The β-carboline Harmine Induces Actin Dynamic Remodeling and Abrogates the Malignant Phenotype in Tumorigenic Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Cell Transfection
2.3. Cell Lysate Preparation
2.4. Alexa 488-Actin Mediated Steady-State Fluorescence Anisotropy Measurement Assay
2.5. Purified Actin Polymerization Measurement Assay
2.6. MTT Toxicity Test
2.7. Immunofluorescence
2.8. Western Blot
2.9. Cell–Cell Aggregation Test
2.10. Measurement of Adhesion Forces between Cells
2.11. Wound Migration Assay
2.12. Apoptosis Assay
2.13. Cell Cycle Analysis
2.14. Anchorage-Independent Growth Assay
3. Results
3.1. Identification of Harmine as a Stimulator of Actin Polymerization Using a Steady-State Fluorescence Anisotropy Assay
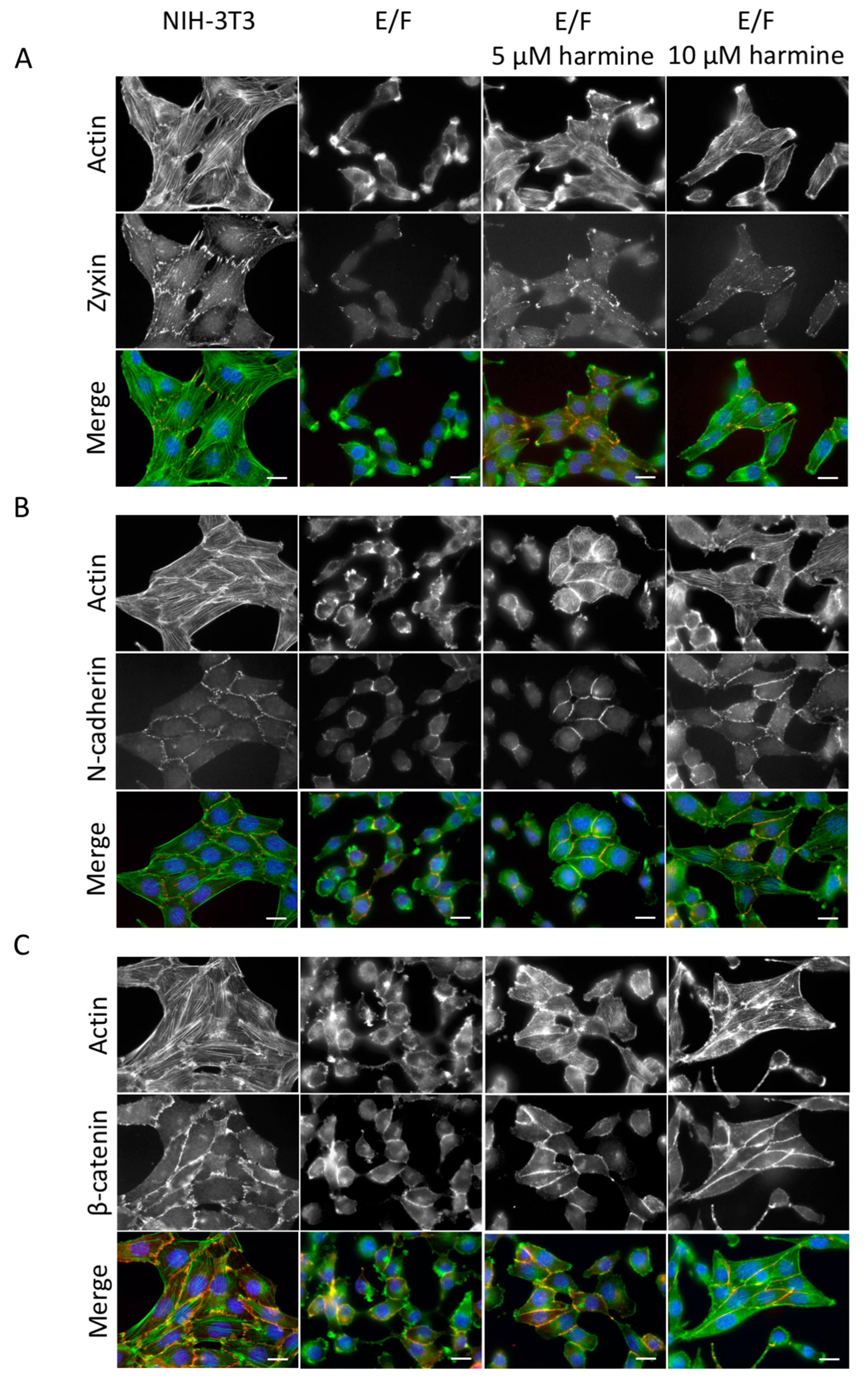
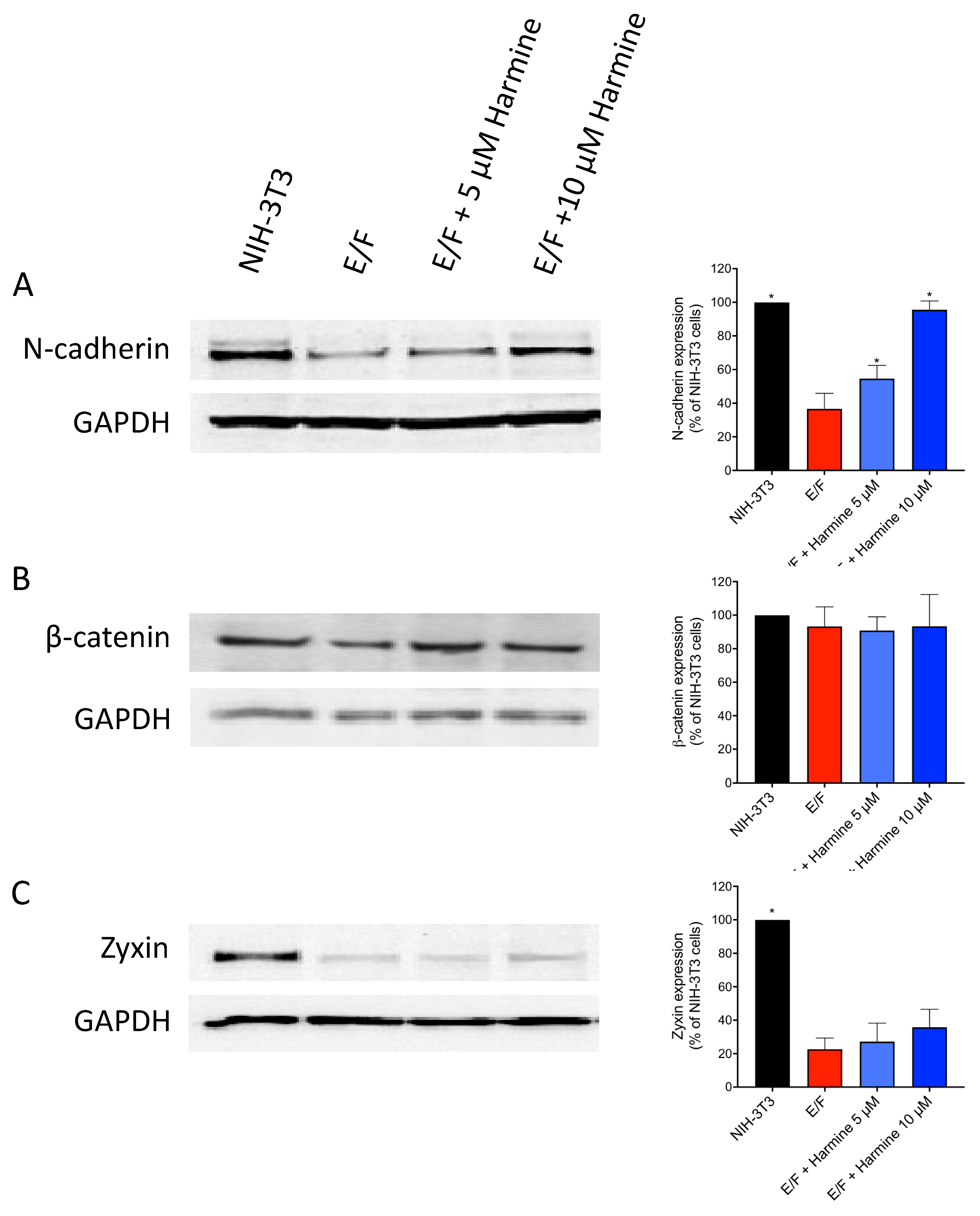
3.2. Harmine Modifies EWS-Fli1-Transformed E/F Cell Morphology and Adhesion Proteins Localization
3.3. Harmine Increases Cell–cell Adhesion Forces in EWS-Fli1-Transformed (E/F) Cells
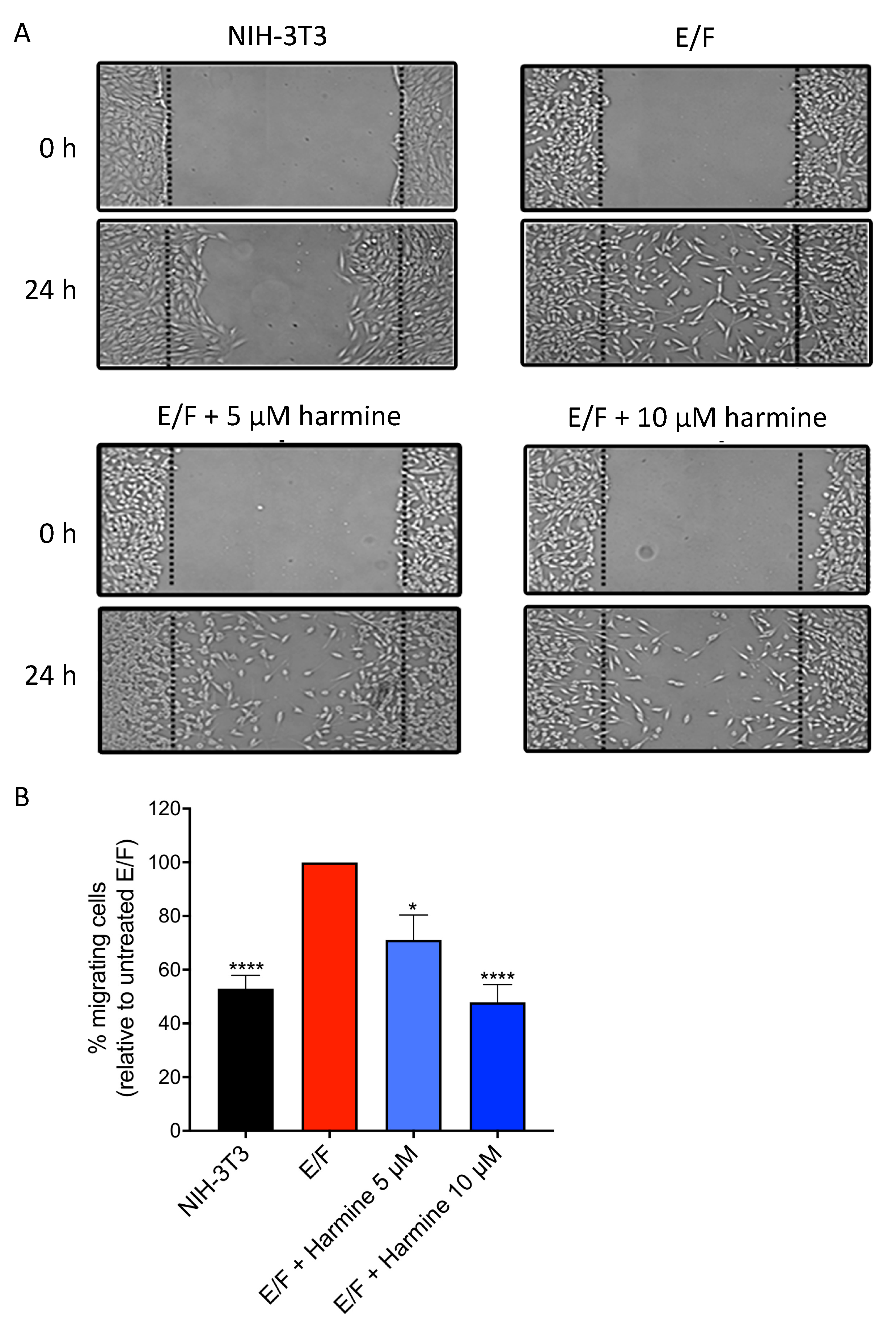
3.4. Harmine Reduces the Intrinsic Motility of EWS-Fli1-Transformed (E/F) Fibroblasts
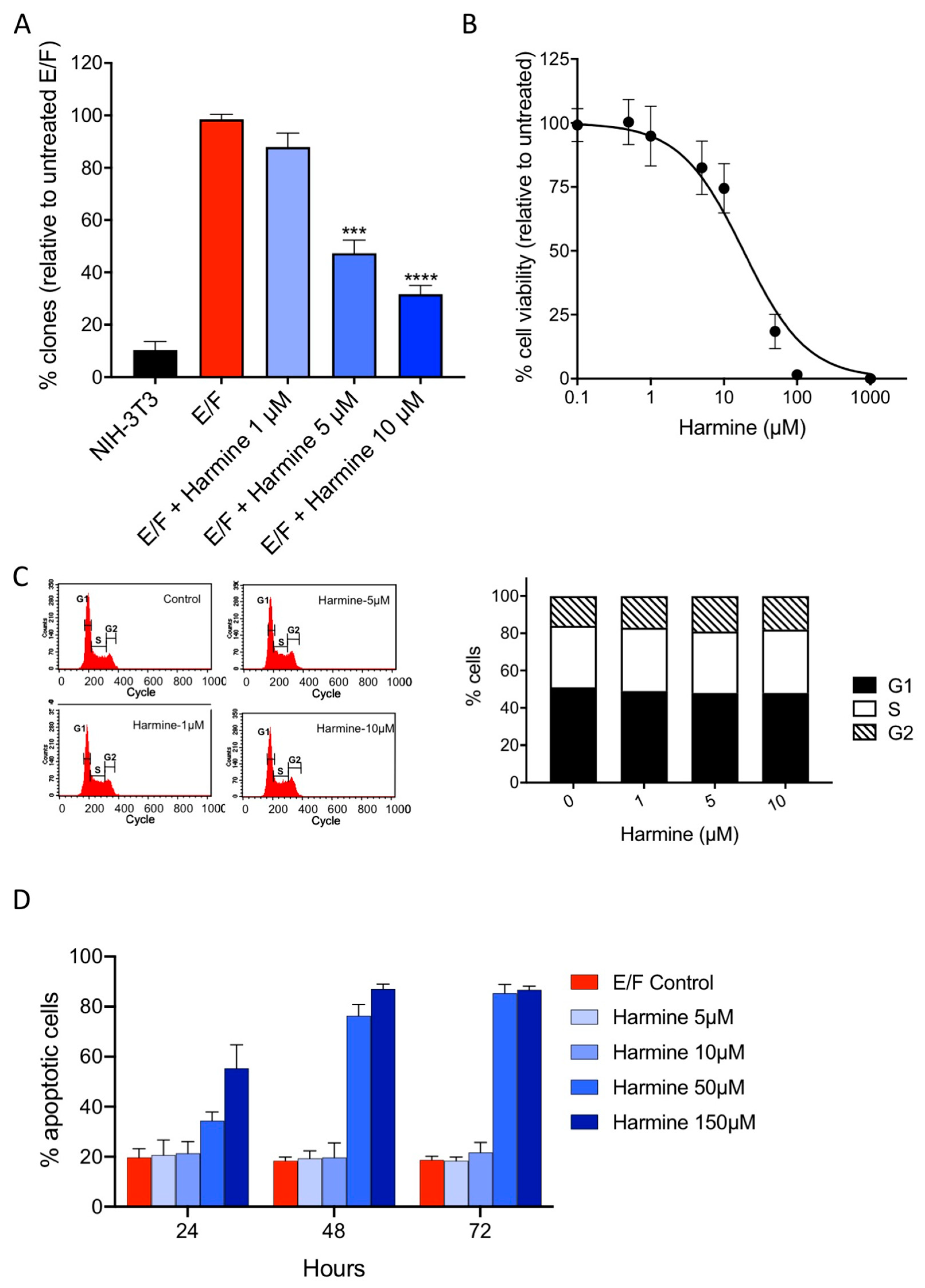
3.5. Harmine Inhibits Anchorage-Independent Growth of EWS-Fli1-Transformed (E/F) Cells at Non-Cytotoxic Concentrations
3.6. Harmine Inhibits Anchorage-Independent Growth and Reverses Tumor Phenotype of B16-F10 Melanoma Cells at Non-Cytotoxic Concentrations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weber, K.; Lazarides, E.; Goldman, R.D.; Vogel, A.; Pollack, R. Localization, and distribution of actin fibers in normal transformed and revertant cells. Cold Spring Harb. Symp. Quant. Biol. 1975, 39, 363–369. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, and actin-binding proteins. A critical evaluation of mechanisms and functions. Ann. Rev. Biochem. 1986, 55, 987–1035. [Google Scholar] [CrossRef]
- Singer, S.J. Intercellular communication, and cell-cell adhesion. Science 1992, 255, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Hitt, A.L.; Luna, E.J. Membrane interactions with the actin cytoskeleton. Curr. Opin. Cell Biol. 1994, 6, 120–130. [Google Scholar] [CrossRef]
- Ben-Ze’ev, A. The cytoskeleton in cancer cells. Biochim. Biophys. Acta 1985, 780, 197–212. [Google Scholar] [CrossRef]
- Pawlak, G.; Helfman, D.M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 2001, 11, 41–47. [Google Scholar] [CrossRef]
- Buda, A.; Pignatelli, M. Cytoskeletal network in colon cancer: From genes to clinical application. Int. J. Biochem. Cell Biol. 2004, 36, 759–765. [Google Scholar] [CrossRef]
- Fincham, V.J.; Brunton, V.G.; Frame, M.C. The SH3 domain directs acto-myosin-dependent targeting of v-Src to focal adhesions via phosphatidylinositol 3-kinase. Mol. Cell. Biol. 2000, 20, 6518–6536. [Google Scholar] [CrossRef]
- Weisberg, E.; Sattler, M.; Ewaniuk, D.S.; Salgia, R. Role of focal adhesion proteins in signal transduction and oncogenesis. Crit. Rev. Oncog. 1997, 8, 343–358. [Google Scholar] [CrossRef]
- Olson, M.F.; Ashworth, A.; Hall, A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 1995, 269, 1270–1272. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Li, N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targets 2004, 4, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Noske, A.; Denkert, C.; Schober, H.; Sers, C.; Zhumabayeva, B.; Weichert, W.; Dietel, M.; Wiechen, K. Loss of Gelsolin expression in human ovarian carcinomas. Eur. J. Cancer 2005, 41, 461–469. [Google Scholar] [CrossRef]
- Asch, H.L.; Head, K.; Dong, Y.; Natoli, F.; Winston, J.S.; Connolly, J.L.; Asch, B.B. Widespread loss of gelsolin in breast cancers of humans, mice, and rats. Cancer Res. 1996, 56, 4841–4845. [Google Scholar]
- Tanaka, M.; Mullauer, L.; Ogiso, Y.; Fujita, H.; Moriya, S.; Furuuchi, K.; Harabayashi, T.; Shinohara, N.; Koyanagi, T.; Kuzumaki, N. Gelsolin: A candidate for suppressor of human bladder cancer. Cancer Res. 1995, 55, 3228–3232. [Google Scholar] [PubMed]
- Prasad, S.C.; Thraves, P.J.; Dritschilo, A.; Kuettel, M.R. Protein expression changes associated with radiation-induced neoplastic progression of human prostate epithelial cells. Electrophoresis 1997, 18, 629–637. [Google Scholar] [CrossRef]
- Dosaka-Akita, H.; Hommura, F.; Fujita, H.; Kinoshita, I.; Nishi, M.; Morikawa, T.; Katoh, H.; Kawakami, Y.; Kuzumaki, N. Frequent loss of gelsolin expression in non-small cell lung cancers of heavy smokers. Cancer Res. 1998, 58, 322–327. [Google Scholar]
- Giganti, A.; Friederich, E. The actin cytoskeleton as a therapeutic target: State of the art and future directions. Prog. Cell Cycle Res. 2003, 5, 511–525. [Google Scholar]
- Bonello, T.T.; Stehn, J.R.; Gunning, P.W. New approaches to targeting the actin cytoskeleton for chemotherapy. Future Med. Chem. 2009, 1, 1311–1331. [Google Scholar] [CrossRef]
- Bousquet, P.F.; Paulsen, L.A.; Fondy, C.; Lipski, K.M.; Loucy, K.J.; Fondy, T.P. Effects of cytochalasin B in culture and in vivo on murine Madison 109 lung carcinoma and on B16 melanoma. Cancer Res. 1990, 50, 1431–1439. [Google Scholar]
- Bubb, M.R.; Senderowicz, A.M.; Sausville, E.A.; Duncan, K.L.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar]
- Stingl, J.; Andersen, R.J.; Emerman, J.T. In vitro screening of crude extracts and pure metabolites obtained from marine invertebrates for the treatment of breast cancer. Cancer Chemother. Pharmacol. 1992, 30, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruta, H.; He, H.; Tikoo, A.; Vuong, T.; Nur, E.K.M. G proteins, phosphoinositides, and actin-cytoskeleton in the control of cancer growth. Microsc. Res. Tech. 1999, 47, 61–66. [Google Scholar] [CrossRef]
- Amsellem, V.; Kryszke, M.H.; Hervy, M.; Subra, F.; Athman, R.; Leh, H.; Brachet-Ducos, C.; Auclair, C. The actin cytoskeleton-associated protein zyxin acts as a tumor suppressor in Ewing tumor cells. Exp. Cell Res. 2005, 304, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Beckerle, M.C. Zyxin: Zinc fingers at sites of cell adhesion. Bioessays 1997, 19, 949–957. [Google Scholar] [CrossRef]
- Spitz, J.A.; Polard, V.; Maksimenko, A.; Subra, F.; Baratti-Elbaz, C.; Meallet-Renault, R.; Pansu, R.B.; Tauc, P.; Auclair, C. Assessment of cellular actin dynamics by measurement of fluorescence anisotropy. Anal. Biochem. 2007, 367, 95–103. [Google Scholar] [CrossRef]
- Lamchouri, F.; Settaf, A.; Cherrah, Y.; Zemzami, M.; Lyoussi, B.; Zaid, A.; Atif, N.; Hassar, M. Antitumour principles from Peganum harmala seeds. Therapie 1999, 54, 753–758. [Google Scholar]
- Iurlo, M.; Leone, G.; Schilstrom, B.; Linner, L.; Nomikos, G.; Hertel, P.; Silvestrini, B.; Svensson, H. Effects of harmine on dopamine output and metabolism in rat striatum: Role of monoamine oxidase-A inhibition. Psychopharmacology 2001, 159, 98–104. [Google Scholar] [CrossRef]
- Chen, Q.; Chao, R.; Chen, H.; Hou, X.; Yan, H.; Zhou, S.; Peng, W.; Xu, A. Antitumor and neurotoxic effects of novel harmine derivatives and structure-activity relationship analysis. Int. J. Cancer 2005, 114, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Walker, S.B.; Pollard, T.D. Pyrene actin: Documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 1983, 4, 253–262. [Google Scholar] [CrossRef]
- Chu, Y.S.; Thomas, W.A.; Eder, O.; Pincet, F.; Perez, E.; Thiery, J.P.; Dufour, S. Force measurements in E-cadherin-mediated cell doublets reveal rapid adhesion strengthened by actin cytoskeleton remodeling through Rac and Cdc42. J. Cell Biol. 2004, 167, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Bachir, A.I.; Horwitz, A.R.; Nelson, W.J.; Bianchini, J.M. Actin-Based Adhesion Modules Mediate Cell Interactions with the Extracellular Matrix and Neighboring Cells. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Cifone, M.A.; Fidler, I.J. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc. Natl. Acad. Sci. USA 1980, 77, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.O.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of cellular cytoskeleton in epithelial-mesenchymal transition process during cancer progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Drees, B.E.; Andrews, K.M.; Beckerle, M.C. Molecular dissection of zyxin function reveals its involvement in cell motility. J. Cell Biol. 1999, 147, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; He, J.; Huang, J.; Yu, T.; Shi, X.; Zhang, T.; Yan, G.; Chen, S.; Peng, C. Harmine induces anticancer activity in breast cancer cells via targeting TAZ. Int. J. Oncol. 2019, 54, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.L.; Ding, K.; Hu, X.; Wu, L.W.; Zhou, D.M.; Rao, M.J.; Lin, N.M.; Zhang, C. DYRK1A inhibition suppresses STAT3/EGFR/Met signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J. Cell. Mol. Med. 2019, 23, 7427–7437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.W.; Zhang, J.K.; Rao, M.; Zhang, Z.Y.; Zhu, H.J.; Zhang, C. Harmine suppresses the proliferation of pancreatic cancer cells and sensitizes pancreatic cancer to gemcitabine treatment. OncoTargets Ther. 2019, 12, 4585–4593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gockler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Sung, J.Y.; Park, J.; Song, W.J.; Chang, S.; Chung, K.C. Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J. Cell Sci. 2012, 125, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chen-Hwang, M.C.; Dolios, G.; Murakami, N.; Padovan, J.C.; Wang, R.; Hwang, Y.W. Mnb/Dyrk1A phosphorylation regulates the interaction of dynamin 1 with SH3 domain-containing proteins. Biochemistry 2004, 43, 10173–10185. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, J.; Guo, Z.; Deng, S.; Du, X.; Zhu, S.; Ye, C.; Shi, Y.S.; Liu, J.J. Endophilin A1 Promotes Actin Polymerization in Dendritic Spines Required for Synaptic Potentiation. Front. Mol. Neurosci. 2018, 11, 177. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Moigne, R.; Subra, F.; Karam, M.; Auclair, C. The β-carboline Harmine Induces Actin Dynamic Remodeling and Abrogates the Malignant Phenotype in Tumorigenic Cells. Cells 2020, 9, 1168. https://doi.org/10.3390/cells9051168
Le Moigne R, Subra F, Karam M, Auclair C. The β-carboline Harmine Induces Actin Dynamic Remodeling and Abrogates the Malignant Phenotype in Tumorigenic Cells. Cells. 2020; 9(5):1168. https://doi.org/10.3390/cells9051168
Chicago/Turabian StyleLe Moigne, Ronan, Frédéric Subra, Manale Karam, and Christian Auclair. 2020. "The β-carboline Harmine Induces Actin Dynamic Remodeling and Abrogates the Malignant Phenotype in Tumorigenic Cells" Cells 9, no. 5: 1168. https://doi.org/10.3390/cells9051168