Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Osteosarcoma
2.1. Low-Grade Osteosarcomas
2.2. High-Grade Osteosarcomas
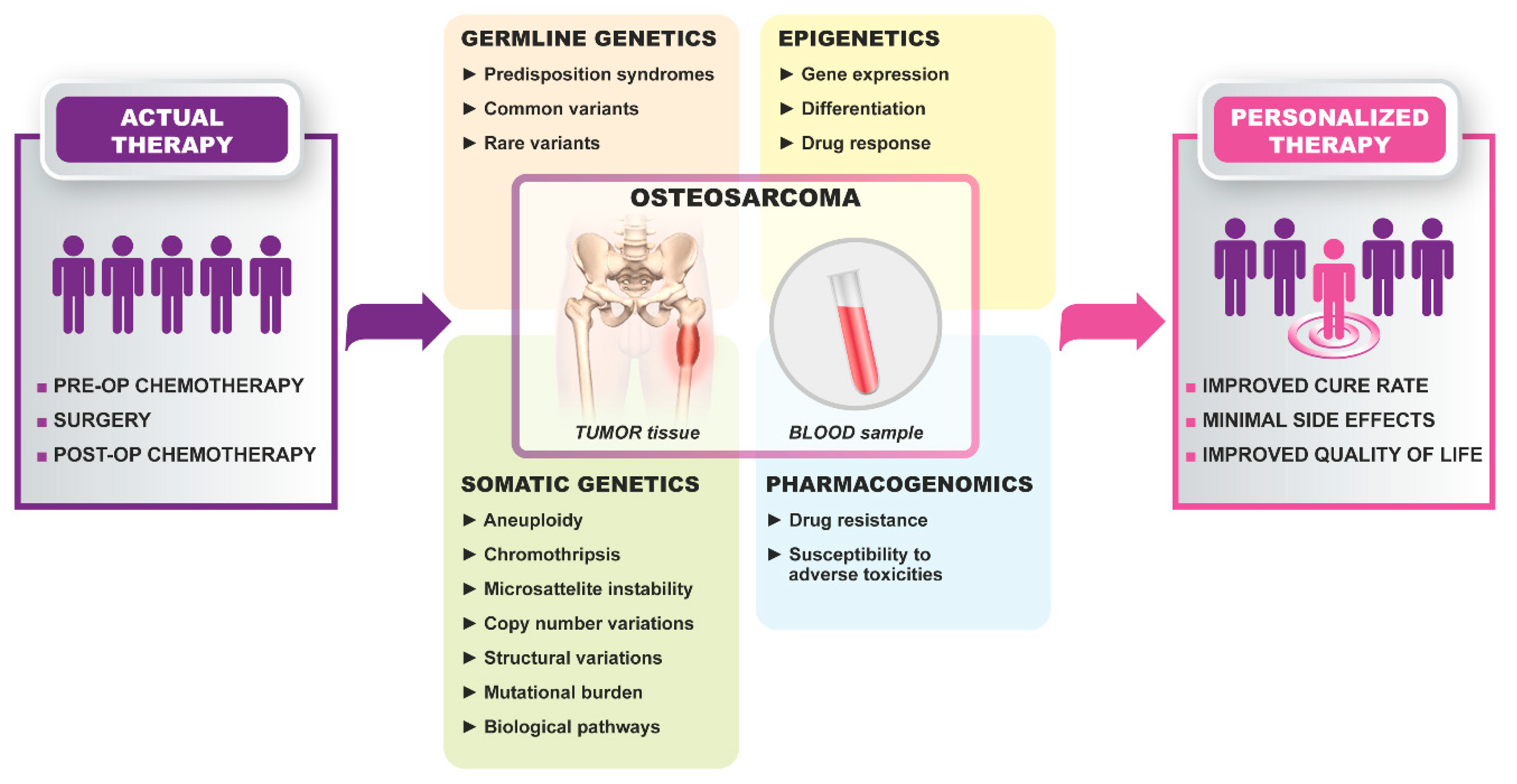
2.2.1. Germline Genetics
2.2.2. Somatic Genetics
2.2.3. Candidate Predictive and Prognostic Biomarkers
2.2.4. Epigenetics
2.2.5. Immunotherapy and Tumor Mutational Burden
3. Chondrosarcoma
3.1. Central Chondrosarcomas
3.2. Secondary Peripheral Chondrosarcomas
3.3. Rare Variants of Chondrosarcoma
4. Ewing Sarcoma
5. Critical Open Issues and Perspectives
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT/CS1 | atypical cartilaginous tumour/chondrosarcoma grade 1 |
| ACVR2A-FN | activin receptor 2A- fibronectin |
| AKT | Serine/Threonine Kinase+ |
| APC | APC regulator of WNT signaling pathway |
| APCDD1 | APC down-regulated 1 |
| Bcl-2 | Apoptosis Regulator |
| BET | the bromodomain and extra terminal domain |
| BRCA1 | breast related cancer antigen 1 |
| BRCA2 | breast related cancer antigen 2 |
| CD99 | CD99 Molecule (XgBloodGroup) |
| CDK4 | cyclin-dependent kinase 4 |
| CDK4/6 | cyclin-dependent kinase 4/6 |
| CDKN2A | cyclin-dependent kinase Inhibitor 2A |
| CR | complete response |
| DAPK1 | death-associated protein kinase 1 |
| DNA/RNA | DeoxyriboNucleic Acid/RiboNucleic Acid |
| DNMTs | DNA methyltransferases |
| EGFR | epidermal growth factor receptor |
| ERCC1 | excision repair cross-complementation group 1 |
| ERCC2 | excision repair cross-complementation group 2 |
| ERG | ETS transcription factor ERG |
| ETS | proto-oncogene 1, transcription factor 1 |
| ETV1 | ETS Variant Transcription Factor 1 |
| ETV4 | ETS Variant Transcription Factor 4 |
| EWS-FLI | Ewing Sarcoma-Friend Leukemia Insertion |
| EWS | Ewing Sarcoma |
| EWSR1 | Ewing sarcoma breakpoint region 1, EWS RNA Binding Protein 1 |
| EWSR1–FLI1 | Ewing sarcoma breakpoint region 1 (EWS RNA Binding Protein 1) - Friend Leukemia Insertion |
| EXT | exostosin glycosyltransferase |
| EZH2 | enhancer of zeste homolog 2 |
| FEV | FEV Transcription Factor, ETS Family Member |
| FUS | FUS RNA Binding Protein |
| GD2 | disialoganglioside 2 |
| HDAC | histone deacetylase |
| HEY1 | Hes Related Family BHLH Transcription Factor With YRPW Motif 1 |
| HGOS | High-grade osteosarcoma |
| HIC1 | HIC ZBTB transcriptional repressor 1 |
| HSPGs | heparan sulphate proteoglycans |
| ICB | immune checkpoint blockade |
| IDH | isocitrate dehydrogenase genes |
| IGF-1 | Insulin-like growth factor 1 |
| IGF-1R | Insulin-like growth factor 1 receptor |
| IGF-2 | Insulin-like growth factor 2 |
| IGF | Insulin-like growth factor |
| IGF-2BP3 | Insulin-like growth factor 2 - mRNA-binding protein 3 |
| IHH | Indian Hedgehog Signaling Molecule |
| INFORM | individualized therapy for relapsed malignancies in childhood |
| IRF2BP2-CDX1 | Interferon Regulatory Factor 2 Binding Protein 2-Caudal Type Homeobox 1 |
| ISG | Italian Sarcoma Group |
| KDR | kinase insert domain receptor |
| KIT | Proto-Oncogene, Receptor Tyrosine Kinase |
| lncRNA | Long non-coding RNA |
| lncRNAs | long non-coding RNAs |
| LSAMP | Limbic System-Associated Membrane Protein |
| LSD1 | Lysine Demethylase 1° |
| MDM2 | MDM2 Proto-Oncogene |
| MGMT | O-6-methylguanine DNA methyltransferase |
| miRNA | microRNA |
| miRNAs | microRNAs |
| MSH2 | mut S homolog 2 |
| MTHFR | 5,10-methylenetetrahydrofolate reductase |
| mTOR | mammalian target of rapamycin |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NCOA2 | Nuclear Receptor Coactivator 2 |
| NER | Nucleotide excision repair |
| NGS | Next-generation sequencing |
| NRAS | NRAS Proto-Oncogene |
| p16 | also known as p16INK4a, cyclin-dependent kinase inhibitor 2A or CDKN2A, multiple tumor suppressor 1 |
| PALB2 | partner and localizer of BRCA2 |
| PARP | poly (ADP-ribose) polymerase |
| PD-1 | programmedcelldeath 1 |
| PD-L1 | programmed death-ligand 1 |
| PI3K/mTOR | phosphatidylinositol 3-kinase/mammalian target of rapamycin |
| PR | partial response |
| RASSF1A | Ras association domain family 1A |
| RB1 | RB TranscriptionalCorepressor 1 |
| RECQL | RecQ-like helicase |
| RHA | RNA helicase A |
| SAHA | suberoylanilide hydroxamic acid |
| SNP | Single nucleotide polymorphism |
| SQSTM1 | Sequestosome 1 |
| SRC | SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| STAG2 | Stromal Antigen 2 |
| SV | structural variations |
| TAF15 | TATA-Box Binding Protein Associated Factor 15 |
| TARGET | Generate Effective Treatments Osteosarcoma project |
| TET | Tet Methylcytosine Dioxygenase |
| TGFβ | transforming growth factor β |
| TIMP3 | TIMP metallopeptidase inhibitor 3 |
| TMB | tumor mutational burden |
| TP53 | Tumor Protein P53 |
| WWOX | WW Domain ContainingOxidoreductase |
| α-KG | α-ketoglutarate |
| 2-HG | 2-hydroxyglutarate |
References
- Fletcher, C.D.; Bridge, J.; Hogendoorn, P.C.; Mertens, F. WHO Classification of tumours of Soft Tissue and Bone, 4th ed.; WHO Press: Geneva, Switzerland, 2013; Volume 5. [Google Scholar]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroy, T.; Chilamakuri, C.S.; Lorenz, S.; Sun, J.; Bruland, O.S.; Myklebost, O.; Meza-Zepeda, L.A. Genome Analysis of Osteosarcoma Progression Samples Identifies FGFR1 Overexpression as a Potential Treatment Target and CHM as a Candidate Tumor Suppressor Gene. PLoS ONE 2016, 11, e0163859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picci, P. Osteosarcomas (OS). In Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions, 2nd ed.; Picci, P., Manfrini, M., Donati, D.M., Gambarotti, M., Righi, A., Vanel, D., Dei Tos, A.P., Eds.; Springer: Cham, Switzerland, 2020; pp. 185–212. [Google Scholar]
- He, X.; Pang, Z.; Zhang, X.; Lan, T.; Chen, H.; Chen, M.; Yang, H.; Huang, J.; Chen, Y.; Zhang, Z.; et al. Consistent Amplification of FRS2 and MDM2 in Low-grade Osteosarcoma: A Genetic Study of 22 Cases With Clinicopathologic Analysis. Am. J. Surg. Pathol. 2018, 42, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef]
- Sandberg, A.A.; Bridge, J.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Osteosarcoma and related tumors. Cancer Genet. Cytogenet. 2003, 145, 1–30. [Google Scholar] [CrossRef]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef]
- Smida, J.; Xu, H.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mosch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Negri, G.L.; Grande, B.M.; Delaidelli, A.; El-Naggar, A.; Cochrane, D.; Lau, C.C.; Triche, T.J.; Moore, R.A.; Jones, S.J.; Montpetit, A.; et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J. Pathol. 2019, 249, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Serra, M. Role of pharmacogenetics of drug-metabolizing enzymes in treating osteosarcoma. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Hattinger, C.M. The pharmacogenomics of osteosarcoma. Pharmacogenomics J. 2017, 17, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Deng, C.; Tian, Y.; Ma, X. Meta-analysis showing that ERCC1 polymorphism is predictive of osteosarcoma prognosis. Oncotarget 2017, 8, 62769–62779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ge, J.; Hong, H.; Bi, L.; Sun, Z. Genetic polymorphisms in ERCC1 and ERCC2 genes are associated with response to chemotherapy in osteosarcoma patients among Chinese population: A meta-analysis. World J. Surg. Oncol. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Hattinger, C.M.; Tavanti, E.; Fanelli, M.; Vella, S.; Picci, P.; Serra, M. Pharmacogenomics of genes involved in antifolate drug response and toxicity in osteosarcoma. Expert Opin. Drug Metab. Toxicol. 2017, 13, 245–257. [Google Scholar] [CrossRef]
- Xie, L.; Guo, W.; Yang, Y.; Ji, T.; Xu, J. More severe toxicity of genetic polymorphisms on MTHFR activity in osteosarcoma patients treated with high-dose methotrexate. Oncotarget 2018, 9, 11465–11476. [Google Scholar] [CrossRef] [Green Version]
- Egas-Bejar, D.; Anderson, P.M.; Agarwal, R.; Corrales-Medina, F.; Devarajan, E.; Huh, W.W.; Brown, R.E.; Subbiah, V. Theranostic Profiling for Actionable Aberrations in Advanced High Risk Osteosarcoma with Aggressive Biology Reveals High Molecular Diversity: The Human Fingerprint Hypothesis. Oncoscience 2014, 1, 167–179. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef]
- Zhou, Q.; Deng, Z.; Zhu, Y.; Long, H.; Zhang, S.; Zhao, J. mTOR/p70S6K signal transduction pathway contributes to osteosarcoma progression and patients’ prognosis. Med. Oncol. 2010, 27, 1239–1245. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Pandya, P.H.; Liu, E.; Chandra, P.; Wang, L.; Murray, M.E.; Carter, J.; Ferguson, M.; Saadatzadeh, M.R.; Bijangi-Visheshsaraei, K.; et al. Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma. BMC Med. Genom. 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ye, Z. Epigenetic alterations in osteosarcoma: Promising targets. Mol. Biol. Rep. 2014, 41, 3303–3315. [Google Scholar] [CrossRef]
- Morrow, J.J.; Khanna, C. Osteosarcoma Genetics and Epigenetics: Emerging Biology and Candidate Therapies. Crit. Rev. Oncog. 2015, 20, 173–197. [Google Scholar] [CrossRef] [Green Version]
- Easwaran, H.; Johnstone, S.E.; Van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012, 22, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Ji, M.; Yang, B.; Chen, Z.; Qiu, J.; Shi, X.; Lu, Z. Quantitative analysis of promoter hypermethylation in multiple genes in osteosarcoma. Cancer 2006, 106, 1602–1609. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59, 798–802. [Google Scholar]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.; Kimchi, A. DAP kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [CrossRef]
- Rosas, S.L.; Koch, W.; da Costa Carvalho, M.G.; Wu, L.; Califano, J.; Westra, W.; Jen, J.; Sidransky, D. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001, 61, 939–942. [Google Scholar] [PubMed]
- Pegg, A.E. Mammalian O6-alkylguanine-DNA alkyltransferase: Regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar] [PubMed]
- Han, W.; Liu, J. Epigenetic silencing of the Wnt antagonist APCDD1 by promoter DNA hyper-methylation contributes to osteosarcoma cell invasion and metastasis. Biochem. Biophys. Res. Commun. 2017, 491, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Kim, H.S.; Kim, H.H.; Kim, W.H.; Lee, S.H. Aberrant methylation of p14ARF gene correlates with poor survival in osteosarcoma. Clin. Orthop. Relat. Res. 2006, 442, 216–222. [Google Scholar] [CrossRef]
- Rathi, A.; Virmani, A.K.; Harada, K.; Timmons, C.F.; Miyajima, K.; Hay, R.J.; Mastrangelo, D.; Maitra, A.; Tomlinson, G.E.; Gazdar, A.F. Aberrant methylation of the HIC1 promoter is a frequent event in specific pediatric neoplasms. Clin. Cancer Res. 2003, 9, 3674–3678. [Google Scholar]
- Chen, W.; Cooper, T.K.; Zahnow, C.A.; Overholtzer, M.; Zhao, Z.; Ladanyi, M.; Karp, J.E.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L.; et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 2004, 6, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Ropke, M.; Boltze, C.; Neumann, H.W.; Roessner, A.; Schneider-Stock, R. Genetic and epigenetic alterations in tumor progression in a dedifferentiated chondrosarcoma. Pathol. Res. Pract. 2003, 199, 437–444. [Google Scholar] [CrossRef]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar]
- Benassi, M.S.; Molendini, L.; Gamberi, G.; Ragazzini, P.; Sollazzo, M.R.; Merli, M.; Asp, J.; Magagnoli, G.; Balladelli, A.; Bertoni, F.; et al. Alteration of pRb/p16/cdk4 regulation in human osteosarcoma. Int. J. Cancer 1999, 84, 489–493. [Google Scholar] [CrossRef]
- Benassi, M.S.; Molendini, L.; Gamberi, G.; Magagnoli, G.; Ragazzini, P.; Gobbi, G.A.; Sangiorgi, L.; Pazzaglia, L.; Asp, J.; Brantsing, C.; et al. Involvement of INK4A gene products in the pathogenesis and development of human osteosarcoma. Cancer 2001, 92, 3062–3067. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Sekine, K.; Hinohara, S.; Namiki, T.; Nobori, T.; Kaneko, Y. Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet. Cytogenet. 2000, 120, 91–98. [Google Scholar] [CrossRef]
- Patino-Garcia, A.; Pineiro, E.S.; Diez, M.Z.; Iturriagagoitia, L.G.; Klussmann, F.A.; Ariznabarreta, L.S. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. J. Pediatr. Hematol. Oncol. 2003, 25, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Palmini, G.; Marini, F.; Brandi, M.L. What Is New in the miRNA World Regarding Osteosarcoma and Chondrosarcoma? Molecules 2017, 22, 417. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wang, G.; Zheng, Y.; Hua, Y.; Cai, Z. Long non-coding RNAs in osteosarcoma. Oncotarget 2017, 8, 20462–20475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattinger, C.M.; Patrizio, M.P.; Tavanti, E.; Luppi, S.; Magagnoli, F.; Picci, P.; Serra, M. Genetic testing for high-grade osteosarcoma: A guide for future tailored treatments? Expert Rev. Mol. Diagn. 2018, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Manara, M.C.; Valente, S.; Cristalli, C.; Nicoletti, G.; Landuzzi, L.; Zwergel, C.; Mazzone, R.; Stazi, G.; Arimondo, P.B.; Pasello, M.; et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol. Cancer Ther. 2018, 17, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Zwergel, C.; Schnekenburger, M.; Sarno, F.; Battistelli, C.; Manara, M.C.; Stazi, G.; Mazzone, R.; Fioravanti, R.; Gros, C.; Ausseil, F.; et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin. Epigenet. 2019, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef]
- Nuytemans, L.; Sys, G.; Creytens, D.; Lapeire, L. NGS-analysis to the rescue: Dual checkpoint inhibition in metastatic osteosarcoma—A case report and review of the literature. Acta Clin. Belg. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Xiao, Q.; Zhou, B.; Dai, Z.; Kang, Y. Prognostic Significance of Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Axial Osteosarcoma. World Neurosurg. 2019, 129, e240–e254. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Ren, T.; Huang, Y.; Sun, K.; Wang, S.; Bao, X.; Liu, K.; Guo, W. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J. Hematol. Oncol. 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmerini, E.; Agostinelli, C.; Picci, P.; Pileri, S.; Marafioti, T.; Lollini, P.L.; Scotlandi, K.; Longhi, A.; Benassi, M.S.; Ferrari, S. Tumoral immune-infiltrate (IF), PD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget 2017, 8, 111836–111846. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Chang, T.C.; Carter, R.A.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; Peng, J.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovee, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Donati, D.M.; Bianchi, G. Chondrosarcomas (CHS). In Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions, 2nd ed.; Picci, P., Manfrini, M., Donati, D.M., Gambarotti, M., Righi, A., Vanel, D., Dei Tos, A.P., Eds.; Springer: Cham, Switzerland, 2020; pp. 157–179. [Google Scholar]
- Grimer, R.J.; Gosheger, G.; Taminiau, A.; Biau, D.; Matejovsky, Z.; Kollender, Y.; San-Julian, M.; Gherlinzoni, F.; Ferrari, C. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur. J. Cancer 2007, 43, 2060–2065. [Google Scholar] [CrossRef] [PubMed]
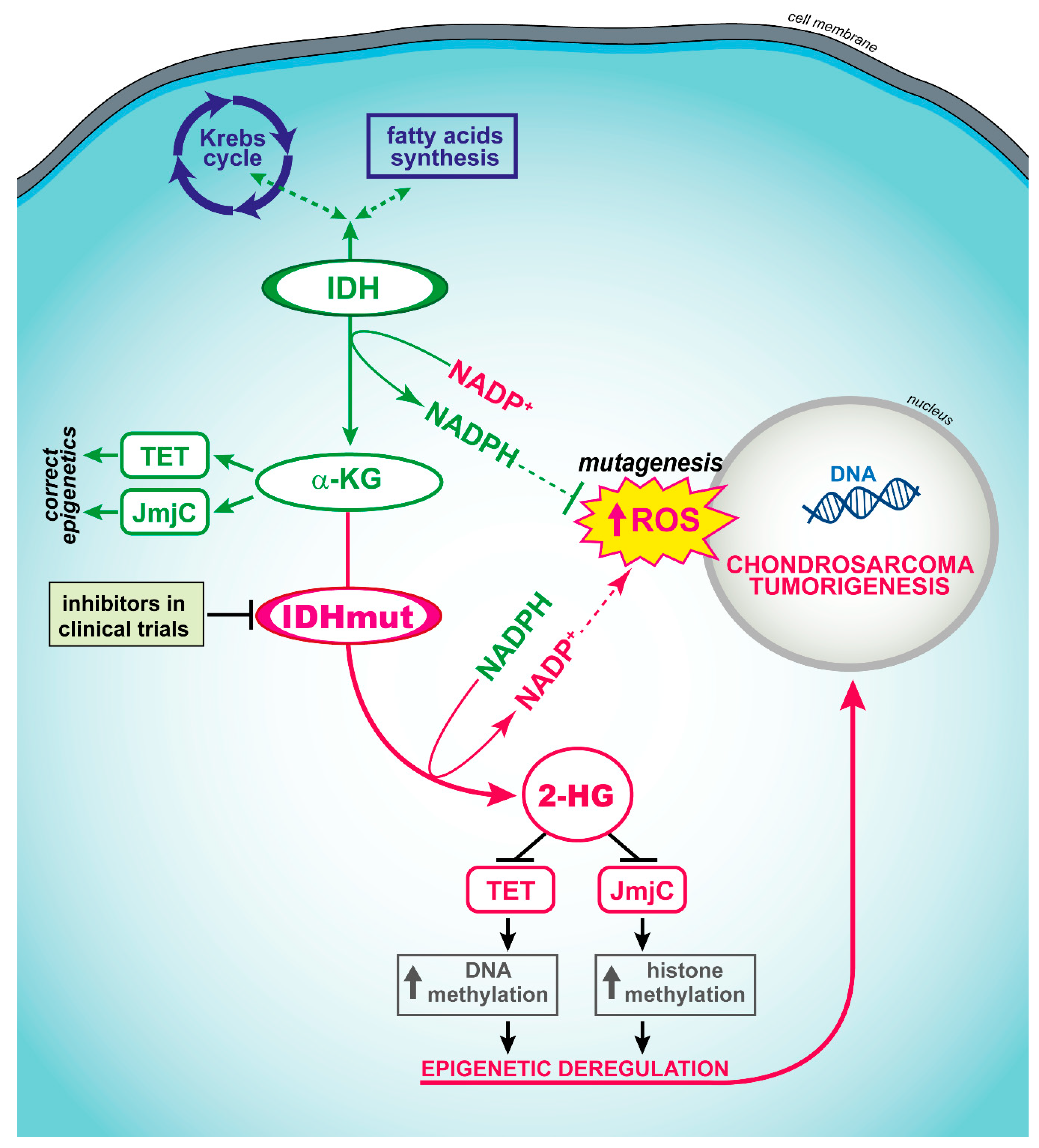
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Verdegaal, S.H.; Bovee, J.V.; Pansuriya, T.C.; Grimer, R.J.; Ozger, H.; Jutte, P.C.; San Julian, M.; Biau, D.J.; van der Geest, I.C.; Leithner, A.; et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicenter study of 161 patients. Oncologist 2011, 16, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.G.; Nafa, K.; Agaram, N.; Zehir, A.; Benayed, R.; Sadowska, J.; Borsu, L.; Kelly, C.; Tap, W.D.; Fabbri, N.; et al. Genomic Profiling Identifies Association of IDH1/IDH2 Mutation with Longer Relapse-Free and Metastasis-Free Survival in High-Grade Chondrosarcoma. Clin. Cancer Res. 2020, 26, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Tallini, G.; Dorfman, H.; Brys, P.; Dal Cin, P.; De Wever, I.; Fletcher, C.D.; Jonson, K.; Mandahl, N.; Mertens, F.; Mitelman, F.; et al. Correlation between clinicopathological features and karyotype in 100 cartilaginous and chordoid tumours. A report from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. J. Pathol. 2002, 196, 194–203. [Google Scholar] [CrossRef]
- Meijer, D.; de Jong, D.; Pansuriya, T.C.; van den Akker, B.E.; Picci, P.; Szuhai, K.; Bovee, J.V. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes Chromosomes Cancer 2012, 51, 899–909. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef]
- Totoki, Y.; Yoshida, A.; Hosoda, F.; Nakamura, H.; Hama, N.; Ogura, K.; Fujiwara, T.; Arai, Y.; Toguchida, J.; Tsuda, H.; et al. Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res. 2014, 24, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Buhnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovee, J.V.; et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amary, M.F.; Ye, H.; Forbes, G.; Damato, S.; Maggiani, F.; Pollock, R.; Tirabosco, R.; Flanagan, A.M. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015, 466, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Fritchie, K.; Wei, S.; Ali, N.; Curless, K.; Shen, T.; Brini, A.T.; Latif, F.; Sumathi, V.; Siegal, G.P.; et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum. Pathol. 2017, 65, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Berzero, G.; Di Stefano, A.L.; Sanson, M. The clinical use of IDH1 and IDH2 mutations in gliomas. Expert Rev. Mol. Diagn. 2018, 18, 1041–1051. [Google Scholar] [CrossRef]
- Hameetman, L.; Rozeman, L.B.; Lombaerts, M.; Oosting, J.; Taminiau, A.H.; Cleton-Jansen, A.M.; Bovee, J.V.; Hogendoorn, P.C. Peripheral chondrosarcoma progression is accompanied by decreased Indian Hedgehog signalling. J. Pathol. 2006, 209, 501–511. [Google Scholar] [CrossRef]
- Addie, R.D.; de Jong, Y.; Alberti, G.; Kruisselbrink, A.B.; Que, I.; Baelde, H.; Bovee, J. Exploration of the chondrosarcoma metabolome; the mTOR pathway as an important pro-survival pathway. J. Bone Oncol. 2019, 15, 100222. [Google Scholar] [CrossRef]
- Schrage, Y.M.; Briaire-de Bruijn, I.H.; de Miranda, N.F.; van Oosterwijk, J.; Taminiau, A.H.; van Wezel, T.; Hogendoorn, P.C.; Bovee, J.V. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Cancer Res. 2009, 69, 6216–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosterwijk, J.G.; Meijer, D.; van Ruler, M.A.; van den Akker, B.E.; Oosting, J.; Krenacs, T.; Picci, P.; Flanagan, A.M.; Liegl-Atzwanger, B.; Leithner, A.; et al. Screening for potential targets for therapy in mesenchymal, clear cell, and dedifferentiated chondrosarcoma reveals Bcl-2 family members and TGFbeta as potential targets. Am. J. Pathol. 2013, 182, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterwijk, J.G.; Herpers, B.; Meijer, D.; Briaire-de Bruijn, I.H.; Cleton-Jansen, A.M.; Gelderblom, H.; van de Water, B.; Bovee, J.V. Restoration of chemosensitivity for doxorubicin and cisplatin in chondrosarcoma in vitro: BCL-2 family members cause chemoresistance. Ann. Oncol. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Bovee, J.V.; Cleton-Jansen, A.M.; Wuyts, W.; Caethoven, G.; Taminiau, A.H.; Bakker, E.; Van Hul, W.; Cornelisse, C.J.; Hogendoorn, P.C. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am. J. Hum. Genet. 1999, 65, 689–698. [Google Scholar] [CrossRef] [Green Version]
- De Andrea, C.E.; Reijnders, C.M.; Kroon, H.M.; de Jong, D.; Hogendoorn, P.C.; Szuhai, K.; Bovee, J.V. Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene 2012, 31, 1095–1104. [Google Scholar] [CrossRef]
- De Andrea, C.E.; Zhu, J.F.; Jin, H.; Bovee, J.V.; Jones, K.B. Cell cycle deregulation and mosaic loss of Ext1 drive peripheral chondrosarcomagenesis in the mouse and reveal an intrinsic cilia deficiency. J. Pathol. 2015, 236, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Fei, L.; Ngoh, C.; Porter, D.E. Chondrosarcoma transformation in hereditary multiple exostoses: A systematic review and clinical and cost-effectiveness of a proposed screening model. J. Bone Oncol. 2018, 13, 114–122. [Google Scholar] [CrossRef]
- McCormick, C.; Leduc, Y.; Martindale, D.; Mattison, K.; Esford, L.E.; Dyer, A.P.; Tufaro, F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat. Genet. 1998, 19, 158–161. [Google Scholar] [CrossRef]
- Hallor, K.H.; Staaf, J.; Bovee, J.V.; Hogendoorn, P.C.; Cleton-Jansen, A.M.; Knuutila, S.; Savola, S.; Niini, T.; Brosjo, O.; Bauer, H.C.; et al. Genomic profiling of chondrosarcoma: Chromosomal patterns in central and peripheral tumors. Clin. Cancer Res. 2009, 15, 2685–2694. [Google Scholar] [CrossRef] [Green Version]
- Cleven, A.H.; Zwartkruis, E.; Hogendoorn, P.C.; Kroon, H.M.; Briaire-de Bruijn, I.; Bovee, J.V. Periosteal chondrosarcoma: A histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology 2015, 67, 483–490. [Google Scholar] [CrossRef]
- Nishio, J.; Reith, J.D.; Ogose, A.; Maale, G.; Neff, J.R.; Bridge, J.A. Cytogenetic findings in clear cell chondrosarcoma. Cancer Genet. Cytogenet. 2005, 162, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Cho, C.H.; Chi, S.G.; Han, C.S.; Ushigome, S.; Unni, K.K. Low incidence of genetic alterations of the p16CDKN2a in clear cell chondrosarcoma. Int. J. Oncol. 2001, 19, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Park, H.R.; Chi, S.G.; Ushigome, S.; Unni, K.K. Overexpression of p53 and absent genetic mutation in clear cell chondrosarcoma. Int. J. Oncol. 2001, 19, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Haugom, L.; Gorunova, L.; Bjerkehagen, B.; Fossa, A.; Guriby, M.; Nome, T.; Lothe, R.A.; et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS ONE 2012, 7, e49705. [Google Scholar] [CrossRef]
- Amary, F.; Perez-Casanova, L.; Ye, H.; Cottone, L.; Strobl, A.C.; Cool, P.; Miranda, E.; Berisha, F.; Aston, W.; Rocha, M.; et al. Synovial chondromatosis and soft tissue chondroma: Extraosseous cartilaginous tumor defined by FN1 gene rearrangement. Mod. Pathol. 2019, 32, 1762–1771. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Jeon, I.S.; Davis, J.N.; Braun, B.S.; Sublett, J.E.; Roussel, M.F.; Denny, C.T.; Shapiro, D.N. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995, 10, 1229–1234. [Google Scholar]
- Peter, M.; Couturier, J.; Pacquement, H.; Michon, J.; Thomas, G.; Magdelenat, H.; Delattre, O. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997, 14, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.L.; O’Sullivan, M.J.; Pallen, C.J.; Hayes, M.; Clarkson, P.W.; Winstanley, M.; Sorensen, P.H.; Nielsen, T.O.; Horsman, D.E. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J. Mol. Diagn. 2007, 9, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, P.H.; Liu, X.F.; Delattre, O.; Rowland, J.M.; Biggs, C.A.; Thomas, G.; Triche, T.J. Reverse transcriptase PCR amplification of EWS/FLI-1 fusion transcripts as a diagnostic test for peripheral primitive neuroectodermal tumors of childhood. Diagn. Mol. Pathol. 1993, 2, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet. 2015, 47, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.; Monument, M.J.; Watkins, W.S.; Smith, R.; Boucher, K.M.; Schiffman, J.D.; Jorde, L.B.; Randall, R.L.; Lessnick, S.L. EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet. 2012, 205, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; LeRoith, D.; Helman, L.J. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Franzetti, G.A.; Laud-Duval, K.; van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; de Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, H.; Meisel-Sharon, S.; Bruchim, I. Oncogenic fusion proteins adopt the insulin-like growth factor signaling pathway. Mol. Cancer 2018, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Favoni, R.E.; Lebovic, G.S.; Lombana, F.; Powell, D.R.; Reynolds, C.P.; Rosen, N. Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation. A potential autocrine growth factor. J. Clin. Investig. 1990, 86, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Benini, S.; Sarti, M.; Serra, M.; Lollini, P.L.; Maurici, D.; Picci, P.; Manara, M.C.; Baldini, N. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: A possible therapeutic target. Cancer Res. 1996, 56, 4570–4574. [Google Scholar]
- Benini, S.; Manara, M.C.; Baldini, N.; Cerisano, V.; Massimo, S.; Mercuri, M.; Lollini, P.L.; Nanni, P.; Picci, P.; Scotlandi, K. Inhibition of insulin-like growth factor I receptor increases the antitumor activity of doxorubicin and vincristine against Ewing’s sarcoma cells. Clin. Cancer Res. 2001, 7, 1790–1797. [Google Scholar]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [Green Version]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Scotlandi, K. IGF system in sarcomas: A crucial pathway with many unknowns to exploit for therapy. J. Mol. Endocrinol. 2018, 61, T45–T60. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Pasello, M.; Manara, M.C.; Toracchio, L.; Sciandra, E.F.; Picci, P.; Scotlandi, K. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 Influences Sensitivity to Anti-IGF System Agents Through the Translational Regulation of IGF1R. Front. Endocrinol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Gellini, M.; Ascione, A.; Flego, M.; Mallano, A.; Dupuis, M.L.; Zamboni, S.; Terrinoni, M.; D’Alessio, V.; Manara, M.C.; Scotlandi, K.; et al. Generation of human single-chain antibody to the CD99 cell surface determinant specifically recognizing Ewing’s sarcoma tumor cells. Curr. Pharm. Biotechnol. 2013, 14, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Guerzoni, C.; Fiori, V.; Terracciano, M.; Manara, M.C.; Moricoli, D.; Pasello, M.; Sciandra, M.; Nicoletti, G.; Gellini, M.; Dominici, S.; et al. CD99 triggering in Ewing sarcoma delivers a lethal signal through p53 pathway reactivation and cooperates with doxorubicin. Clin. Cancer Res. 2015, 21, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotlandi, K.; Perdichizzi, S.; Bernard, G.; Nicoletti, G.; Nanni, P.; Lollini, P.L.; Curti, A.; Manara, M.C.; Benini, S.; Bernard, A.; et al. Targeting CD99 in association with doxorubicin: An effective combined treatment for Ewing’s sarcoma. Eur. J. Cancer 2006, 42, 91–96. [Google Scholar] [CrossRef]
- Manara, M.C.; Pasello, M.; Scotlandi, K. CD99: A Cell Surface Protein with an Oncojanus Role in Tumors. Genes 2018, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Pasello, M.; Manara, M.C.; Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal. 2018, 12, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Sciandra, M.; Flashner, B.; Gelmez, E.; Kayraklioglu, N.; Allegakoen, D.V.; Petro, J.R.; Conn, E.J.; Hour, S.; Han, J.; et al. Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99. Oncogene 2018, 37, 2181–2196. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, D.J.; Hontecillas-Prieto, L.; Rodriguez-Nunez, P.; Pascual-Pasto, G.; Vila-Ubach, M.; Garcia-Mejias, R.; Robles, M.J.; Tirado, O.M.; Mora, J.; Carcaboso, A.M.; et al. The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 2018, 9, 31397–31410. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Spriano, F.; Chung, E.Y.L.; Gaudio, E.; Tarantelli, C.; Cascione, L.; Napoli, S.; Jessen, K.; Carrassa, L.; Priebe, V.; Sartori, G.; et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin. Cancer Res. 2019, 25, 5167–5176. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, P.; Likhotvorik, R.; Baig, N.; Cropper, J.; Carlson, R.; Kurmasheva, R.; Sridhar, S. Nanoformulation of Talazoparib Increases Maximum Tolerated Doses in Combination With Temozolomide for Treatment of Ewing Sarcoma. Front. Oncol. 2019, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Rau, R.E.; Berg, S.L.; Liu, X.; Minard, C.G.; Bishop, A.J.R.; Romero, J.C.; Hicks, M.J.; Nelson, M.D., Jr.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr. Blood Cancer 2020, 67, e28073. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Salvador-Barbero, B.; Alvarez-Fernandez, M.; Zapatero-Solana, E.; El Bakkali, A.; Menendez, M.D.C.; Lopez-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| Losartan + Sunitinib | Sunitinib: multi-target inhibition of RTK | HGOS | NCT03900793 | USA | phase I (08/2019–02/2025) |
| Famitinib plus Camrelizumab (SHR-1210) or Famitinib alone or Famitinib plus Ifosfamide | Famitinib: multi-target inhibition of RTK, including SCFR (c-Kit), VEGFR2 and 3, PDGFR, Flt1 and Flt3 Camrelizumab: inhibition of PD-1 immune checkpoint | advanced HGOS | NCT04044378 | China | phase I/II (08/2019–09/2022) |
| Pazopanib hydrochloride (Votrient®) with oral Topotecan hydrochloride | Inhibition of VEGFR-1, -2, -3, PDGFR-α and -β, and KIT (Pazopanib) | recurrent or metastatic HGOS | NCT02357810 | USA | phase II (02/2015–06/2022) |
| Apatinib (YN968D1) in combination with chemotherapy | Inhibition of VEGFR2 | HGOS with pulmonary metastasis | NCT03742193 | China | phase II (03/2019–09/2022) |
| Regorafenib (BAY 73-4506, commercial name Stivarga) | Multi-kinase inhibitor targeting VEGFR2, TIE2, PDGFR-beta, FGFR, KIT, RET, and RAF | HGOS, Ewing sarcoma | NCT02048371 (SARC024) | USA | phase II (07/2014–12/2020) |
| Regorafenib (BAY 73-4506, commercial name Stivarga) | Multi-kinase inhibitor targeting VEGFR2, TIE2, PDGFR-beta, FGFR, KIT, RET, and RAF | metastatic bone sarcomas (HGOS, Ewing sarcoma, chondrosarcoma) | NCT02389244 (REGOBONE) | France | phase II (09/2014–03/2023) |
| Cabozantinib-S-Malate (Cabometyx; Cometriq) | Inhibition of MET, VEGFR2, AXL and RET | recurrent, refractory, or newly diagnosed sarcomas, including HGOS | NCT02867592 | USA | phase II (05/2017–06/2020) |
| Erdafitinib (JNJ-42756493) | FGFR inhibitor with negative effects on angiogenesis | relapsed or refractory advanced solid tumors, including HGOS | NCT03210714 (The Pediatric MATCH Screening Trial) | USA | phase II (11/2017–12/2024) |
| Palbociclib (PD-0332991, trade name Ibrance) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | relapsed or refractory advanced solid tumors, including refractory HGOS | NCT03526250 (The Pediatric MATCH Screening Trial) | USA | phase II (06/2018–06/2025) |
| Abemaciclib (Verzenios) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | recurrent or refractory solid tumors, including HGOS and Ewing sarcoma | NCT02644460 | USA | phase I (02/2016–12/2020) |
| Abemaciclib (Verzenios) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | advanced HGOS and chondrosarcoma | NCT04040205 | USA | phase II (10/2019–09/2024) |
| Samotolisib (LY3023414) | Inhibition of PI3K/AKT/mTOR pathway | relapsed or refractory advanced solid tumors, including HGOS | NCT03213678 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Berzosertib (M6620; VX-970; Captisol®) | Selective inhibitor of ATR | HGOS | NCT03718091 | USA | phase II (01/2019–04/2025) |
| Olaparib (AZD-2281, MK-7339 trade name Lynparza®) | Inhibition of PARP1, opposing DNA repair, in patients with hereditary BRCA1 or BRCA2 mutations | relapsed or refractory advanced solid tumors, including HGOS | NCT03233204 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Ulixertinib (BVD-523; VRT752271) | Inhibition of ERK1/2 kinases, belonging to the MAPK pathway | relapsed or refractory advanced solid tumors, including recurrent HGOS | NCT03698994 (The Pediatric MATCH Screening Trial) | USA | phase II (10/2018–12/2025) |
| Vemurafenib (PLX40321; Zelboraf®) | Inhibition of the mutated B-Raf protein, interrupting its stimulation of cell growth | relapsed or refractory advanced solid tumors, including HGOS | NCT03220035 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–12/2023) |
| Larotrectinib (ARRY-470; LOXO-101; Vitrakvi®) | Inhibition of tropomyosin kinase receptors TrkA, TrkB, and TrkC | relapsed or refractory advanced solid tumors, including HGOS | NCT03213704 | USA | phase II (07/2017–09/2024) |
| 9-ING-41 with Gemcitabine, Doxorubicin, Lomustine, Carboplatin, Nab paclitaxel, Paclitaxel | 9-ING-41: inhibition of GSK-3 | advanced cancers, including HGOS | NCT03678883 | USA | phase I/II (01/2019–11/2022) |
| Tazemetostat (EPZ-6438) | Inhibition of the activity of human polycomb repressive complex 2 -containing wild-type histone-lysine N-methyltransferases EZH1 and EZH2 | advanced cancers, including HGOS and Ewing sarcoma | NCT03213665 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Avelumab (Bavencio®) | Targeting PD-L1 | recurrent or progressive HGOS | NCT03006848 | USA | phase II (02/2017–01/2023) |
| ZKAB001 (STI-1014; STI-A1014) | Targeting PD-L1 | recurrent or refractory HGOS | NCT03676985 | China | phase I/II (10/2018–06/2023) |
| Nivolumab (Opdivo®) with or without Azacitidine | Targeting PD-1 (Nivolumab) | recurrent HGOS | NCT03628209 | USA | phase I/II (07/2019–07/2022) |
| Pembrolizumab (MK3475) combined with metronomic Cyclophosphamide | Targeting PD-1 in association with metronomic chemotherapy | advanced sarcomas | NCT02406781 | France | phase II (06/2015–06/2023) |
| Pepinemab (VX15/2503) | Targeting Semaphorin-4D (SEMA4D), also known as Cluster of Differentiation 100 (CD100), which binds to CD72 to activate B cells and dendritic cells | recurrent and refractory HGOS | NCT03320330 | USA | phase I/II (01/2018–09/2021) |
| 4th generation safety-engineered CAR T cells targeting sarcomas | 4th generation safety-engineered CAR T cells targeting sarcoma surface antigens | sarcomas including HGOS and Ewing sarcoma | NCT03356782 | China | phase I (12/2017–12/2020) |
| EGFR806 CAR T cell immunotherapy | second generation EGFR-specific CAR T cells, which have been genetically modified to express either the EGFR receptor alone (EGFR806CAR(2G)-EGFRt) or in addition also the CD19 receptor (CD19CAR(2G)-T2A-HER2tG) | recurrent or refractory solid tumors, including HGOS and Ewing sarcoma | NCT03618381 | USA | phase I (06/2019–06/2036) |
| C7R-GD2.CAR T cell immunotherapy | relapsed or refractory GD2-positive tumors, including HGOS and Ewing sarcoma | NCT03635632 | USA | phase I (04/2019–12/2037) | |
| Humanized Monoclonal Antibody 3F8 (Hu3F8) combined with GM-CSF | Targeting GD2 with the humanized antibody Hu3F8 | recurrent HGOS | NCT02502786 | USA | phase II (07/2015–07/2021) |
| Humanized anti-GD2 bispecific antibody Hu3F8-BsAb | Targeting GD2 with the bispecific antibody Hu3F8-BsAb | HGOS | NCT03860207 | USA | phase I/II (02/2019–02/2022) |
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| Sirolimus and Cyclophosphamide | Inhibition of mTOR signalling | Conventional, Mesenchymal and Dedifferentiated Chondrosarcomas | NCT02821507 (COSYMO) | Netherlands Spain | phase II (06/2014–06/2021) |
| FT 2102 (Olutasidenib) | Inhibitor of mutant IDH1 | Chondrosarcoma | NCT03684811 | USA | phase II (11/2018–04/2022) |
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| SP-2577 (Seclidemstat) | Inhibition of the LSD1 epigenetic enzyme | Ewing Sarcoma | NCT03600649 | USA | phase I (06/2018–12/2021) |
| INCB059872 | Inhibition of the LSD1 epigenetic enzyme | relapsed or refractory Ewing sarcoma | NCT03514407 | USA Italy Spain UK | phase I (06/2018–06/2021) |
| Niraparib and Temozolomide and/or Irinotecan | Niraparib: inhibition of PARP | previously treated, incurable Ewing sarcoma | NCT02044120 | USA UK | phase I (05/2014–04/2021) |
| Pbi-shRNA™ EWS/FLI1 Type 1 LPX | Targeting EWS/FLI1 type 1 fusion transcript | advanced Ewing sarcoma | NCT02736565 | USA | phase I (10/2016–02/2022) |
| TK216 | Inhibition of the between EWS-FLI1 and RNA helicase A through binding to EWS-FLI1 | relapsed or refractory Ewing sarcoma | NCT02657005 | USA | phase I (08/2016–06/2021) |
| Vorinostat (Zolinza) in combination with chemotherapy | Inhibition of HDAC | Ewing Sarcoma | NCT04308330 | USA | phase I (03/2017–12/2022) |
| Olaparib and Temozolomide | Inhibition of PARP | Ewing Sarcoma | NCT01858168 | USA | phase I (07/2013–07/2024) |
| Anlotinib and Irinotecan | Multi-target inhibition of RTK, including VEGFR2 and VEGFR3 | metastatic Ewing Sarcoma | NCT03416517 | China | phase I (01/2018–12/2020) |
| Palbociclib combined with chemotherapy | Inhibition of CDK4/6 | Ewing Sarcoma | NCT03709680 | USA | phase I (05/2019–04/2024) |
| Palbociclib plus Ganitumab | Palbociclib: inhibition of CDK4/6 Ganitumab: inhibition of IGF-1R | Ewing Sarcoma | NCT04129151 | USA | phase II (12/2019–08/2022) |
| Eribulin Mesylate | Inhibition polymerization of tubulin subunits impairing the EWS-FLI1 mediated microtubule stabilization | Ewing Sarcoma | NCT03441360 | USA | phase II (04/2018–06/2020) |
| Eribulin Mesylate | Inhibition polymerization of tubulin subunits impairing the EWS-FLI1 mediated microtubule stabilization | Ewing Sarcoma | NCT03245450 | France Germany Greece Italy Spain UK | phase II (08/2017–09/2021) |
| Nivolumab (Opdivo®) plus ABI-009 (Nab-rapamycin; Nab-sirolimus) | Targeting PD-1 (Nivolumab) and mTOR (ABI-009) | Ewing sarcoma | NCT03190174 | USA | phase I/II (08/2017–04/2021) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scotlandi, K.; Hattinger, C.M.; Pellegrini, E.; Gambarotti, M.; Serra, M. Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells 2020, 9, 968. https://doi.org/10.3390/cells9040968
Scotlandi K, Hattinger CM, Pellegrini E, Gambarotti M, Serra M. Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells. 2020; 9(4):968. https://doi.org/10.3390/cells9040968
Chicago/Turabian StyleScotlandi, Katia, Claudia Maria Hattinger, Evelin Pellegrini, Marco Gambarotti, and Massimo Serra. 2020. "Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors" Cells 9, no. 4: 968. https://doi.org/10.3390/cells9040968
APA StyleScotlandi, K., Hattinger, C. M., Pellegrini, E., Gambarotti, M., & Serra, M. (2020). Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells, 9(4), 968. https://doi.org/10.3390/cells9040968