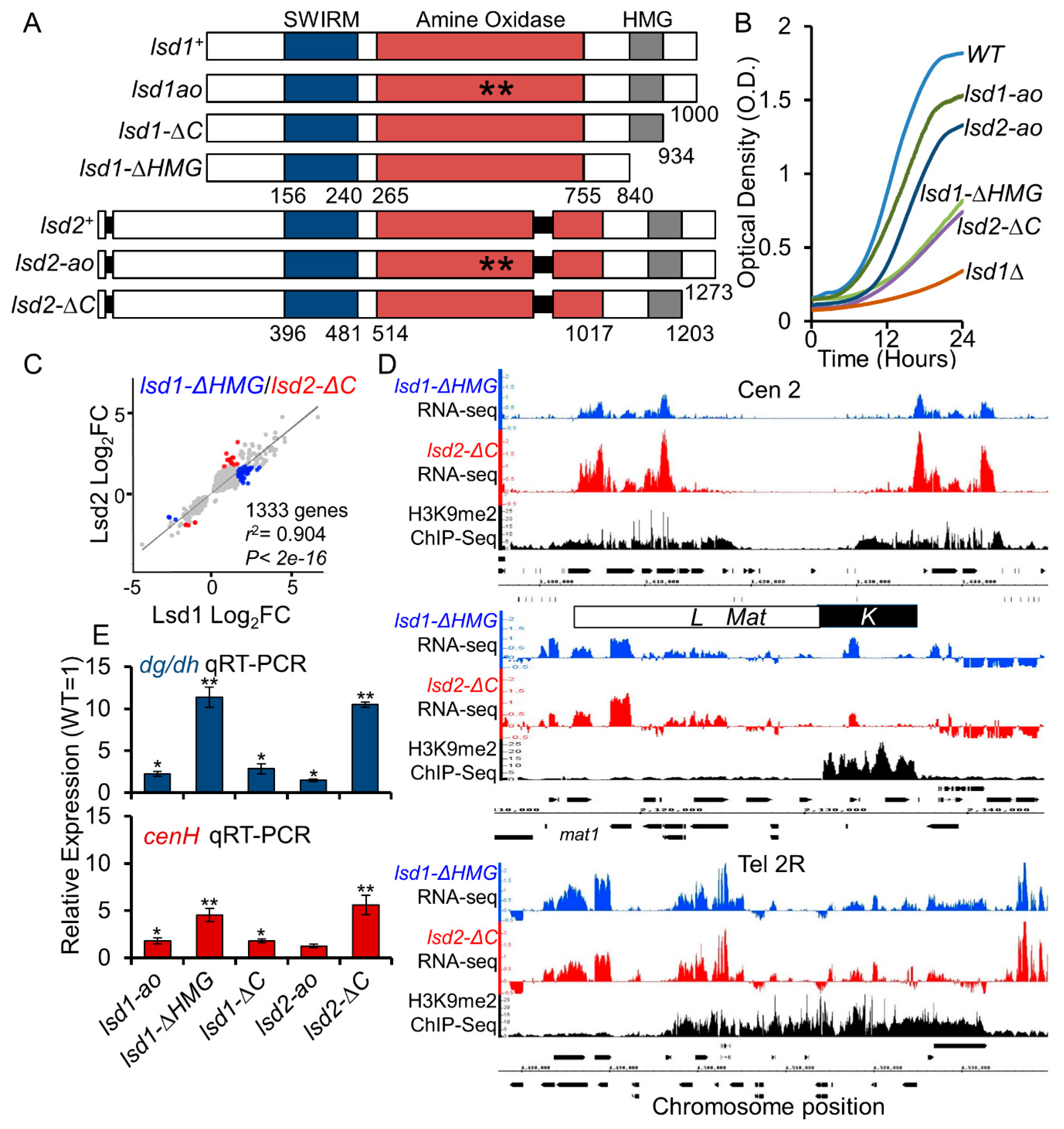
3.2. The HMG-Box Domains Are Essential for Lsd1 and Lsd2 Functions
3.3. Lsd1 and Lsd2 C-Terminal Mutants Affect Genome-Wide Gene Expression and Display Defective Silencing at Constitutive Heterochromatic Regions
3.4. C-Terminal Mutants of Lsd1 and Lsd2 Retain Some Enzymatic Activities
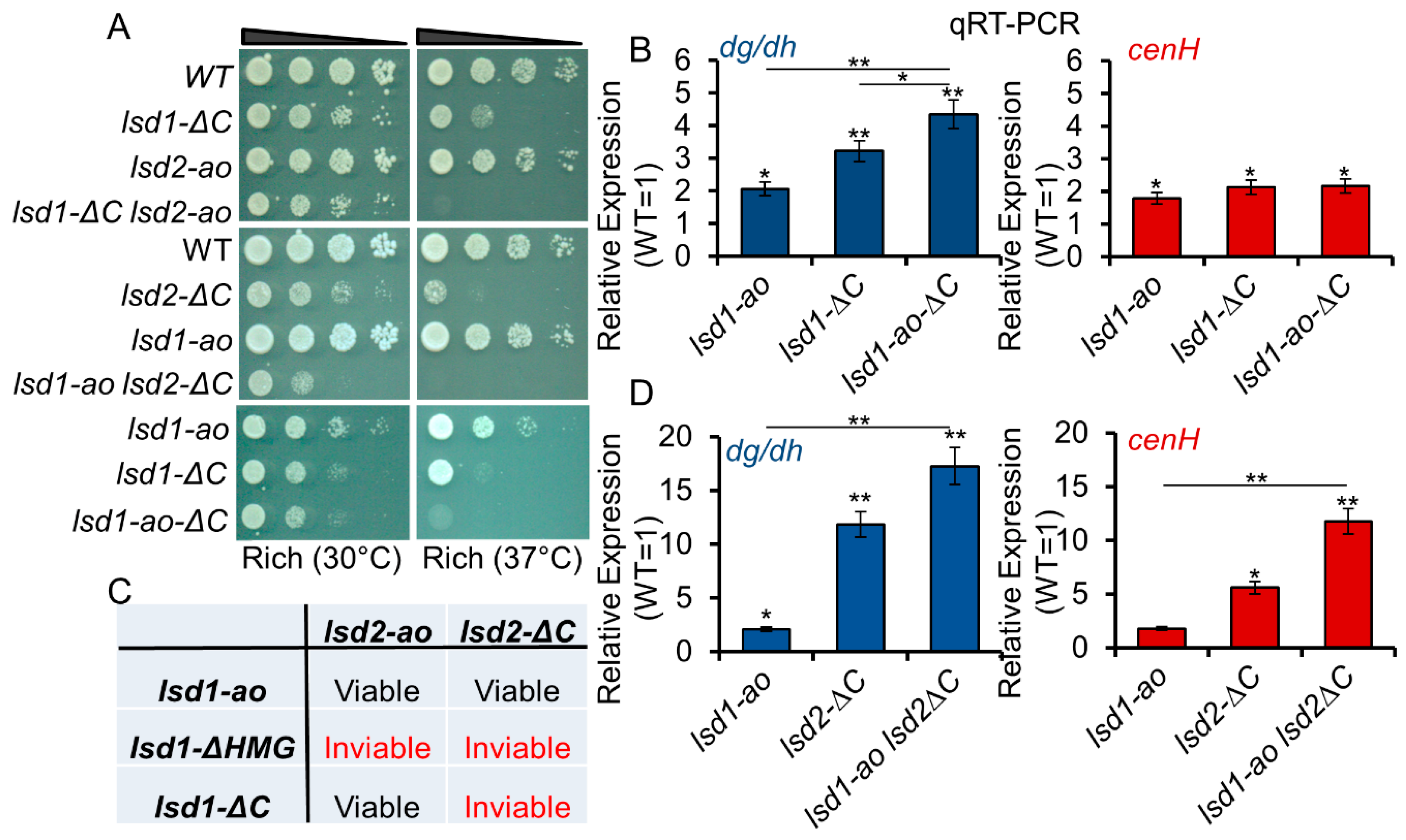
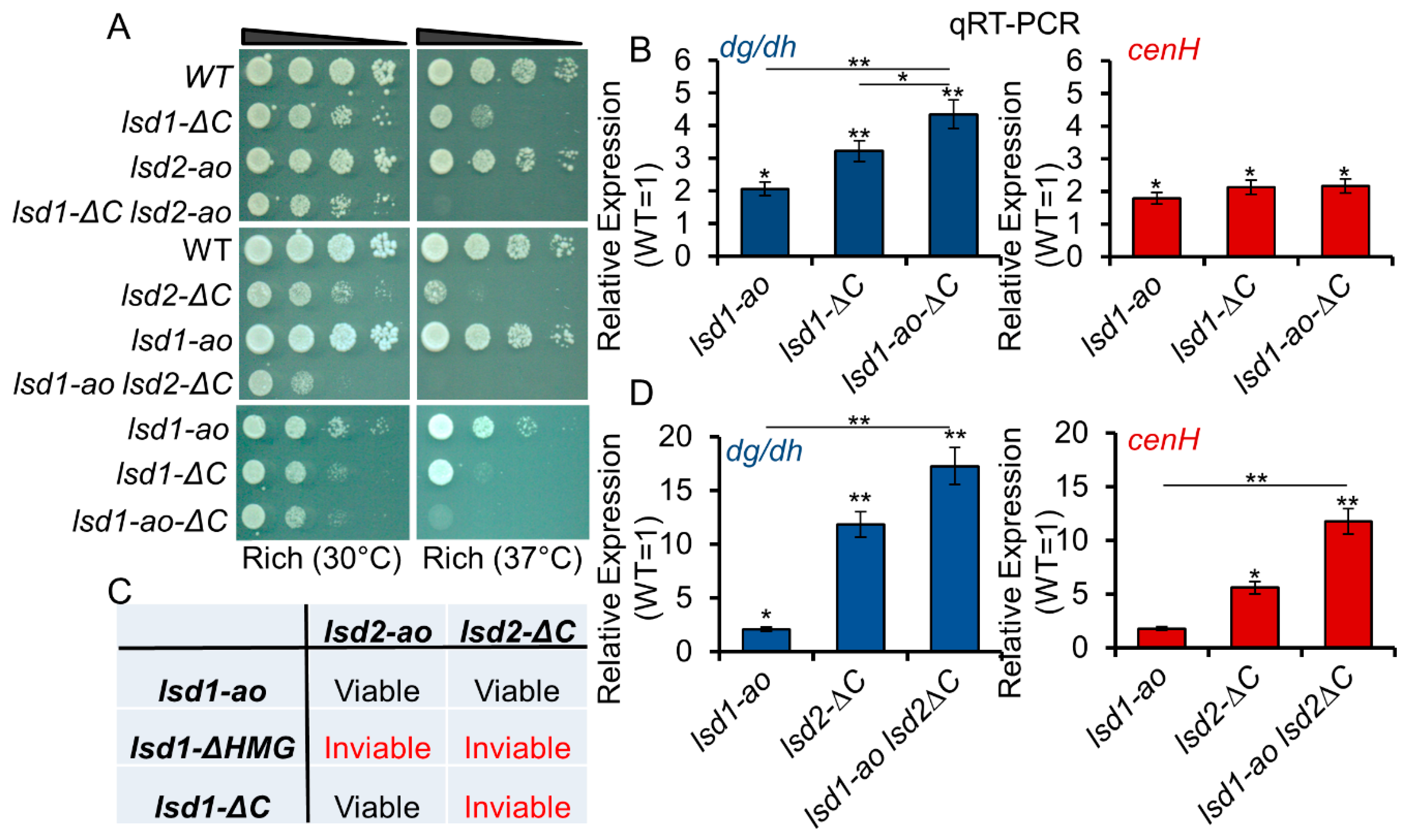
3.5. Lsd1 and Lsd2 Serve Overlapping, but Divergent, Functions
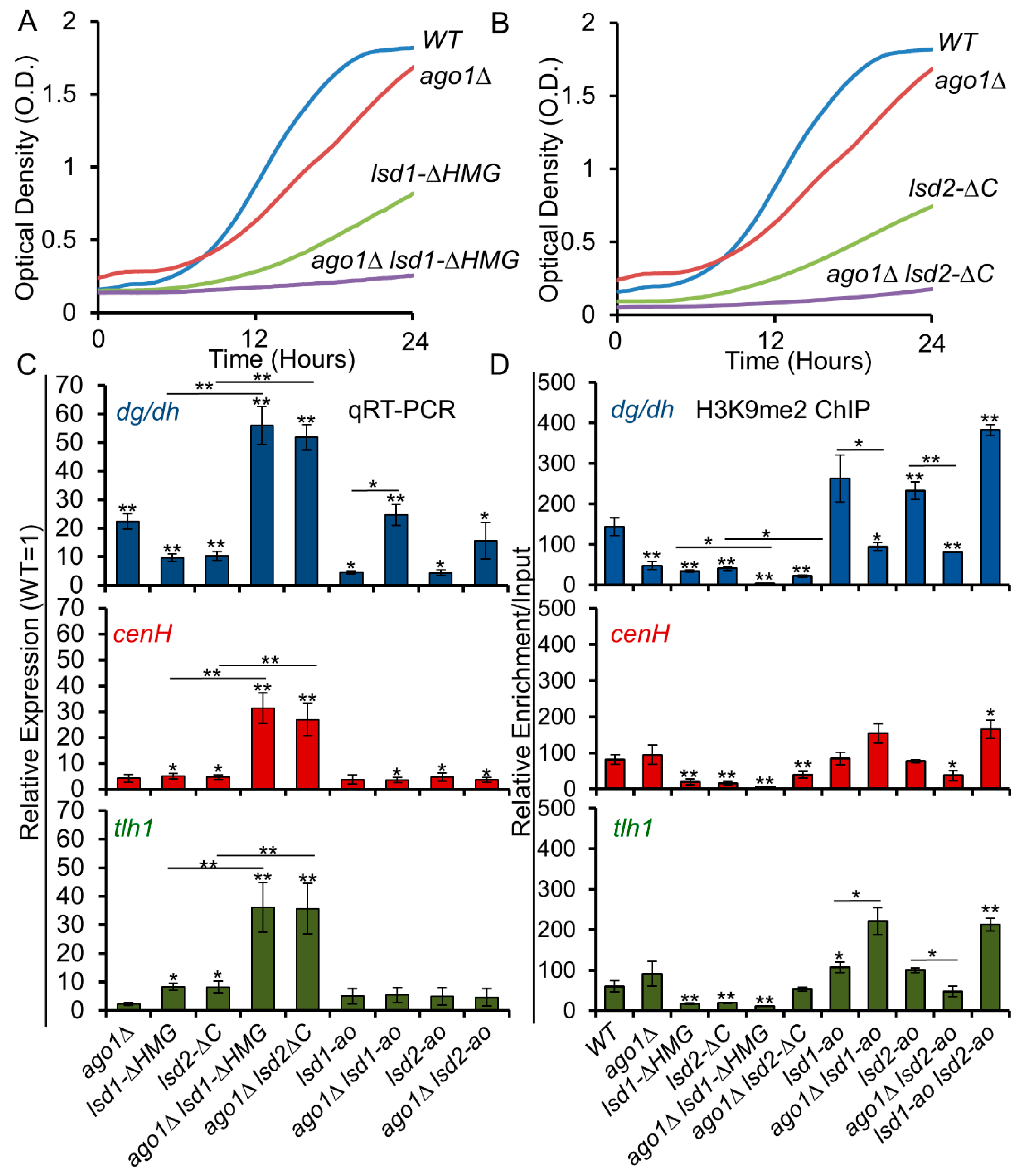
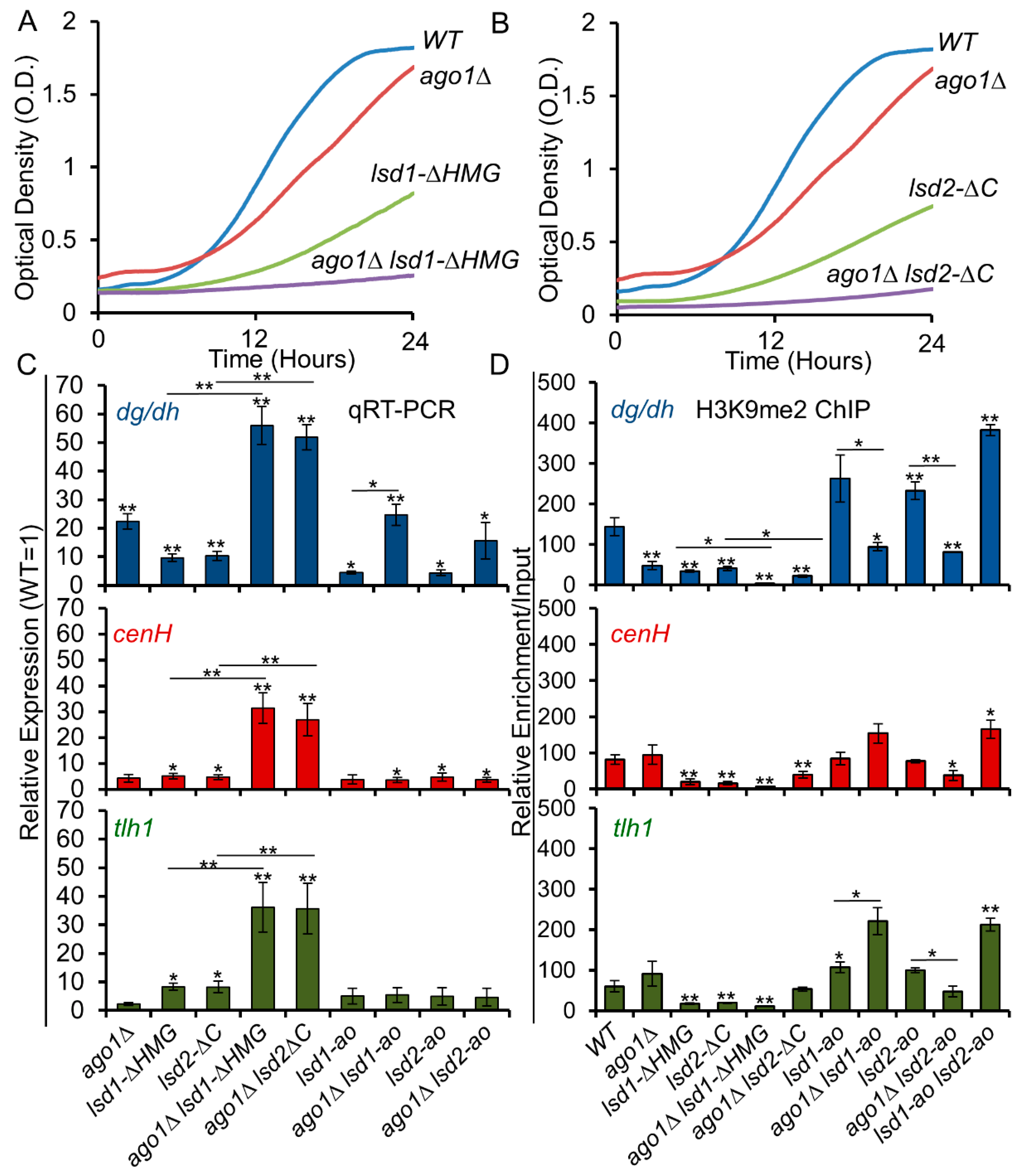
3.6. C-Terminal Mutants of Lsd1 and Lsd2 Exacerbate the Silencing Defects of ago1Δ
3.7. The Lysine Demethylase Activity of Lsd Proteins Antagonize RNAi
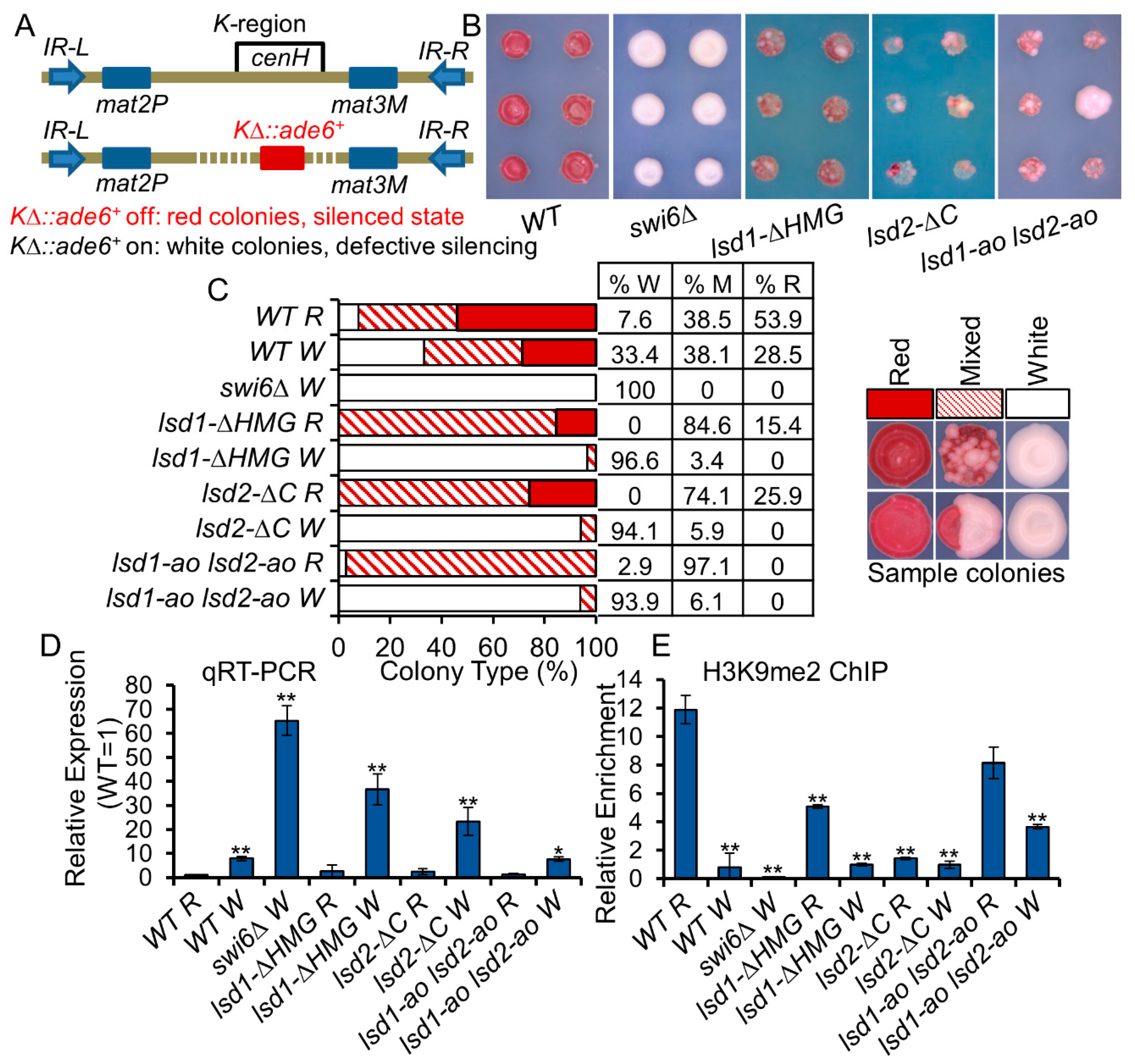
3.8. Lsd1/2 Play Roles in the Maintenance and Re-Establishment of Heterochromatin at the Mat Locus via an RNAi-Independent Mechanism(s)
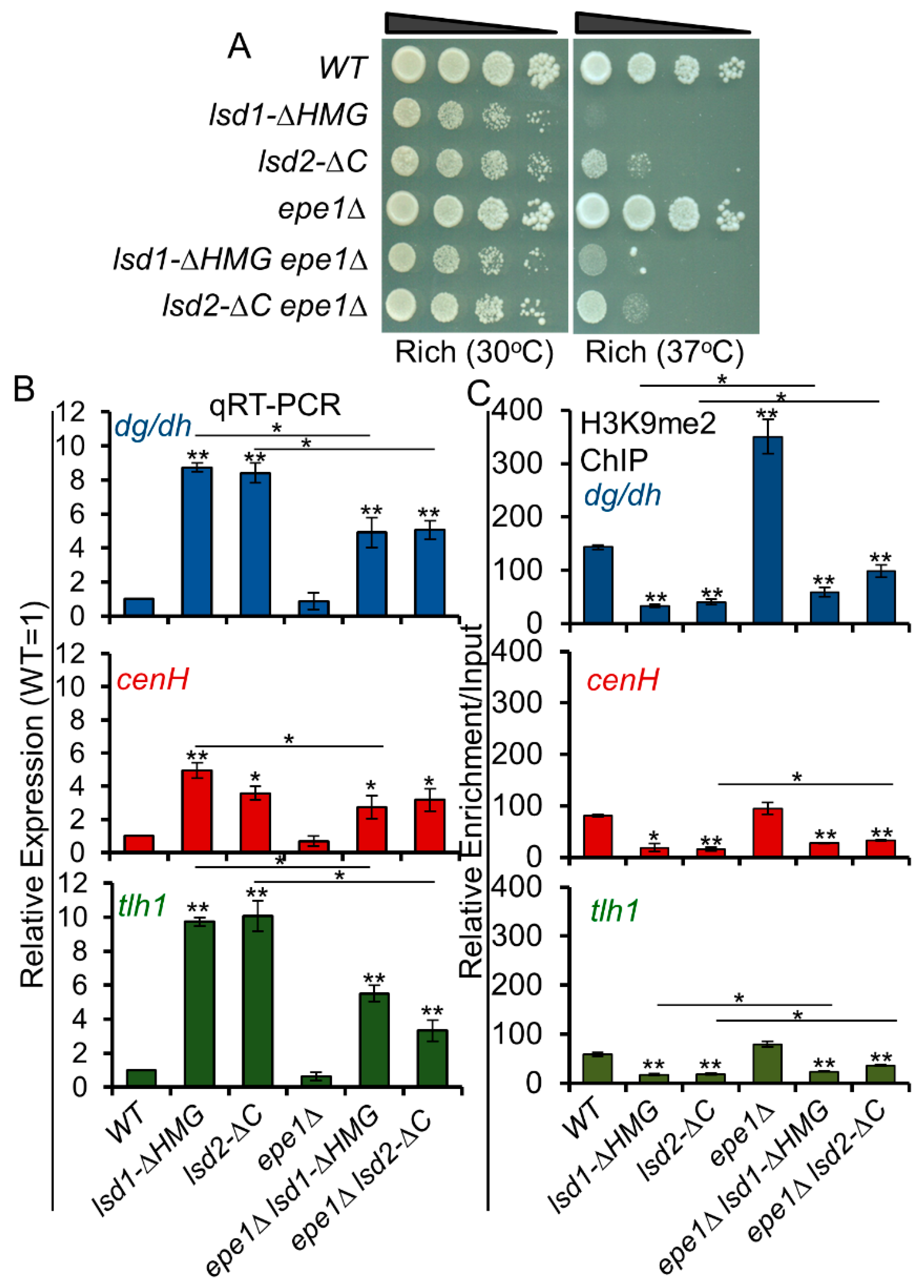
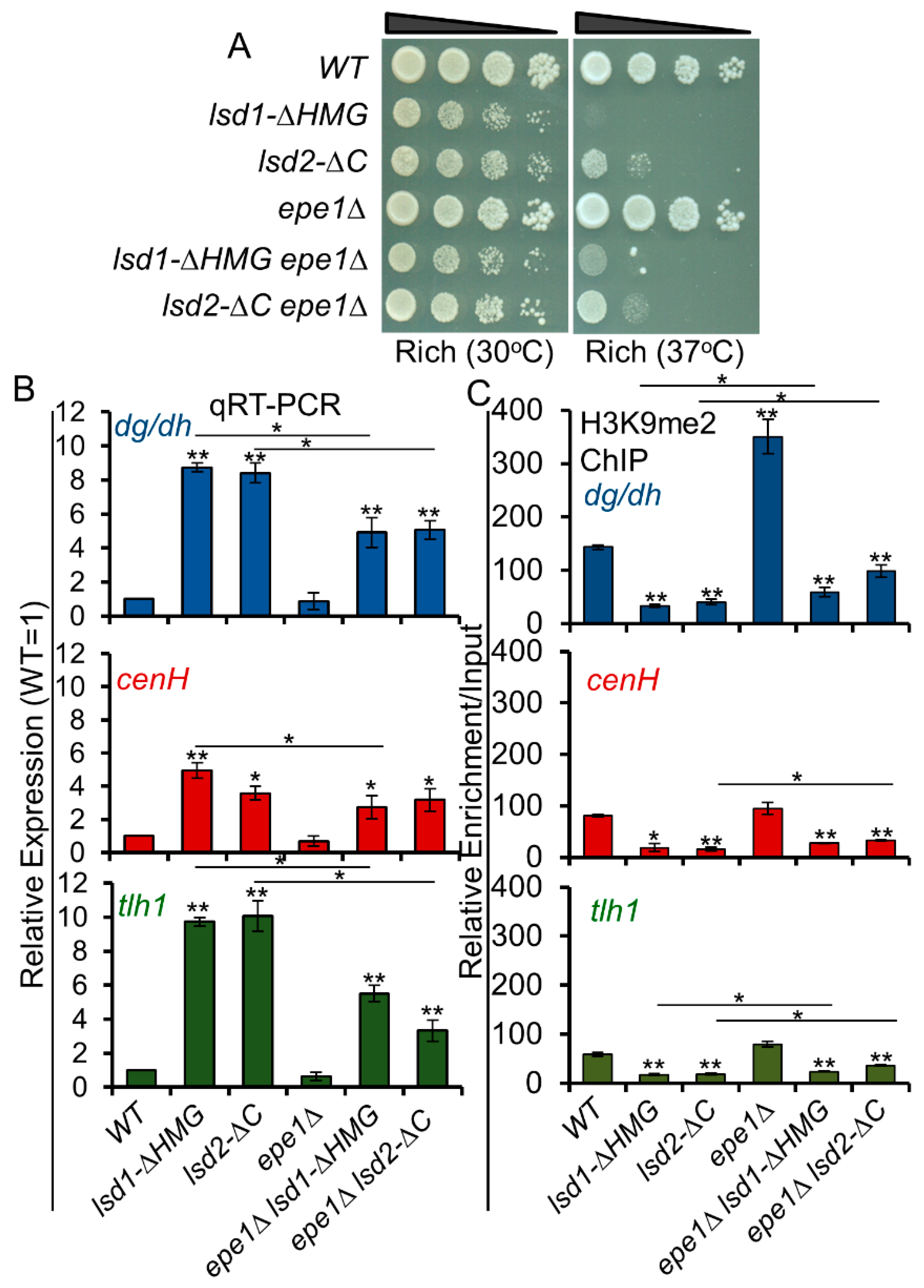
3.9. Loss of Epe1 Suppresses the Silencing Defects of Lsd1 and Lsd2 Mutants
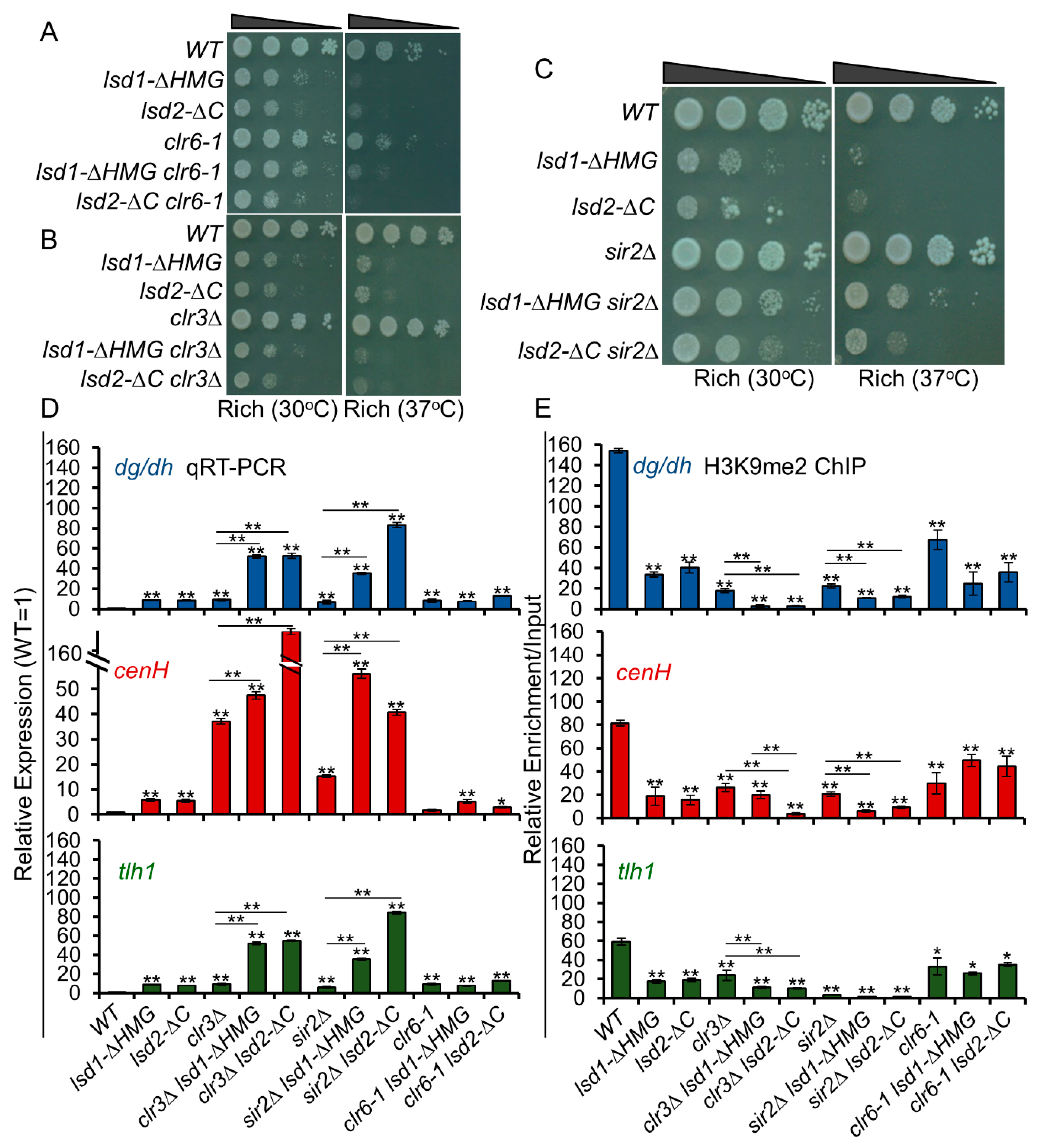
3.10. Lsd1 and Lsd2 Coordinate with Clr3 and Sir2, but Not with Clr6, in Transcriptional Repression at Heterochromatic Regions
4. Discussion
4.1. The N-Terminus of Lsd2
4.2. Mutual Regulation of Lsd1 and Lsd2
4.3. The Catalytic Activities of Lsd Proteins Antagonize RNAi
4.4. The Role of Lsd Proteins in Maintenance and Re-Establishment of Epigenetic Silencing
4.5. The Roles of LSD Proteins beyond Their Catalytic Activities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 128, 802-e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, E.I.; Reinberg, D. Histones: Annotating chromatin. Annu. Rev. Genet. 2009, 43, 559–599. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [Green Version]
- Shogren-Knaak, M.; Peterson, C.L. Switching on chromatin: Mechanistic role of histone H4-K16 acetylation. Cell Cycle 2006, 5, 1361–1365. [Google Scholar] [CrossRef]
- Huisinga, K.L.; Brower-Toland, B.; Elgin, S.C. The contradictory definitions of heterochromatin: Transcription and silencing. Chromosoma 2006, 115, 110–122. [Google Scholar] [CrossRef]
- Grewal, S.I.; Elgin, S.C. Transcription and RNA interference in the formation of heterochromatin. Nature 2007, 447, 399–406. [Google Scholar] [CrossRef]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Noma, K.; Allis, C.D.; Grewal, S.I. Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 2001, 293, 1150–1155. [Google Scholar] [CrossRef]
- Cam, H.P.; Sugiyama, T.; Chen, E.S.; Chen, X.; FitzGerald, P.C.; Grewal, S.I. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 2005, 37, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Djupedal, I.; Portoso, M.; Spahr, H.; Bonilla, C.; Gustafsson, C.M.; Allshire, R.C.; Ekwall, K. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 2005, 19, 2301–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.S.; Zhang, K.; Nicolas, E.; Cam, H.P.; Zofall, M.; Grewal, S.I. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature 2008, 451, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, M.R.; Verdel, A.; Colmenares, S.U.; Gerber, S.A.; Gygi, S.P.; Moazed, D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell 2004, 119, 789–802. [Google Scholar] [CrossRef]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Provost, P.; Dishart, D.; Doucet, J.; Frendewey, D.; Samuelsson, B.; Radmark, O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002, 21, 5864–5874. [Google Scholar] [CrossRef] [Green Version]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004, 303, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Partridge, J.F.; Scott, K.S.; Bannister, A.J.; Kouzarides, T.; Allshire, R.C. cis-acting DNA from fission yeast centromeres mediates histone H3 methylation and recruitment of silencing factors and cohesin to an ectopic site. Curr. Biol. 2002, 12, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Mosch, K.; Fischle, W.; Grewal, S.I. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 2008, 15, 381–388. [Google Scholar] [CrossRef]
- Schalch, T.; Job, G.; Noffsinger, V.J.; Shanker, S.; Kuscu, C.; Joshua-Tor, L.; Partridge, J.F. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell 2009, 34, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.J.; Villen, J.; Gerace, E.L.; Gygi, S.P.; Moazed, D. A cullin E3 ubiquitin ligase complex associates with Rik1 and the Clr4 histone H3-K9 methyltransferase and is required for RNAi-mediated heterochromatin formation. RNA Biol. 2005, 2, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvine, D.V.; Zaratiegui, M.; Tolia, N.H.; Goto, D.B.; Chitwood, D.H.; Vaughn, M.W.; Joshua-Tor, L.; Martienssen, R.A. Argonaute slicing is required for heterochromatic silencing and spreading. Science 2006, 313, 1134–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noma, K.; Sugiyama, T.; Cam, H.; Verdel, A.; Zofall, M.; Jia, S.; Moazed, D.; Grewal, S.I. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat. Genet. 2004, 36, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Giles, K.E.; Ghirlando, R.; Felsenfeld, G. Maintenance of a constitutive heterochromatin domain in vertebrates by a Dicer-dependent mechanism. Nat. Cell Biol. 2010, 12, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martienssen, R.; Moazed, D. RNAi and heterochromatin assembly. Cold Spring Harb. Perspect. Biol. 2015, 7, a019323. [Google Scholar] [CrossRef] [PubMed]
- Yigit, E.; Batista, P.J.; Bei, Y.; Pang, K.M.; Chen, C.C.; Tolia, N.H.; Joshua-Tor, L.; Mitani, S.; Simard, M.J.; Mello, C.C. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell 2006, 127, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Noma, K.; Grewal, S.I. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science 2004, 304, 1971–1976. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Zhang, K.; Zofall, M.; Chen, E.; Grewal, S.I. Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat. Struct. Mol. Biol. 2011, 18, 1132–1138. [Google Scholar] [CrossRef] [Green Version]
- Tucker, J.F.; Ohle, C.; Schermann, G.; Bendrin, K.; Zhang, W.; Fischer, T.; Zhang, K. A Novel Epigenetic Silencing Pathway Involving the Highly Conserved 5’-3’ Exoribonuclease Dhp1/Rat1/Xrn2 in Schizosaccharomyces pombe. PLoS Genet. 2016, 12, e1005873. [Google Scholar] [CrossRef] [Green Version]
- Chalamcharla, V.R.; Folco, H.D.; Dhakshnamoorthy, J.; Grewal, S.I. Conserved factor Dhp1/Rat1/Xrn2 triggers premature transcription termination and nucleates heterochromatin to promote gene silencing. Proc. Natl. Acad. Sci. USA 2015, 112, 15548–15555. [Google Scholar] [CrossRef] [Green Version]
- Alper, B.J.; Job, G.; Yadav, R.K.; Shanker, S.; Lowe, B.R.; Partridge, J.F. Sir2 is required for Clr4 to initiate centromeric heterochromatin assembly in fission yeast. EMBO J. 2013, 32, 2321–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscaino, A.; Lejeune, E.; Audergon, P.; Hamilton, G.; Pidoux, A.; Allshire, R.C. Distinct roles for Sir2 and RNAi in centromeric heterochromatin nucleation, spreading and maintenance. EMBO J. 2013, 32, 1250–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekwall, K. Genome-wide analysis of HDAC function. Trends Genet. 2005, 21, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, E.; Lee, M.G.; Hakimi, M.A.; Cam, H.P.; Grewal, S.I.; Shiekhattar, R. Fission yeast homologs of human histone H3 lysine 4 demethylase regulate a common set of genes with diverse functions. J. Biol. Chem. 2006, 281, 35983–35988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.R.; Burns, G.; Mata, J.; Volpe, T.A.; Martienssen, R.A.; Bahler, J.; Thon, G. Global effects on gene expression in fission yeast by silencing and RNA interference machineries. Mol. Cell. Biol. 2005, 25, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Cam, H.P.; Noma, K.; Ebina, H.; Levin, H.L.; Grewal, S.I. Host genome surveillance for retrotransposons by transposon-derived proteins. Nature 2008, 451, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, N.; Noma, K.; Isaac, S.; Kahan, T.; Grewal, S.I.; Cohen, A. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol. Cell. Biol. 2003, 23, 4356–4370. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Zaratiegui, M.; Villen, J.; Vaughn, M.W.; Verdel, A.; Huarte, M.; Shi, Y.; Gygi, S.P.; Moazed, D.; Martienssen, R.A.; et al. pombe LSD1 homologs regulate heterochromatin propagation and euchromatic gene transcription. Mol. Cell 2007, 26, 89–101. [Google Scholar] [CrossRef]
- Trewick, S.C.; Minc, E.; Antonelli, R.; Urano, T.; Allshire, R.C. The JmjC domain protein Epe1 prevents unregulated assembly and disassembly of heterochromatin. EMBO J. 2007, 26, 4670–4682. [Google Scholar] [CrossRef] [Green Version]
- Aygun, O.; Mehta, S.; Grewal, S.I. HDAC-mediated suppression of histone turnover promotes epigenetic stability of heterochromatin. Nat. Struct. Mol. Biol. 2013, 20, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, S.; Asano, T.; Kanoh, J.; Ishikawa, F. Transcription-induced chromatin association of RNA surveillance factors mediates facultative heterochromatin formation in fission yeast. Genes Cells 2013, 18, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tadeo, X.; Hou, H.; Tu, P.G.; Thompson, J.; Yates, J.R.; Jia, S. Epe1 recruits BET family bromodomain protein Bdf2 to establish heterochromatin boundaries. Genes Dev. 2013, 27, 1886–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrier, L.; Taglini, F.; Barrales, R.R.; Webb, S.; Urano, T.; Braun, S.; Bayne, E.H. Global regulation of heterochromatin spreading by Leo1. Open Biol. 2015, 5, 150045. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huarte, M.; Zaratiegui, M.; Vaughn, M.W.; Shi, Y.; Martienssen, R.; Cande, W.Z. Lid2 is required for coordinating H3K4 and H3K9 methylation of heterochromatin and euchromatin. Cell 2008, 135, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Fischle, W.; Sugiyama, T.; Allis, C.D.; Grewal, S.I. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol. Cell 2005, 20, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Watts, B.R.; Wittmann, S.; Wery, M.; Gautier, C.; Kus, K.; Birot, A.; Heo, D.H.; Kilchert, C.; Morillon, A.; Vasiljeva, L. Histone deacetylation promotes transcriptional silencing at facultative heterochromatin. Nucleic Acids Res. 2018, 46, 5426–5440. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Gordon, M.; Holt, D.G.; Panigrahi, A.; Wilhelm, B.T.; Erdjument-Bromage, H.; Tempst, P.; Bahler, J.; Cairns, B.R. Genome-wide dynamics of SAPHIRE, an essential complex for gene activation and chromatin boundaries. Mol. Cell. Biol. 2007, 27, 4058–4069. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Sawada, J.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Cowger, J.J.; Zhao, Q.; Isovic, M.; Torchia, J. Biochemical characterization of the zinc-finger protein 217 transcriptional repressor complex: Identification of a ZNF217 consensus recognition sequence. Oncogene 2007, 26, 3378–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, J.M.; Link, J.E.; Morgan, B.S.; Heller, F.J.; Hargrove, A.E.; McCafferty, D.G. KDM1 class flavin-dependent protein lysine demethylases. Biopolymers 2015, 104, 213–246. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Chen, F.; Dong, Z.; Hu, D.; Barbera, A.J.; Clark, E.A.; Fang, J.; Yang, Y.; Mei, P.; Rutenberg, M.; et al. LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Mol. Cell 2013, 49, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Van Essen, D.; Zhu, Y.; Saccani, S. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol. Cell 2010, 39, 750–760. [Google Scholar] [CrossRef]
- Fang, R.; Barbera, A.J.; Xu, Y.; Rutenberg, M.; Leonor, T.; Bi, Q.; Lan, F.; Mei, P.; Yuan, G.C.; Lian, C.; et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 2010, 39, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Karytinos, A.; Forneris, F.; Profumo, A.; Ciossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 2009, 284, 17775–17782. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Collins, R.E.; De Cegli, R.; Alpatov, R.; Horton, J.R.; Shi, X.; Gozani, O.; Cheng, X.; Shi, Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 2007, 448, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Opel, M.; Lando, D.; Bonilla, C.; Trewick, S.C.; Boukaba, A.; Walfridsson, J.; Cauwood, J.; Werler, P.J.; Carr, A.M.; Kouzarides, T.; et al. Genome-wide studies of histone demethylation catalysed by the fission yeast homologues of mammalian LSD1. PLoS ONE 2007, 2, e386. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Roseaulin, L.; Schurra, C.; Waxin, H.; Lambert, S.; Zaratiegui, M.; Martienssen, R.A.; Arcangioli, B. Lsd1 and lsd2 control programmed replication fork pauses and imprinting in fission yeast. Cell Rep. 2012, 2, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, S.; Klar, A.; Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991, 194, 795–823. [Google Scholar] [PubMed]
- Bahler, J.; Wu, J.Q.; Longtine, M.S.; Shah, N.G.; McKenzie, A., III; Steever, A.B.; Wach, A.; Philippsen, P.; Pringle, J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 1998, 14, 943–951. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Vo, T.V.; Thillainadesan, G.; Holla, S.; Zhang, B.; Jiang, Y.; Lv, M.; Xu, Z.; Wang, C.; Balachandran, V.; et al. A conserved dimer interface connects ERH and YTH family proteins to promote gene silencing. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Hagan, I.M. Chromatin and Cell Wall Staining of Schizosaccharomyces pombe. Cold Spring Harb. Protoc. 2016, 2016, pdb–prot091025. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, H.; Ding, D.Q.; Haraguchi, T.; Hiraoka, Y. Microscopic Observation of Living Cells Stained with Fluorescent Probes. Cold Spring Harb. Protoc. 2017, 2017, pdb-prot079848. [Google Scholar] [CrossRef] [PubMed]
- Marayati, B.F.; Drayton, A.L.; Tucker, J.F.; Huckabee, R.H.; Anderson, A.M.; Pease, J.B.; Zeyl, C.W.; Zhang, K. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics 2018, 209, 967–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malarkey, C.S.; Churchill, M.E. The high mobility group box: The ultimate utility player of a cell. Trends Biochem. Sci. 2012, 37, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grewal, S.I.; Klar, A.J. Chromosomal inheritance of Epigenetic states in fission yeast during mitosis and meiosis. Cell 1996, 86, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Moazed, D. DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science 2017, 356, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Wang, X.; Moazed, D. Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 2018, 558, 615–619. [Google Scholar] [CrossRef]
- Zofall, M.; Grewal, S.I. Swi6/HP1 recruits a JmjC domain protein to facilitate transcription of heterochromatic repeats. Mol. Cell 2006, 22, 681–692. [Google Scholar] [CrossRef]
- Bao, K.; Shan, C.M.; Moresco, J.; Yates, J.; Jia, S. Anti-silencing factor Epe1 associates with SAGA to regulate transcription within heterochromatin. Genes Dev. 2019, 33, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Audergon, P.N.; Catania, S.; Kagansky, A.; Tong, P.; Shukla, M.; Pidoux, A.L.; Allshire, R.C. Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science 2015, 348, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Kim, T.Y.; Kim, M.S.; Kim, M.A.; Park, S.H.; Jang, Y.K. Nuclear import of human histone lysine-specific demethylase LSD1. J. Biochem. 2014, 156, 305–313. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.B.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, B.; Shan, Z.; Nie, J.; Deng, R.; Xiong, R.; Tsun, A.; Pan, W.; Zhao, H.; Chen, L.; et al. Histone demethylase LSD1 restricts influenza A virus infection by erasing IFITM3-K88 monomethylation. PLoS Pathog. 2017, 13, e1006773. [Google Scholar] [CrossRef]
- Sakane, N.; Kwon, H.S.; Pagans, S.; Kaehlcke, K.; Mizusawa, Y.; Kamada, M.; Lassen, K.G.; Chan, J.; Greene, W.C.; Schnoelzer, M.; et al. Activation of HIV transcription by the viral Tat protein requires a demethylation step mediated by lysine-specific demethylase 1 (LSD1/KDM1). PLoS Pathog. 2011, 7, e1002184. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, X.; Yang, H.; Xu, Y. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol. Cell 2015, 58, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A.; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018, 115, E4179–E4188. [Google Scholar] [CrossRef] [Green Version]
- Wissmann, M.; Yin, N.; Muller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Gunther, T.; Buettner, R.; et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Grewal, S.I. RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 2010, 20, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Hall, I.M.; Shankaranarayana, G.D.; Noma, K.; Ayoub, N.; Cohen, A.; Grewal, S.I. Establishment and maintenance of a heterochromatin domain. Science 2002, 297, 2232–2237. [Google Scholar] [CrossRef] [Green Version]
- Jih, G.; Iglesias, N.; Currie, M.A.; Bhanu, N.V.; Paulo, J.A.; Gygi, S.P.; Garcia, B.A.; Moazed, D. Unique roles for histone H3K9me states in RNAi and heritable silencing of transcription. Nature 2017, 547, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, P.S.; Larkin, M.; Thillainadesan, G.; Dhakshnamoorthy, J.; Balachandran, V.; Xiao, H.; Wellman, C.; Chatterjee, R.; Wheeler, D.; Grewal, S.I.S. Iron homeostasis regulates facultative heterochromatin assembly in adaptive genome control. Nat. Struct. Mol. Biol. 2018, 25, 372–383. [Google Scholar] [CrossRef]
- Sugiyama, T.; Cam, H.P.; Sugiyama, R.; Noma, K.; Zofall, M.; Kobayashi, R.; Grewal, S.I. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell 2007, 128, 491–504. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marayati, B.F.; Tucker, J.F.; De La Cerda, D.A.; Hou, T.-C.; Chen, R.; Sugiyama, T.; Pease, J.B.; Zhang, K. The Catalytic-Dependent and -Independent Roles of Lsd1 and Lsd2 Lysine Demethylases in Heterochromatin Formation in Schizosaccharomyces pombe. Cells 2020, 9, 955. https://doi.org/10.3390/cells9040955
Marayati BF, Tucker JF, De La Cerda DA, Hou T-C, Chen R, Sugiyama T, Pease JB, Zhang K. The Catalytic-Dependent and -Independent Roles of Lsd1 and Lsd2 Lysine Demethylases in Heterochromatin Formation in Schizosaccharomyces pombe. Cells. 2020; 9(4):955. https://doi.org/10.3390/cells9040955
Chicago/Turabian StyleMarayati, Bahjat F., James F. Tucker, David A. De La Cerda, Tien-Chi Hou, Rong Chen, Tomoyasu Sugiyama, James B. Pease, and Ke Zhang. 2020. "The Catalytic-Dependent and -Independent Roles of Lsd1 and Lsd2 Lysine Demethylases in Heterochromatin Formation in Schizosaccharomyces pombe" Cells 9, no. 4: 955. https://doi.org/10.3390/cells9040955
APA StyleMarayati, B. F., Tucker, J. F., De La Cerda, D. A., Hou, T.-C., Chen, R., Sugiyama, T., Pease, J. B., & Zhang, K. (2020). The Catalytic-Dependent and -Independent Roles of Lsd1 and Lsd2 Lysine Demethylases in Heterochromatin Formation in Schizosaccharomyces pombe. Cells, 9(4), 955. https://doi.org/10.3390/cells9040955