Evidence for Cytoprotective Effect of Carbon Monoxide Donor in the Development of Acute Esophagitis Leading to Acute Esophageal Epithelium Lesions

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design, Animals, Chemicals, and Drugs
2.2. Determination of EBF Level, Macroscopic and Microscopic Assessment of Esophageal Lesion Score and Biological Samples Collection
2.3. Esophageal Lesions Histology, Mucus Production, and Selected Proteins Distribution Assessment Preparation
2.4. Determination of Esophageal mRNA Expression for Selected Genes by Real-Time PCR
2.5. Determination of Proteins Expression in Esophageal Mucosa by Western Blot
2.6. Measurement of COHb Content in Blood Samples by Gas Chromatography (GC)
2.7. Determination of PGE2 and 8-hydroxy-deoxyguanozine (8-OHdG) Concentration in Esophageal Mucosa
2.8. Assessment of Serum Content of Inflammatory Markers by Luminex Microbeads Fluorescent Assays
2.9. Statistical Analysis
3. Results
3.1. The Effect of Pretreatment with CORM-2 on Esophageal Lesions, EBF, and Mucus Secretion and Protein Distribution of Nrf-2, HMOX-1 within Esophageal Mucosa
3.2. Alterations in Esophageal mRNA and/or Protein Expression of HMOX-1, HMOX-2 and Nrf-2 and in Blood Level of COHb in Rats with RE Pretreated with Vehicle or CORM-2
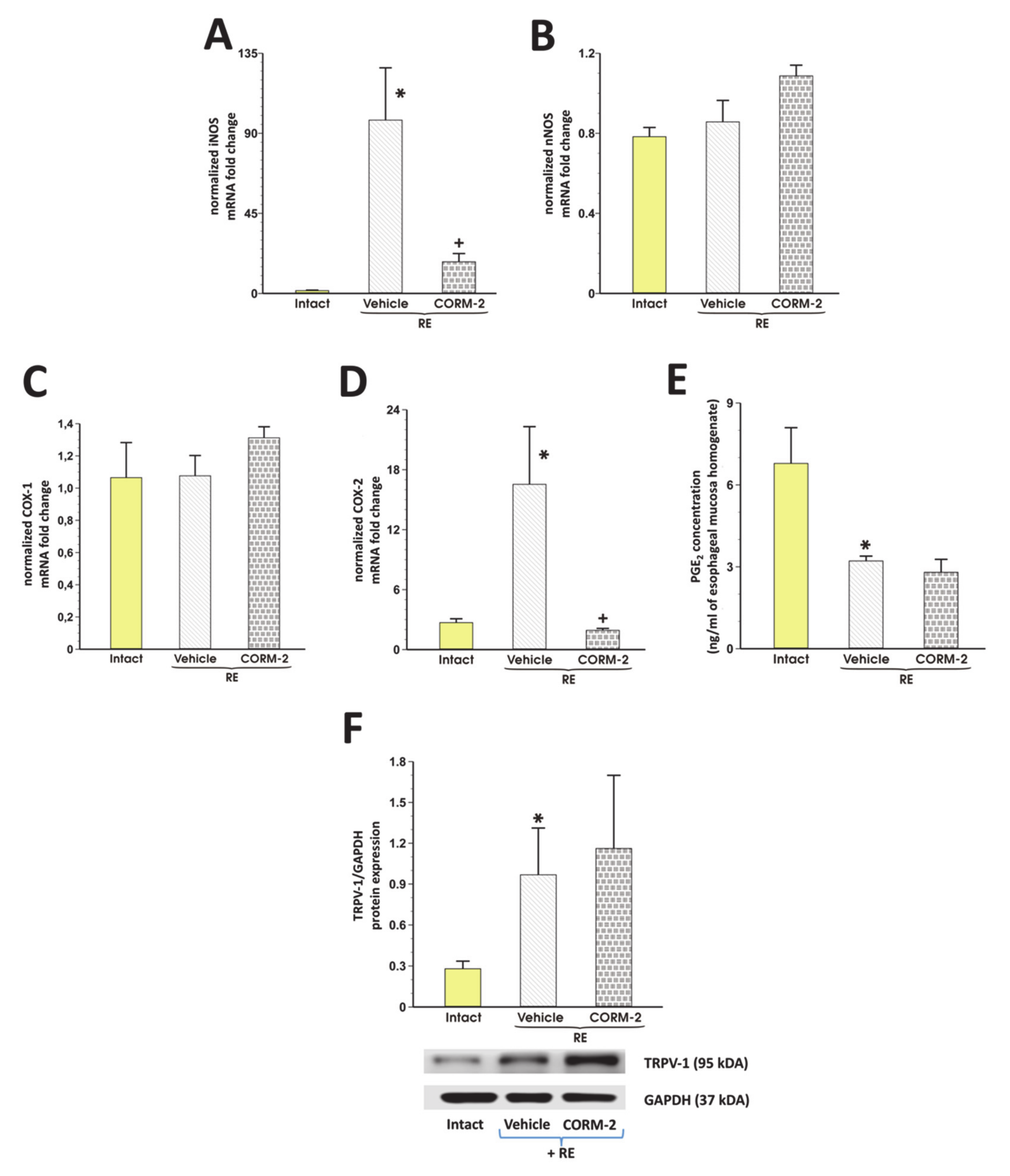
3.3. Involvement of Endogenous NO/NOS, PG/COX, TRPV1 and Afferent Sensory Neurons in CORM-2 Mediated Esophagoprotection and Upregulation of EBF
3.4. The Effect of CORM-2 on Hypoxia and Oxidation Observed in Esophageal Mucosa with RE
3.5. CORM-2-Mediated Modulation of Systemic and Local Inflammatory Response in Rats Exposed to RE
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Schneider, J.L.; Zhao, W.K.; Corley, D.A. Aspirin and nonsteroidal anti-inflammatory drug use and the risk of Barrett’s esophagus. Dig. Dis. Sci. 2015, 60, 436–443. [Google Scholar] [CrossRef]
- Yang, L.; Francois, F.; Pei, Z. Molecular pathways: Pathogenesis and clinical implications of microbiome alteration in esophagitis and Barrett esophagus. Clin. Cancer Res. 2012, 18, 2138–2144. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Jeong, J.B.; Kim, J.W.; Koh, S.J.; Kim, B.G.; Lee, K.L.; Chang, M.S.; Im, J.P.; Kang, H.W.; Shin, C.M. Clinical and endoscopic characteristics of drug-induced esophagitis. World J. Gastroenterol. 2014, 20, 10994–10999. [Google Scholar] [CrossRef] [PubMed]
- Grossi, L.; Ciccaglione, A.F.; Marzio, L. Esophagitis and its causes: Who is “guilty” when acid is found “not guilty”? World J. Gastroenterol. 2017, 23, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Bazin, C.; Benezech, A.; Alessandrini, M.; Grimaud, J.C.; Vitton, V. Esophageal Motor Disorders Are a Strong and Independant Associated Factor of Barrett’s Esophagus. J. Neurogastroenterol. Motil. 2018, 24, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, R.F. The role of acid and bile reflux in oesophagitis and Barrett’s metaplasia. Biochem. Soc. Trans. 2010, 38, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, J.E. Role of the gastric refluxate in gastroesophageal reflux disease: Acid, weak acid and bile. Am. J. Med. Sci. 2009, 338, 89–95. [Google Scholar] [CrossRef]
- Sun, D.; Wang, X.; Gai, Z.; Song, X.; Jia, X.; Tian, H. Bile acids but not acidic acids induce Barrett’s esophagus. Int. J. Clin. Exp. Pathol. 2015, 8, 1384–1392. [Google Scholar]
- Souza, R.F.; Huo, X.; Mittal, V.; Schuler, C.M.; Carmack, S.W.; Zhang, H.Y.; Zhang, X.; Yu, C.; Hormi–Carver, K.; Genta, R.M.; et al. Gastroesophageal Reflux Might Cause Esophagitis Through a Cytokine-Mediated Mechanism Rather Than Caustic Acid Injury. Gastroenterology 2009, 137, 1776–1784. [Google Scholar] [CrossRef]
- Souza, R.F. From Reflux Esophagitis to Esophageal Adenocarcinoma. Dig. Dis. 2016, 34, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Que, J.; Garman, K.S.; Souza, R.F.; Spechler, S.J. Pathogenesis and Cells of Origin of Barrett’s Esophagus. Gastroenterology 2019, 157, 349–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triadafilopoulos, G. Acid and bile reflux in Barrett’s esophagus: A tale of two evils. Gastroenterology 2001, 121, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Ianaro, A.; de Nucci, G. Gaseous Mediators in Gastrointestinal Mucosal Defense and Injury. Dig. Dis. Sci. 2017, 62, 2223–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magierowska, K.; Brzozowski, T.; Magierowski, M. Emerging role of carbon monoxide in regulation of cellular pathways and in the maintenance of gastric mucosal integrity. Pharmacol. Res. 2018, 129, 56–64. [Google Scholar] [CrossRef]
- Cacanyiova, S.; Majzunova, M.; Golas, S.; Berenyiova, A. The role of perivascular adipose tissue and endogenous hydrogen sulfide in vasoactive responses of isolated mesenteric arteries in normotensive and spontaneously hypertensive rats. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Gromotowicz-Poplawska, A.; Kloza, M.; Aleksiejczuk, M.; Marcinczyk, N.; Szemraj, J.; Kozlowska, H.; Chabielska, E. Nitric oxide as a modulator in platelet- and endothelium-dependent antithrombotic effect of eplerenone in diabetic rats. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Berenyiova, A.; Drobna, M.; Cebova, M.; Kristek, F.; Cacanyiova, S. Changes in the vasoactive effects of nitric oxide, hydrogen sulfide and the structure of the rat thoracic aorta: The role of age and essential hypertension. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Babu, D.; Motterlini, R.; Lefebvre, R.A. CO and CO-releasing molecules (CO-RMs) in acute gastrointestinal inflammation. Br. J. Pharmacol. 2015, 172, 1557–1573. [Google Scholar] [CrossRef] [Green Version]
- Magierowski, M.; Magierowska, K.; Hubalewska-Mazgaj, M.; Surmiak, M.; Sliwowski, Z.; Wierdak, M.; Kwiecien, S.; Chmura, A.; Brzozowski, T. Cross-talk between hydrogen sulfide and carbon monoxide in the mechanism of experimental gastric ulcers healing, regulation of gastric blood flow and accompanying inflammation. Biochem. Pharmacol. 2018, 149, 131–142. [Google Scholar] [CrossRef]
- Takeuchi, K.; Aihara, E.; Kimura, M.; Dogishi, K.; Hara, T.; Hayashi, S. Gas mediators involved in modulating duodenal HCO3(-) secretion. Curr. Med. Chem. 2012, 19, 43–54. [Google Scholar] [CrossRef]
- Rattan, S.; Fan, Y.P.; Chakder, S. Mechanism of inhibition of VIP-induced LES relaxation by heme oxygenase inhibitor zinc protoporphyrin IX. Am. J. Physiol. 1999, 276, G138–G145. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol. Cell Physiol. 2017, 312, C302–C313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Ju, T.J.; Kim, Y.W.; Dan, J.M.; Kim, J.Y.; Kim, Y.D.; Seo, J.S.; Park, S.Y. Hemin, heme oxygenase-1 inducer, attenuates immobilization-induced skeletal muscle atrophy in mice. Life Sci. 2013, 92, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Magierowski, M.; Magierowska, K.; Hubalewska-Mazgaj, M.; Sliwowski, Z.; Ginter, G.; Pajdo, R.; Chmura, A.; Kwiecien, S.; Brzozowski, T. Carbon monoxide released from its pharmacological donor, tricarbonyldichlororuthenium (II) dimer, accelerates the healing of pre-existing gastric ulcers. Br. J. Pharmacol. 2017, 174, 3654–3668. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Sasahira, T.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Inhibition of heme oxygenase-1 by zinc protoporphyrin IX reduces tumor growth of LL/2 lungcancer in C57BL mice. Int. J. Cancer. 2007, 120, 500–505. [Google Scholar] [CrossRef]
- Magierowska, K.; Magierowski, M.; Surmiak, M.; Adamski, J.; Mazur-Bialy, A.I.; Pajdo, R.; Sliwowski, Z.; Kwiecien, S.; Brzozowski, T. The Protective Role of Carbon Monoxide (CO) Produced by Heme Oxygenases and Derived from the CO-Releasing Molecule CORM-2 in the Pathogenesis of Stress-Induced Gastric Lesions: Evidence for Non-Involvement of Nitric Oxide (NO). Int. J. Mol. Sci. 2016, 17, 442. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Kilbourn, R.G. Nitric oxide synthase inhibitors: Amino acids. Methods Enzymol. 1996, 268, 375–392. [Google Scholar]
- Suemaru, K.; Yoshikawa, M.; Aso, H.; Watanabe, M. TRPV1 mediates the anticonvulsant effects of acetaminophen in mice. Epilepsy Res. 2018, 145, 153–159. [Google Scholar] [CrossRef]
- Kwiecien, S.; Magierowska, K.; Magierowski, M.; Surmiak, M.; Hubalewska-Mazgaj, M.; Pajdo, R.; Sliwowski, Z.; Chmura, A.; Wojcik, D.; Brzozowski, T. Role of sensory afferent nerves, lipid peroxidation and antioxidative enzymes in the carbon monoxide-induced gastroprotection against stress ulcerogenesis. J. Physiol. Pharmacol. 2016, 67, 717–729. [Google Scholar]
- Konturek, P.C.; Brzozowska, I.; Targosz, A.; Pawlik, M.; Kania, J.; Hess, T.; Kwiecien, S.; Konturek, S.J.; Reiter, R.J.; Brzozowski, T. Esophagoprotection mediated by exogenous and endogenous melatonin in an experimental model of reflux esophagitis. J. Pineal Res. 2013, 55, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Korbut, E.; Hubalewska-Mazgaj, M.; Surmiak, M.; Chmura, A.; Bakalarz, D.; Buszewicz, G.; Wójcik, D.; Śliwowski, Z.; Ginter, G.; et al. Oxidative gastric mucosal damage induced by ischemia/reperfusion and the mechanisms of its prevention by carbon monoxide-releasing tricarbonyldichlororuthenium (II) dimer. Free Radic Biol. Med. 2019, 145, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Wojcik, D.; Chmura, A.; Bakalarz, D.; Wierdak, M.; Kwiecien, S.; Sliwowski, Z.; Brzozowski, T.; Magierowski, M. Alterations in Gastric Mucosal Expression of Calcitonin Gene-Related Peptides, Vanilloid Receptors, and Heme Oxygenase-1 Mediate Gastroprotective Action of Carbon Monoxide against Ethanol-Induced Gastric Mucosal Lesions. Int. J. Mol. Sci. 2018, 19, E2960. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Bakalarz, D.; Wójcik, D.; Chmura, A.; Hubalewska-Mazgaj, M.; Licholai, S.; Korbut, E.; Kwiecien, S.; Sliwowski, Z.; Ginter, G.; et al. Time-dependent course of gastric ulcer healing and molecular markers profile modulated by increased gastric mucosal content of carbon monoxide released from its pharmacological donor. Biochem. Pharmacol. 2019, 163, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Peinnequin, A.; Mouret, C.; Birot, O.; Alonso, A.; Mathieu, J.; Clarençon, D.; Agay, D.; Chancerelle, Y.; Multon, E. Rat pro-inflammatory cytokine and cytokine related mRNA quantification by real-time polymerase chain reaction using SYBR green. BMC Immunol. 2004, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Boeckxstaens, G.E. Review article: The pathophysiology of gastro-oesophageal reflux disease. Aliment. Pharmacol. Ther. 2007, 26, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Bognár, L.; Vereczkei, A.; Papp, A.; Jancsó, G.; Horváth, Ö.P. Gastroesophageal Reflux Disease Might Induce Certain-Supposedly Adaptive-Changes in the Esophagus: A Hypothesis. Dig. Dis. Sci. 2018, 63, 2529–2535. [Google Scholar] [CrossRef]
- Sayin, S.I.; Baumeister, T.; Wang, T.C.; Quante, M. Origins of Metaplasia in the Esophagus: Is This a GE Junction Stem Cell Disease? Dig. Dis. Sci. 2018, 63, 2013–2021. [Google Scholar] [CrossRef]
- Boulton, R.A.; Usselmann, B.; Mohammed, I.; Jankowski, J. Barrett’s esophagus: Environmental influences in the progression of dysplasia. World J. Surg. 2003, 27, 1014–1017. [Google Scholar] [CrossRef]
- Souza, R.F. Reflux esophagitis and its role in the pathogenesis of Barrett’s metaplasia. J. Gastroenterol. 2017, 52, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Tack, J.; Pandolfino, J.E. Pathophysiology of gastroesophageal reflux disease. Gastroenterology 2018, 154, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fang, J.; Liao, L.; Nakamura, H.; Maeda, H. Styrene-maleic acid copolymer-encapsulated CORM2, a water-soluble carbon monoxide (CO) donor with a constant CO-releasing property, exhibits therapeutic potential for inflammatory bowel disease. J. Control. Release 2014, 187, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Naito, Y.; Uchiyama, K.; Suzuki, T.; Hirata, I.; Mizushima, K.; Tsuboi, H.; Hayashi, N.; Handa, O.; Ishikawa, T.; et al. Carbon monoxide liberated from carbon monoxide-releasing molecule exerts an anti-inflammatory effect on dextran sulfate sodium-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Magierowska, K.; Magierowski, M.; Hubalewska-Mazgaj, M.; Adamski, J.; Surmiak, M.; Sliwowski, Z.; Kwiecien, S.; Brzozowski, T. Carbon Monoxide (CO) Released from Tricarbonyldichlororuthenium (II) Dimer (CORM-2) in Gastroprotection against Experimental Ethanol-Induced Gastric Damage. PLoS ONE 2015, 10, e0140493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Zhao, L.; Li, N.; Duan, H.; Liu, H.; Pu, F.; Zhang, G.; Zhou, E.-M.; Xiao, S. Carbon Monoxide Inhibits Porcine Reproductive and Respiratory Syndrome Virus Replication by the Cyclic GMP/Protein Kinase G and NF-κB Signaling Pathway. J. Virol. 2016, 91. [Google Scholar] [CrossRef] [Green Version]
- Koçer, G.; Nasircilar Ülker, S.; Şentürk, Ü.K. The contribution of carbon monoxide to vascular tonus. Microcirculation 2018, 25, e12495. [Google Scholar] [CrossRef]
- Kim, H.H.; Choi, S. Therapeutic Aspects of Carbon Monoxide in Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, E2381. [Google Scholar] [CrossRef] [Green Version]
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Brzozowska, I.; Pawlik, T. Role of prostaglandins in gastroprotection and gastric adaptation. J. Physiol. Pharmacol. 2005, 56, 33–55. [Google Scholar]
- Magierowski, M.; Hubalewska-Mazgaj, M.; Magierowska, K.; Wojcik, D.; Sliwowski, Z.; Kwiecien, S.; Brzozowski, T. Nitric oxide, afferent sensory nerves, and antioxidative enzymes in the mechanism of protection mediated by tricarbonyldichlororuthenium(II) dimer and sodium hydrosulfide against aspirin-induced gastric damage. J. Gastroenterol. 2018, 53, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Akiba, Y.; Mizumori, M.; Kuo, M.; Ham, M.; Guth, P.H.; Engel, E.; Kaunitz, J.D. CO2 chemosensing in rat oesophagus. Gut 2008, 57, 1654–1664. [Google Scholar] [CrossRef]
- Peles, S.; Medda, B.K.; Zhihong Zhang Banerjee, B.; Lehmann, A.; Shaker, R.; Sengupta, J.N. Differential Effects of TRPV1 Antagonists in Acid-induced Excitation of Esophageal Vagal Afferent Fibers of Rats. Neuroscience 2009, 161, 515–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, F.; Cheng, L.; Harnett, K.M.; Chak, A.; Cooper, G.S.; Isenberg, G.; Ray, M.; Katz, J.A.; Catanzaro, A.; O’Shea, R.; et al. Gastroesophageal reflux disease-associated esophagitis induces endogenous cytokine production leading to motor abnormalities. Gastroenterology 2007, 132, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tselepis, C.; Perry, I.; Dawson, C.; Hardy, R.; Darnton, S.J.; McConkey, C.; Stuart, R.C.; Wright, N.; Harrison, R.; Jankowski, J.; et al. Tumour necrosis factor-α in Barrett’s oesophagus: A potential novel mechanism of action. Oncogene 2002, 21, 6071–6081. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, M.; Fujiwara, Y.; Takashima, T.; Hayakawa, T.; Sasaki, E.; Shiba, M.; Watanabe, T.; Tominaga, K.; Oshitani, N.; Matsumoto, T.; et al. Increased expression of cytokines and adhesion molecules in rat chronic esophagitis. Digestion 2003, 68, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, G.; Han, X.; Liu, J.; Li, G.-X.; Zou, D.-W.; Li, Z.-S. Inhibition of p38 MAPK activation attenuates esophageal mucosal damage in a chronic model of reflux esophagitis. Neurogastroenterol. Motil. 2015, 27, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, P.; Nelson, V.M.; Manley, S.; Wellner, M.; Floer, M.; Binion, D.G.; Shaker, R. Effect of curcumin on acidic pH-induced expression of IL-6 and IL-8 in human esophageal epithelial cells (HET-1A): Role of PKC, MAPKs, and NF-κB. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G388–G398. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, H.; Hu, Y.; Djukic, Z.; Tevebaugh, W.; Shaheen, N.J.; Orlando, R.C.; Hu, J.; Chen, X. Gastroesophageal reflux activates the NF-κB pathway and impairs esophageal barrier function in mice. Am. J. Physiol. Liver Physiol. 2013, 305, G58–65. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Gong, Z.; Ma, J.; Su, H.; Guo, T.; Cai, H.; Chen, Q.; Zhao, X.; Qi, J.; Du, J. Interleukin-1 receptor antagonist inhibits angiogenesis in gastric cancer. Int. J. Clin. Oncol. 2018, 23, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Shen, Z.; Liu, Z.; Gao, L.; Han, Z.; Yu, S.; Kang, M. IL-1RA suppresses esophageal cancer cell growth by blocking IL-1α. J. Clin. Lab. Anal. 2019, 33, e22903. [Google Scholar] [CrossRef] [Green Version]
- Krebs, D.L.; Hilton, D.J. SOCS: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113, 2813–2819. [Google Scholar] [PubMed]
- Zafra, M.P.; Cancelliere, N.; Del Rio, P.R.; Ruiz-García, M.; Estevez, L.; Andregnette, V.; Sánchez-Garcia, S.; Fiandor, A.; Collantes, E.; Sastre, J.; et al. Misregulation of suppressors of cytokine signaling in eosinophilic esophagitis. J. Gastroenterol. 2012, 48, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Karin, M. “The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation”. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, J.H.; Yun, J.A.; Cha, J.H.; Kim, Y.; Won, M.H.; Kim, K.W.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Heme oxygenase metabolites improve astrocytic mitochondrial function via a Ca2+-dependent HIF-1α/ERRα circuit. PLoS ONE 2018, 13, e0202039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovater, M.B.; Ying, W.Z.; Agarwal, A.; Sanders, P.W. Nitric oxide and carbon monoxide antagonize TGF-β through ligand-independent internalization of TβR1/ALK5. Am. J. Physiol. Renal. Physiol. 2014, 307, F727–F735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Lee, S.K.; Lim, C.R.; Park, H.W.; Liu, F.; Kim, S.J.; Kim, B.C. Heme oxygenase-1/carbon monoxide axis suppresses transforming growth factor-β1-induced growth inhibition by increasing ERK1/2-mediated phosphorylation of Smad3 at Thr-179 in human hepatocellular carcinoma cell lines. Biochem. Biophys. Res. Commun. 2018, 498, 609–615. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magierowska, K.; Bakalarz, D.; Wójcik, D.; Korbut, E.; Danielak, A.; Głowacka, U.; Pajdo, R.; Buszewicz, G.; Ginter, G.; Surmiak, M.; et al. Evidence for Cytoprotective Effect of Carbon Monoxide Donor in the Development of Acute Esophagitis Leading to Acute Esophageal Epithelium Lesions. Cells 2020, 9, 1203. https://doi.org/10.3390/cells9051203
Magierowska K, Bakalarz D, Wójcik D, Korbut E, Danielak A, Głowacka U, Pajdo R, Buszewicz G, Ginter G, Surmiak M, et al. Evidence for Cytoprotective Effect of Carbon Monoxide Donor in the Development of Acute Esophagitis Leading to Acute Esophageal Epithelium Lesions. Cells. 2020; 9(5):1203. https://doi.org/10.3390/cells9051203
Chicago/Turabian StyleMagierowska, Katarzyna, Dominik Bakalarz, Dagmara Wójcik, Edyta Korbut, Aleksandra Danielak, Urszula Głowacka, Robert Pajdo, Grzegorz Buszewicz, Grzegorz Ginter, Marcin Surmiak, and et al. 2020. "Evidence for Cytoprotective Effect of Carbon Monoxide Donor in the Development of Acute Esophagitis Leading to Acute Esophageal Epithelium Lesions" Cells 9, no. 5: 1203. https://doi.org/10.3390/cells9051203