Functional Characterization of Neurofilament Light Splicing and Misbalance in Zebrafish

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Zebrafish Lines and Microinjections
2.2. In Vivo Live Imaging and Timelapse
2.3. Imaris Filament Tracer
2.4. Acridine Orange Staining
2.5. Touch-Evoked Escape Response (TEER)
2.6. FACS (Fluorescence-Activated Cell Sorting)
2.7. Cell Culture
2.8. Reverse Transcription and PCR
2.9. Western Blot
2.10. Statistical Analysis
3. Results
3.1. The Zebrafish Homolog neflb Encodes Two Splice Variants, neflbE3 and neflbE4, Differently Expressed during the Development
3.2. neflbE3/E4 Misbalance Results in a Strong and Specific Motor Phenotype
3.3. The Motor Phenotype Associated with the Alteration of neflbE3 Expression Correlates with Atrophy of Motor Axons
3.4. neflbE4 Participates in Motor Neuron Axonal Growth, Whereas neflbE3 Fails to Polymerize Normally and Forms Aggregates
3.5. neflb Splice Variants Have Different Assembly Properties in SW13vim- Cells
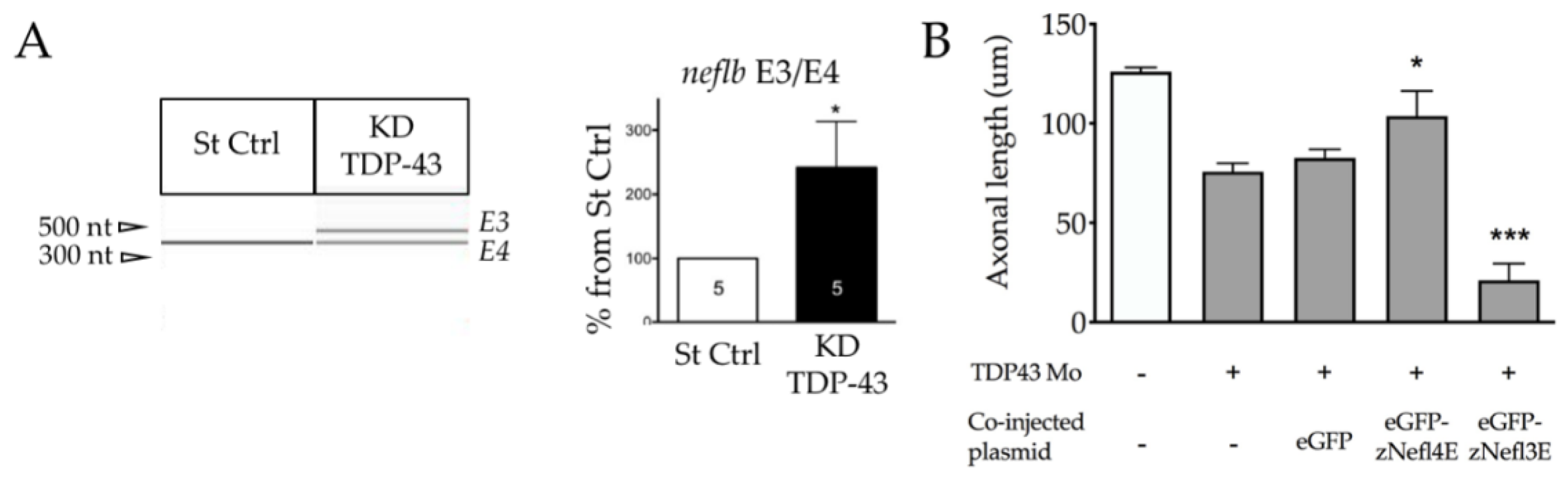
3.6. TDP-43 Regulates neflb Splicing in Zebrafish
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, P.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: A major determinant of axonal caliber. Proc. Natl. Acad. Sci. USA 1987, 84, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Rao, M.V.; Nixon, R.A. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Leung, C.L.; Liem, R.K. Characterization of interactions between the neurofilament triplet proteins by the yeast two-hybrid system. J. Biol. Chem. 1996, 271, 14041–14044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.; Smith, K.E.; O’Brien, B.J.; Angelides, K.J. Characterization of mammalian neurofilament triplet proteins. Subunit stoichiometry and morphology of native and reconstituted filaments. J. Biol. Chem. 1985, 260, 10736–10747. [Google Scholar] [PubMed]
- Hisanaga, S.I.; Matsuoka, Y.; Nishizawa, K.; Saito, T.; Inagaki, M.; Hirokawa, N. Phosphorylation of native and reassembled neurofilaments composed of NF-L, NF-M, and NF-H by the catalytic subunit of cAMP-dependent protein kinase. Mol. Biol. Cell 1994, 5, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streifel, T.D.; Avalos, R.T.; Cohlberg, J.A. CAMP-dependent phosphorylation of neurofilament proteins NF-L and NF-M inhibits their coassembly into filaments in vitro. Biochem. Biophys. Res. Commun. 1996, 222, 646–651. [Google Scholar] [CrossRef]
- Balin, B.J.; Lee, V.M.Y. Individual neurofilament subunits reassembled in vitro exhibit unique biochemical, morphological and immunological properties. Brain Res. 1991, 556, 196–208. [Google Scholar] [CrossRef]
- Ching, G.Y.; Liem, R.K.H. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J. Cell Biol. 1993, 122, 1323–1335. [Google Scholar] [CrossRef]
- Lee, M.K.; Xu, Z.; Wong, P.C.; Cleveland, D.W. Neurofilaments are obligate heteropolymers in vivo. J. Cell Biol. 1993, 122, 1337–1350. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Sershen, H.; Basavarajappa, B.S.; Smiley, J.F.; Hashim, A.; Bleiwas, C.; Berg, M.; Guifoyle, D.N.; Subbanna, S.; Darji, S.; et al. Neurofilament light interaction with GluN1 modulates neurotransmission and schizophrenia-associated behaviors. Transl. Psychiatry 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobsiger, C.S.; Cleveland, D.W. Neurofilaments: Organization and function in neurons. In Encyclopedia of Neuroscience; Elsevier Ltd.: Amsterdam, The Netherlands, 2009; pp. 433–436. ISBN 9780080450469. [Google Scholar]
- Mersiyanova, I.V.; Perepelov, A.V.; Polyakov, A.V.; Sitnikov, V.F.; Dadali, E.L.; Oparin, R.B.; Petrin, A.N.; Evgrafov, O.V. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 2000, 67, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordanova, A.; De Jonghe, P.; Boerkoel, C.F.; Takashima, H.; De Vriendt, E.; Ceuterick, C.; Martin, J.J.; Butler, I.J.; Mancias, P.; Papasozomenos, S.C.; et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain 2003, 126, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J. Neurofilament metabolism in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 169, 170–177. [Google Scholar] [CrossRef]
- Horga, A.; Laurà, M.; Jaunmuktane, Z.; Jerath, N.U.; Gonzalez, M.A.; Polke, J.M.; Poh, R.; Blake, J.C.; Liu, Y.T.; Wiethoff, S.; et al. Genetic and clinical characteristics of NEFL-Related Charcot-Marie-Tooth disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 575–585. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, R. Intermediate Charcot-Marie-Tooth disease. Neurosci. Bull. 2014, 30, 999–1009. [Google Scholar] [CrossRef]
- Sainio, M.T.; Ylikallio, E.; Mäenpää, L.; Lahtela, J.; Mattila, P.; Auranen, M.; Palmio, J.; Tyynismaa, H. Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy. Neurol. Genet. 2018, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gentil, B.J.; Minotti, S.; Beange, M.; Baloh, R.H.; Julien, J.P.; Durham, H.D. Normal role of the low-molecular-weight neurofilament protein in mitochondrial dynamics and disruption in Charcot-Marie-Tooth disease. FASEB J. 2012, 26, 1194–1203. [Google Scholar] [CrossRef]
- Sasaki, T.; Gotow, T.; Shiozaki, M.; Sakaue, F.; Saito, T.; Julien, J.P.; Uchiyama, Y.; Hisanaga, S.I. Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot-Marie-Tooth disease mutants. Hum. Mol. Genet. 2006, 15, 943–952. [Google Scholar] [CrossRef] [Green Version]
- Didonna, A.; Opal, P. The role of neurofilament aggregation in neurodegeneration: Lessons from rare inherited neurological disorders. Mol. Neurodegener. 2019, 14, 19. [Google Scholar] [CrossRef]
- Munoz, D.G.; Greene, C.; Perl, D.P.; Selkoe, D.J. Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J. Neuropathol. Exp. Neurol. 1988, 47, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mizusawa, H.; Matsumoto, S.; Yen, S.H.; Hirano, A.; Rojas-Corona, R.R.; Donnenfeld, H. Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989, 79, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Verde, F.; Steinacker, P.; Weishaupt, J.H.; Kassubek, J.; Oeckl, P.; Halbgebauer, S.; Tumani, H.; Von Arnim, C.A.F.; Dorst, J.; Feneberg, E.; et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 157–164. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Gaiani, A.; Martinelli, I.; Bello, L.; Querin, G.; Puthenparampil, M.; Ruggero, S.; Toffanin, E.; Cagnin, A.; Briani, C.; Pegoraro, E.; et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: Neurofilament light chain levels in definite subtypes of disease. JAMA Neurol. 2017, 74, 525–532. [Google Scholar] [CrossRef]
- Poesen, K.; De Schaepdryver, M.; Stubendorff, B.; Gille, B.; Muckova, P.; Wendler, S.; Prell, T.; Ringer, T.M.; Rhode, H.; Stevens, O.; et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017, 88, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; Bourefis, A.R.; Kabashi, E. Diagnostic challenge and neuromuscular junction contribution to ALS pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.K.Y.; He, B.P.; Strong, M.J. Characterization of neuronal intermediate filament protein expression in cervical spinal motor neurons in sporadic amyotrophic lateral sclerosis (ALS). J. Neuropathol. Exp. Neurol. 2000, 59, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Côté, F.; Collard, J.F.; Julien, J.P. Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: A mouse model of amyotrophic lateral sclerosis. Cell 1993, 73, 35–46. [Google Scholar] [CrossRef]
- Xu, Z.; Cork, L.C.; Griffin, J.W.; Cleveland, D.W. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell 1993, 73, 23–33. [Google Scholar] [CrossRef]
- Julien, J.-P.; Côté, F.; Collard, J.-F. Mice overexpressing the human neurofilament heavy gene as a model of ALS. Neurobiol. Aging 1995, 16, 487–490. [Google Scholar] [CrossRef]
- Collard, J.F.; Côté, F.; Julien, J.P. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995, 375, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; Hosler, B.A.; Cortelli, P.; De Jong, P.J.; Yoshinaga, Y.; Haines, J.L. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 547, 1205–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef]
- Leblond, C.S.; Kaneb, H.M.; Dion, P.A.; Rouleau, G.A. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 91–101. [Google Scholar] [CrossRef]
- Strong, M.J.; Volkening, K.; Hammond, R.; Yang, W.; Strong, W.; Leystra-Lantz, C.; Shoesmith, C. TDP-43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol. Cell. Neurosci. 2007, 35, 320–327. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, J.; Emmett, W.; Fratta, P.; Isaacs, A.M.; Plagnol, V. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med. Genom. 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Gwee, S.S.L.; Radford, R.A.W.; Chow, S.; Syal, M.D.; Morsch, M.; Formella, I.; Lee, A.; Don, E.K.; Badrock, A.P.; Cole, N.J.; et al. Aurora kinase B regulates axonal outgrowth and regeneration in the spinal motor neurons of developing zebrafish. Cell. Mol. Life Sci. 2018, 75, 4269–4285. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Velde, C.V.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2009, 19, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef]
- Lattante, S.; Ciura, S.; Rouleau, G.A.; Kabashi, E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 2015, 31, 263–273. [Google Scholar] [CrossRef]
- Robinson, K.J.; Yuan, K.C.; Don, E.K.; Hogan, A.L.; Winnick, C.G.; Tym, M.C.; Lucas, C.W.; Shahheydari, H.; Watchon, M.; Blair, I.P.; et al. Motor neuron abnormalities correlate with impaired movement in zebrafish that express mutant superoxide dismutase 1. Zebrafish 2019, 16, 8–14. [Google Scholar] [CrossRef]
- Laboissonniere, L.A.; Smith, C.L.; Mesenbrink, J.; Chowdhury, R.; Burney, A.; Lang, M.; Sierra, M.; Stark, A.; Maldonado-Casalduc, G.; Muller, M.; et al. ALS-associated genes display CNS expression in the developing zebrafish. Gene Expr. Patterns 2018, 30, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.H.; Liu, H.F.; Li, Y.W.; Lu, C.S.; Shih, H.Y.; Chiu, C.C.; Lin, S.J.; Huang, Y.C.; Cheng, Y.C. C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp. Neurol. 2018, 304, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ji, D.; Yang, Q.; Li, M.; Ma, Z.; Zhang, S.; Li, H. NEFLb impairs early nervous system development via regulation of neuron apoptosis in zebrafish. J. Cell. Physiol. 2019, 234, 11208–11218. [Google Scholar] [CrossRef]
- Flanagan-Steet, H.; Fox, M.A.; Meyer, D.; Sanes, J.R. Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development 2005, 132, 4471–4481. [Google Scholar] [CrossRef] [Green Version]
- Akerboom, J.; Chen, T.W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderón, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef]
- Böhm, U.L.; Prendergast, A.; Djenoune, L.; Figueiredo, S.N.; Gomez, J.; Stokes, C.; Kaiser, S.; Suster, M.; Kawakami, K.; Charpentier, M.; et al. CSF-contacting neurons regulate locomotion by relaying mechanical stimuli to spinal circuits. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Asakawa, K.; Suster, M.L.; Mizusawa, K.; Nagayoshi, S.; Kotani, T.; Urasaki, A.; Kishimoto, Y.; Hibi, M.; Kawakami, K. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc. Natl. Acad. Sci. USA 2008, 105, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Kwan, K.M.; Fujimoto, E.; Grabher, C.; Mangum, B.D.; Hardy, M.E.; Campbell, D.S.; Parant, J.M.; Yost, H.J.; Kanki, J.P.; Chien, C.B. The Tol2kit: A multisite gateway-based construction Kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007, 236, 3088–3099. [Google Scholar] [CrossRef]
- Diane, E.; Handy Joseph Loscalzo, R.C. Global analysis of hematopoietic and vascular endothelial gene expression by tissue specific microarray profiling in zebrafish. Bone 2011, 23, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gentil, B.J.; Mushynski, W.E.; Durham, H.D. Heterogeneity in the properties of NEFL mutants causing Charcot-Marie-Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. Int. J. Biochem. Cell Biol. 2013, 45, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Campanari, M.-L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; De Calbiac, H.; Le Ber, I.; Brice, A.; Ciura, S.; Kabashi, E. Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum. Mol. Genet. 2015, 24, 1682–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Gentil, B.J.; Tibshirani, M.; Durham, H.D. Neurofilament dynamics and involvement in neurological disorders. Cell Tissue Res. 2015, 360, 609–620. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, L.; Selzer, M.E. Assembly properties of lamprey neurofilament subunits and their expression after spinal cord transection. J. Comp. Neurol. 2011, 519, 3657–3671. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Rao, M.V.; Sasaki, T.; Chen, Y.; Kumar, A.; Liem, R.K.H.; Eyer, J.; Peterson, A.C.; Julien, J.P.; Nixon, R.A. α-internexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J. Neurosci. 2006, 26, 10006–10019. [Google Scholar] [CrossRef]
- Yuan, A.; Sasaki, T.; Kumar, A.; Peterhoff, C.M.; Rao, M.V.; Liem, R.K.; Julien, J.P.; Nixon, R.A. Peripherin is a subunit of peripheral nerve neurofilaments: Implications for differential vulnerability of cns and peripheral nervous system axons. J. Neurosci. 2012, 32, 8501–8508. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Couillard-Després, S.; Julien, J.P. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp. Neurol. 1997, 148, 299–316. [Google Scholar] [CrossRef]
- Križ, J.; Zhu, Q.; Julien, J.P.; Padjen, A.L. Electrophysiological properties of axons in mice lacking neurofilament subunit genes: Disparity between conduction velocity and axon diameter in absence of NF-H. Brain Res. 2000, 885, 32–44. [Google Scholar] [CrossRef]
- Meier, J.; Couillard-Després, S.; Jacomy, H.; Gravel, C.; Julien, J.P. Extra neurofilament NF-L subunits rescue motor neuron disease caused by overexpression of the human NF-H gene in mice. J. Neuropathol. Exp. Neurol. 1999, 58, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, A.; Ishihara, T.; Miyashita, A.; Kuwabara, M.; Kato, T.; Watanabe, N.; Yamahira, A.; Kondo, C.; Yokoseki, A.; Takahashi, M.; et al. Alteration of POLDIP3 splicing associated with loss of function of TDP-43 in tissues affected with ALS. PLoS ONE 2012, 7, e43120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaux, C.; De Aguilar, J.L.G.; Dupuis, L. Unmasking the skiptic task of TDP-43. EMBO J. 2018, 37, e99645. [Google Scholar] [CrossRef]
- Julien, J.P.; Grosveld, F.; Yazdanbaksh, K.; Flavell, D.; Meijer, D.; Mushynski, W. The structure of a human neurofilament gene (NF-L): A unique exon-intron organization in the intermediate filament gene family. BBA-Gene Struct. Expr. 1987, 909, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Hurst, J.; Flavell, D.; Julien, J.P.; Meijer, D.; Mushynski, W.; Grosveld, F. The human neurofilament gene (NEFL) is located on the short arm of chromosome 8. Cytogenet. Genome Res. 1987, 45, 30–32. [Google Scholar] [CrossRef] [Green Version]
- Fasani, F.; Bocquet, A.; Robert, P.; Peterson, A.; Eyer, J. The amount of neurofilaments aggregated in the cell body is controlled by their increased sensitivity to trypsin-like proteases. J. Cell Sci. 2004, 117, 861–869. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demy, D.L.; Campanari, M.L.; Munoz-Ruiz, R.; Durham, H.D.; Gentil, B.J.; Kabashi, E. Functional Characterization of Neurofilament Light Splicing and Misbalance in Zebrafish. Cells 2020, 9, 1238. https://doi.org/10.3390/cells9051238
Demy DL, Campanari ML, Munoz-Ruiz R, Durham HD, Gentil BJ, Kabashi E. Functional Characterization of Neurofilament Light Splicing and Misbalance in Zebrafish. Cells. 2020; 9(5):1238. https://doi.org/10.3390/cells9051238
Chicago/Turabian StyleDemy, Doris Lou, Maria Letizia Campanari, Raphael Munoz-Ruiz, Heather D. Durham, Benoit J. Gentil, and Edor Kabashi. 2020. "Functional Characterization of Neurofilament Light Splicing and Misbalance in Zebrafish" Cells 9, no. 5: 1238. https://doi.org/10.3390/cells9051238