Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics


Abstract
:1. Introduction
- MP structure and function in ECM
- MPs in cell signaling
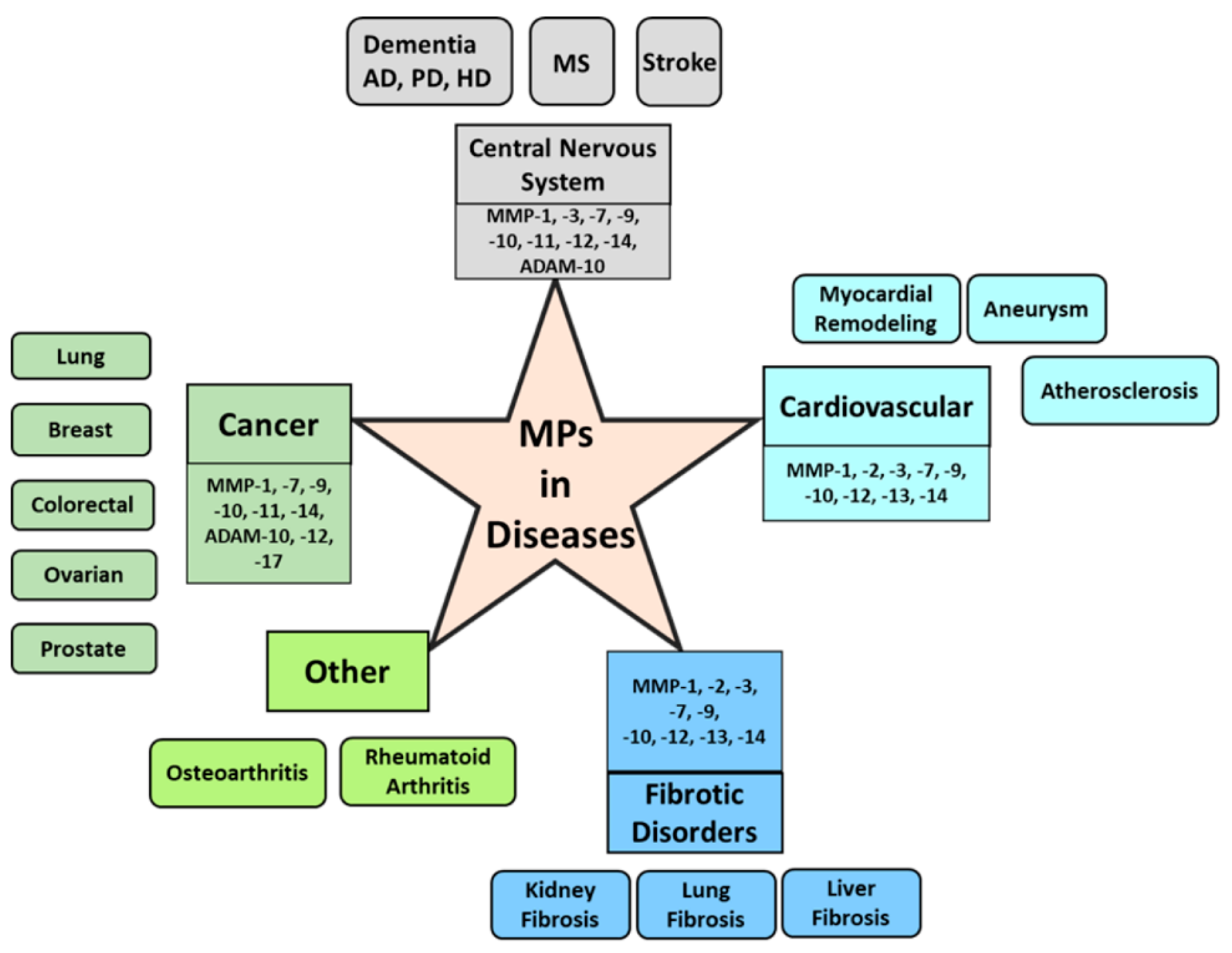
- MPs in cancer
- MPs in central nervous system and neurodegenerative diseases
- MPs in cardiovascular diseases
- MPs in fibrosis and other diseases
- MMP inhibition for developing therapeutics
- MMP-responsive drugs and drug delivery tools
- Conclusion and future directions
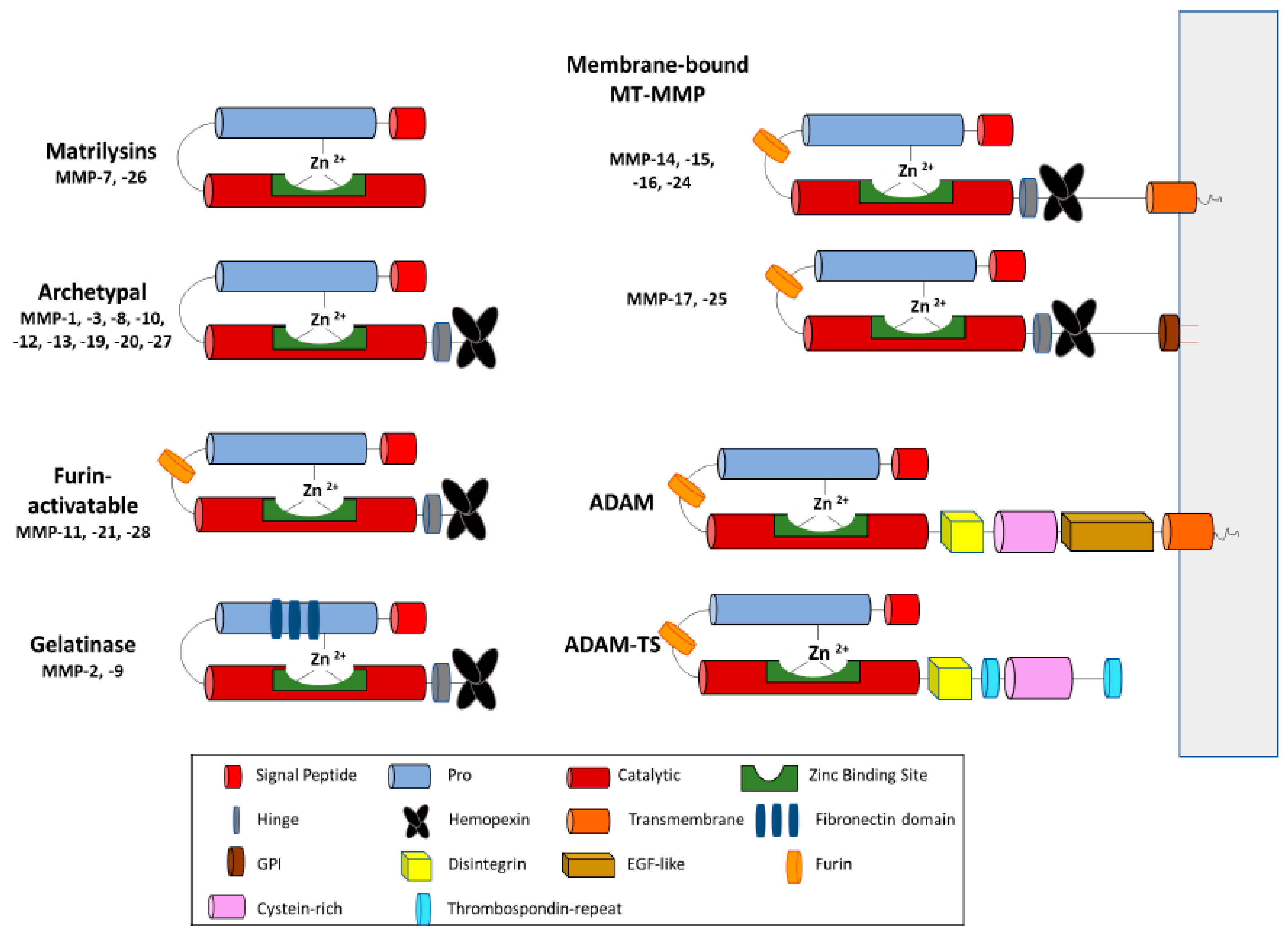
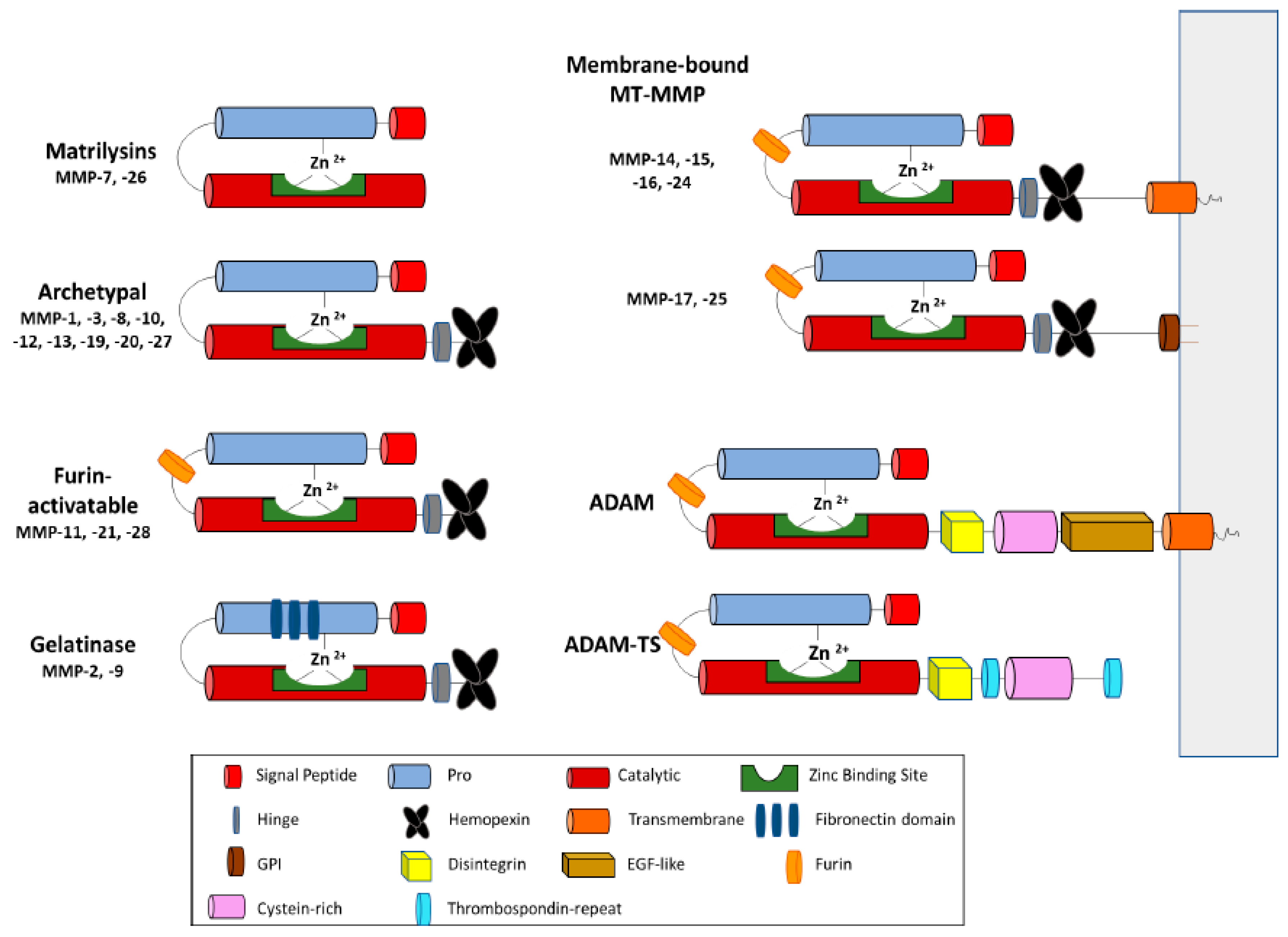
2. MP Structure and Function in ECM
3. MMP Regulation in the ECM
4. MPs and Their Inhibitors in Cell Signaling
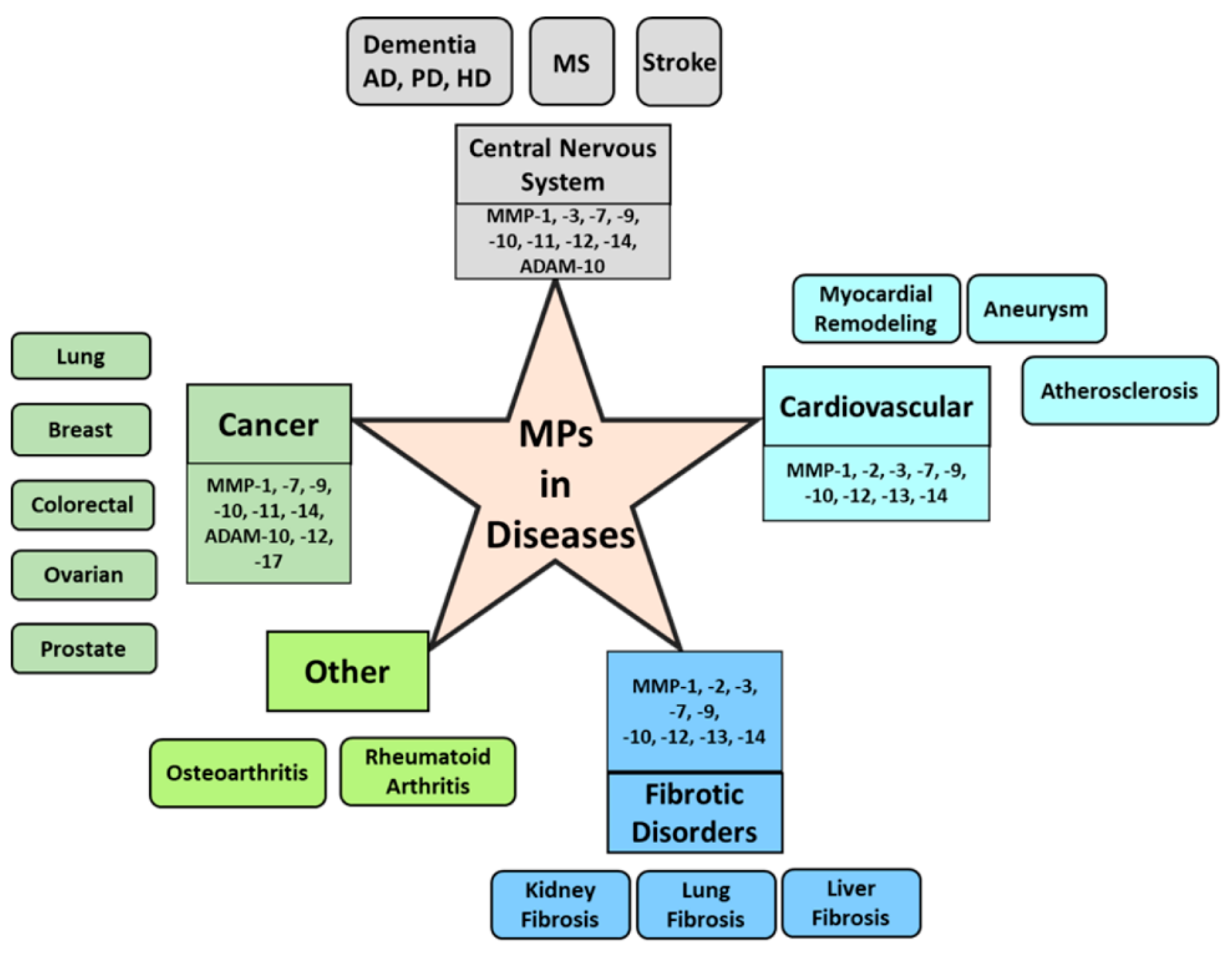
5. MPs in Cancer
6. MPs in Central Nervous System and Neurodegenerative Diseases
7. MPs in Cardiovascular Diseases
8. MPs in Fibrosis and Other Diseases
9. MMP Inhibition for Developing Therapeutics
9.1. Small Molecules
9.2. Peptides
9.3. Protein-Based MP Inhibitors
9.3.1. Antibodies
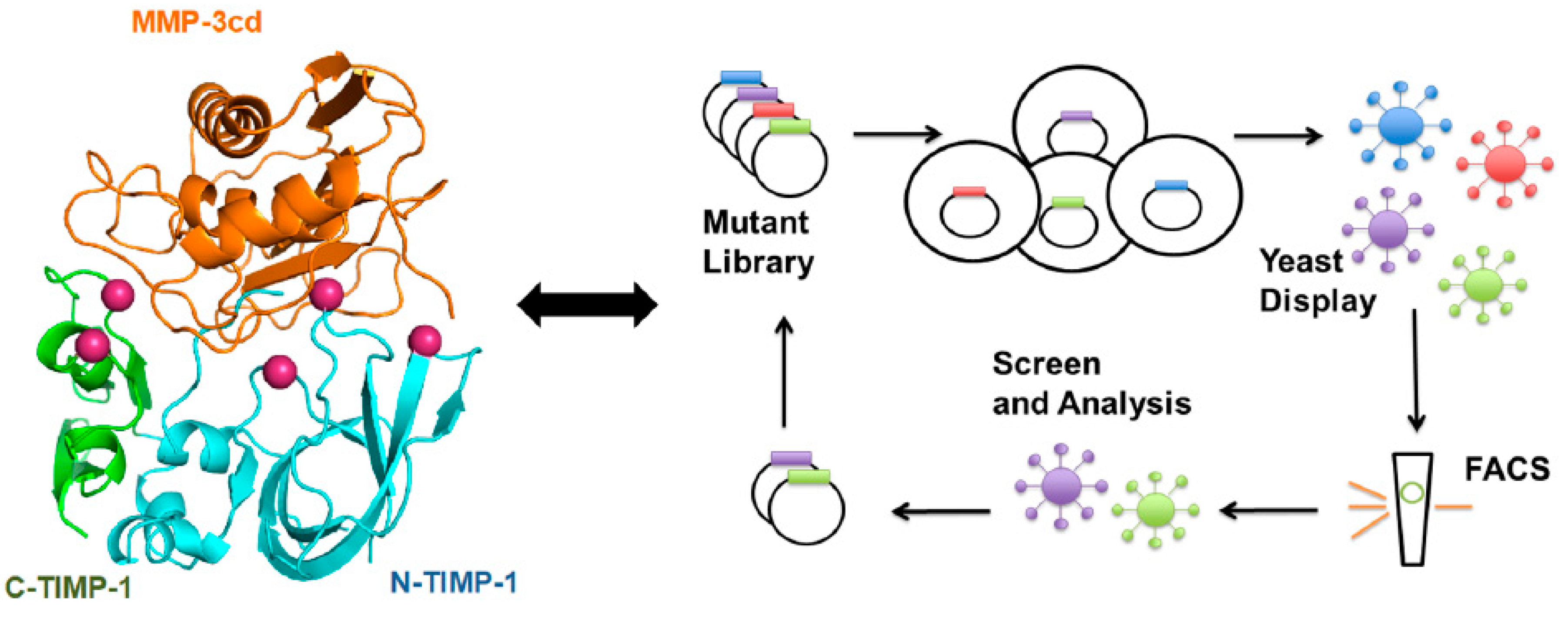
9.3.2. TIMPs
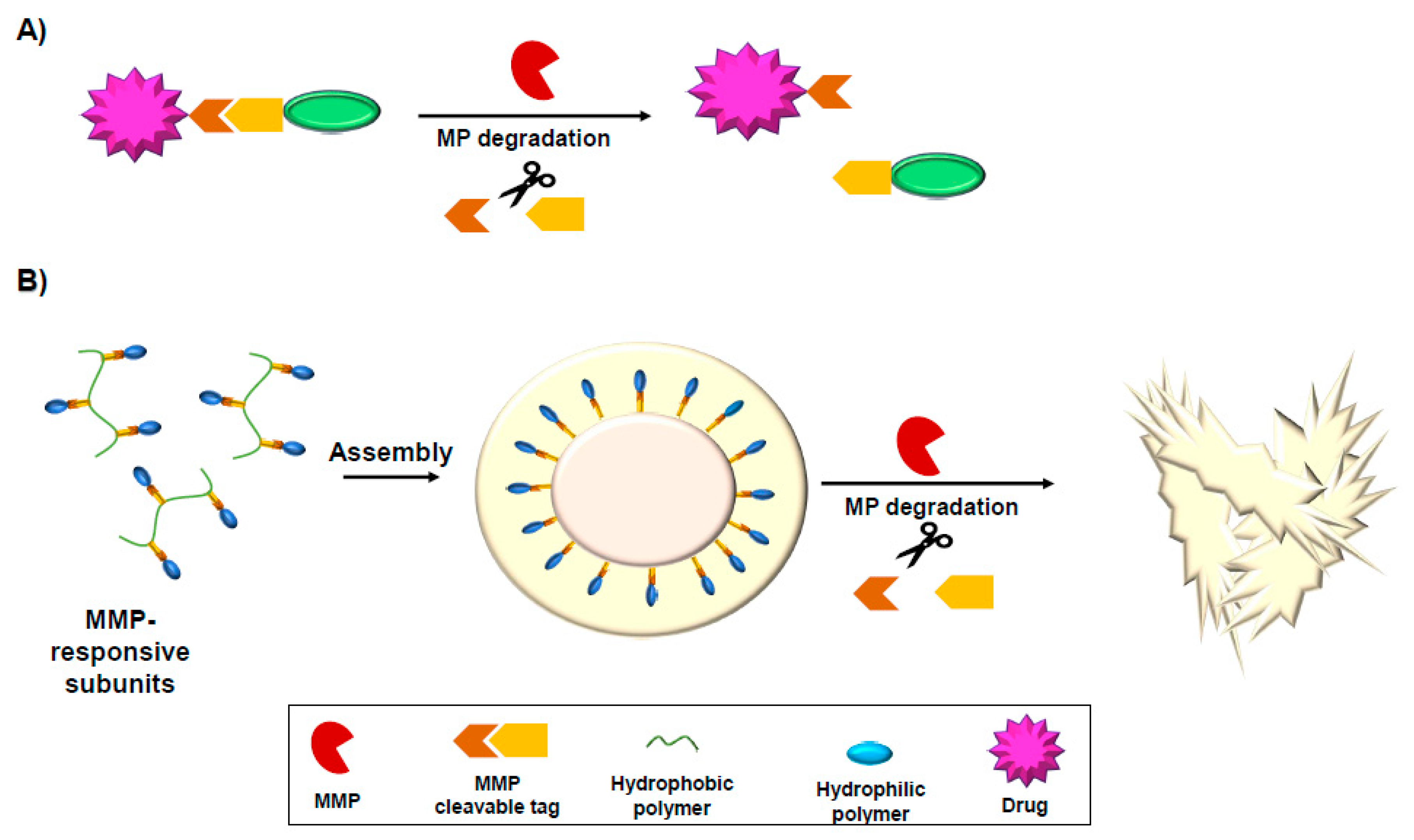
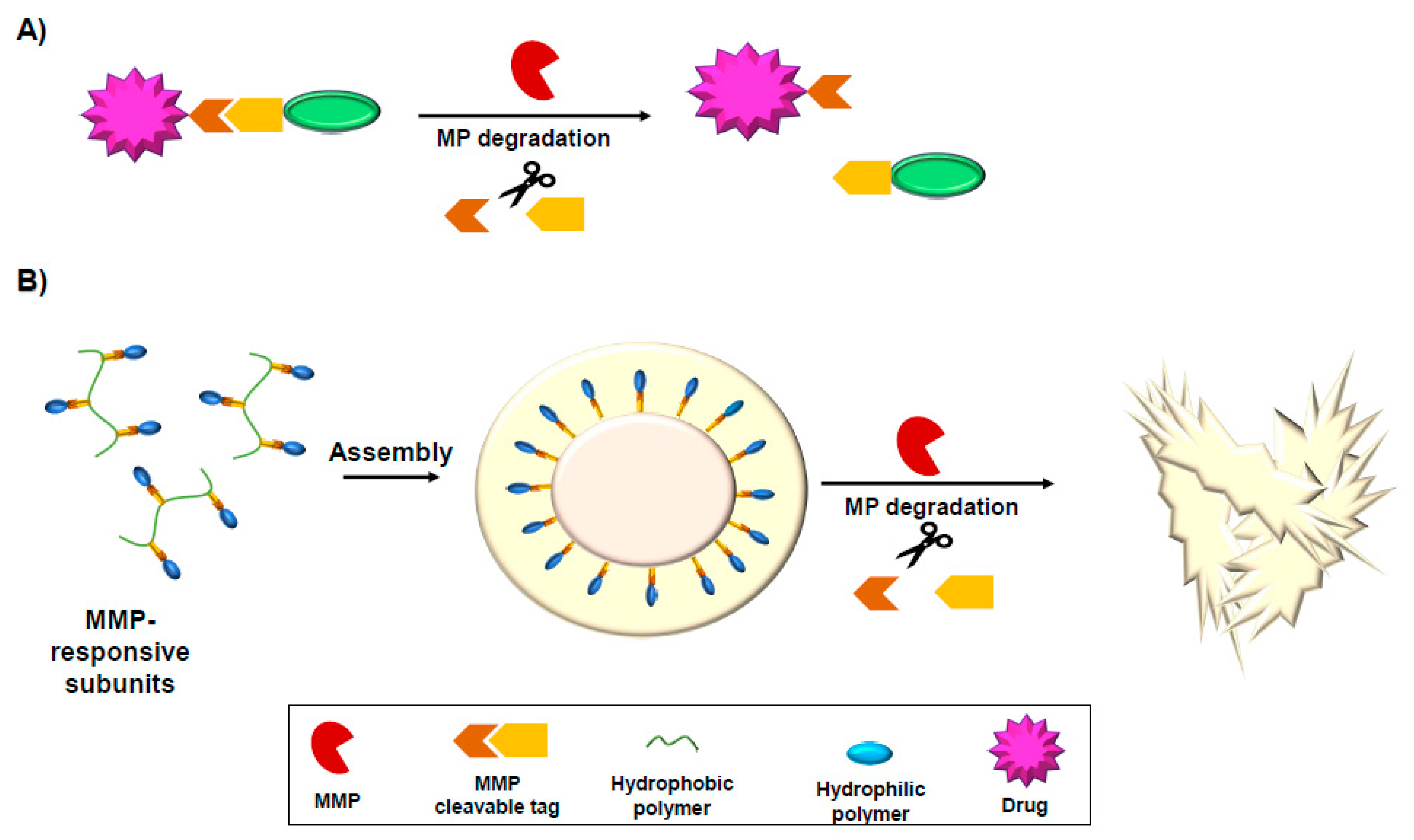
10. MMP-Activated Drugs and Drug Delivery Tools
10.1. MMP-Responsive Prodrugs
10.2. MMP-Responsive Hydrogels
10.3. MMP-Responsive Nanoparticles
11. Discussion and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Przemyslaw, L.; Boguslaw, H.A.; Elzbieta, S.; Malgorzata, S.M. ADAM and ADAMTS family proteins and their role in the colorectal cancer etiopathogenesis. BMB Rep. 2013, 46, 139–150. [Google Scholar] [CrossRef]
- Baker, A.H.; Edwards, D.R.; Murphy, G. Metalloproteinase inhibitors: Biological actions and therapeutic opportunities. J. Cell Sci. 2002, 115, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Noel, A.; Gutierrez-Fernandez, A.; Sounni, N.E.; Behrendt, N.; Maquoi, E.; Lund, I.K.; Cal, S.; Hoyer-Hansen, G.; Lopez-Otin, C. New and paradoxical roles of matrix metalloproteinases in the tumor microenvironment. Front. Pharmacol. 2012, 3, 140. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Tay, F.R.; Yiu, C.K.Y. The past, present and future perspectives of matrix metalloproteinase inhibitors. Pharmacol. Ther. 2020, 207, 107465. [Google Scholar] [CrossRef]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [Green Version]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Ahmad, R.; Kaur, G. Matrix metalloproteinases and cancer—Roles in threat and therapy. Asian Pac. J. Cancer Prev. 2014, 15, 1085–1091. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Srivastava, S.K.; Chaudhuri, T.K.; Upadhyay, G. Multifaceted role of matrix metalloproteinases (MMPs). Front. Mol. Biosci. 2015, 2, 19. [Google Scholar] [CrossRef]
- Rivera, S.; Garcia-Gonzalez, L.; Khrestchatisky, M.; Baranger, K. Metalloproteinases and their tissue inhibitors in Alzheimer’s disease and other neurodegenerative disorders. Cell Mol. Life Sci. 2019, 76, 3167–3191. [Google Scholar] [CrossRef]
- Khalil, R.A. Matrix Metalloproteinses and Tissue Remodeling in Health and Disease: Cardiovascular Remodeling. Prog. Mol. Biol. Transl. 2017, 147, 1–308. [Google Scholar]
- Azevedo, A.; Prado, A.F.; Antonio, R.C.; Issa, J.P.; Gerlach, R.F. Matrix metalloproteinases are involved in cardiovascular diseases. Basic Clin. Pharm. Toxicol. 2014, 115, 301–314. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix Metalloproteinases as Therapeutic Targets for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharm. Res. 2020, 152, 104591. [Google Scholar] [CrossRef]
- Tauro, J.R.; Gemeinhart, R.A. Matrix metalloprotease triggered delivery of cancer chemotherapeutics from hydrogel matrixes. Bioconjugate Chem. 2005, 16, 1133–1139. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. (Landmark Ed.) 2015, 20, 1144–1163. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.L.; Jin, G.; Cao, R.; Zhang, S.; Cao, Y.; Zhou, Z. MT1-MMP sheds LYVE-1 on lymphatic endothelial cells and suppresses VEGF-C production to inhibit lymphangiogenesis. Nat. Commun. 2016, 7, 10824. [Google Scholar] [CrossRef] [Green Version]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Isupov, M.; Lindqvist, Y.; Schneider, G.; Tryggvason, K. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science 1999, 284, 1667–1670. [Google Scholar] [CrossRef]
- Eckhard, U.; Huesgen, P.F.; Schilling, O.; Bellac, C.L.; Butler, G.S.; Cox, J.H.; Dufour, A.; Goebeler, V.; Kappelhoff, R.; Keller, U.A.D.; et al. Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 2016, 49, 37–60. [Google Scholar] [CrossRef]
- Shogan, B.D.; Belogortseva, N.; Luong, P.M.; Zaborin, A.; Lax, S.; Bethel, C.; Ward, M.; Muldoon, J.P.; Singer, M.; An, G.; et al. Collagen degradation and MMP9 activation by Enterococcus faecalis contribute to intestinal anastomotic leak. Sci. Transl. Med. 2015, 7, 286ra268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, J.; Falconer, A.M.D.; Wilkinson, D.J.; Europe-Finner, G.N.; Litherland, G.J.; Rowan, A.D. Protein kinase D3 modulates MMP1 and MMP13 expression in human chondrocytes. PLoS ONE 2018, 13, e0195864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopka, J.A.; DeBaun, M.R.; Chang, W.; Dragoo, J.L. The Intracellular Effect of Relaxin on Female Anterior Cruciate Ligament Cells. Am. J. Sports Med. 2016, 44, 2384–2392. [Google Scholar] [CrossRef] [PubMed]
- Nukuda, A.; Sasaki, C.; Ishihara, S.; Mizutani, T.; Nakamura, K.; Ayabe, T.; Kawabata, K.; Haga, H. Stiff substrates increase YAP-signaling-mediated matrix metalloproteinase-7 expression. Oncogenesis 2015, 4, e165. [Google Scholar] [CrossRef] [Green Version]
- Andreini, C.; Banci, L.; Bertini, I.; Elmi, S.; Rosato, A. Comparative analysis of the ADAM and ADAMTS families. J. Proteome Res. 2005, 4, 881–888. [Google Scholar] [CrossRef]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef]
- Batra, J.; Radisky, E.S. Tissue inhibitors of metalloproteinases (TIMPs): Inhibition of Zn-dependent metallopeptidases. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; John Wiley & Sons: Chichester, UK, 2014. [Google Scholar]
- Gomis-Ruth, F.X.; Maskos, K.; Betz, M.; Bergner, A.; Huber, R.; Suzuki, K.; Yoshida, N.; Nagase, H.; Brew, K.; Bourenkov, G.P.; et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 1997, 389, 77–81. [Google Scholar] [CrossRef]
- Murphy, G.; Houbrechts, A.; Cockett, M.I.; Williamson, R.A.; O’Shea, M.; Docherty, A.J. The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 1991, 30, 8097–8102. [Google Scholar] [CrossRef]
- Huang, W.; Suzuki, K.; Nagase, H.; Arumugam, S.; Van Doren, S.R.; Brew, K. Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 1996, 384, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Raeeszadeh-Sarmazdeh, M.; Greene, K.A.; Sankaran, B.; Downey, G.P.; Radisky, D.C.; Radisky, E.S. Directed evolution of the metalloproteinase inhibitor TIMP-1 reveals that its N- and C-terminal domains cooperate in matrix metalloproteinase recognition. J. Biol. Chem. 2019, 294, 9476–9488. [Google Scholar] [CrossRef]
- Hutton, M.; Willenbrock, F.; Brocklehurst, K.; Murphy, G. Kinetic analysis of the mechanism of interaction of full-length TIMP-2 and gelatinase A: Evidence for the existence of a low-affinity intermediate. Biochemistry 1998, 37, 10094–10098. [Google Scholar] [CrossRef]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2043–2055. [Google Scholar] [CrossRef]
- Butler, G.S.; Apte, S.S.; Willenbrock, F.; Murphy, G. Human tissue inhibitor of metalloproteinases 3 interacts with both the N- and C-terminal domains of gelatinases A and B. Regulation by polyanions. J. Biol. Chem. 1999, 274, 10846–10851. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.P.; Willenbrock, F.; Docherty, A.J.; Eaton, D.; Murphy, G. Analysis of the role of the COOH-terminal domain in the activation, proteolytic activity, and tissue inhibitor of metalloproteinase interactions of gelatinase B. J. Biol. Chem. 1994, 269, 14967–14973. [Google Scholar]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Morgunova, E.; Tuuttila, A.; Bergmann, U.; Tryggvason, K. Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc. Natl. Acad. Sci. USA 2002, 99, 7414–7419. [Google Scholar] [CrossRef] [Green Version]
- Strongin, A.Y.; Collier, I.; Bannikov, G.; Marmer, B.L.; Grant, G.A.; Goldberg, G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995, 270, 5331–5338. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, G.I.; Strongin, A.; Collier, I.E.; Genrich, L.T.; Marmer, B.L. Interaction of 92-kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. J. Biol. Chem. 1992, 267, 4583–4591. [Google Scholar]
- Ogata, Y.; Itoh, Y.; Nagase, H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J. Biol. Chem. 1995, 270, 18506–18511. [Google Scholar] [CrossRef] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Ardi, V.C.; Van den Steen, P.E.; Opdenakker, G.; Schweighofer, B.; Deryugina, E.I.; Quigley, J.P. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J. Biol. Chem. 2009, 284, 25854–25866. [Google Scholar] [CrossRef] [Green Version]
- Amour, A.; Knight, C.G.; Webster, A.; Slocombe, P.M.; Stephens, P.E.; Knauper, V.; Docherty, A.J.P.; Murphy, G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000, 473, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Loechel, F.; Fox, J.W.; Murphy, G.; Albrechtsen, R.; Wewer, U.M. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem. Biophys. Res. Commun. 2000, 278, 511–515. [Google Scholar] [CrossRef]
- Wayne, G.J.; Deng, S.J.; Amour, A.; Borman, S.; Matico, R.; Carter, H.L.; Murphy, G. TIMP-3 inhibition of ADAMTS-4 (Aggrecanase-1) is modulated by interactions between aggrecan and the C-terminal domain of ADAMTS-4. J. Biol. Ogical Chem. 2007, 282, 20991–20998. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, M.; Tortorella, M.; Nagase, H.; Brew, K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 2001, 276, 12501–12504. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, G.; Bacchetti, T.; Banach, M.; Simental-Mendia, L.E.; Sahebkar, A. Impact of Statin Therapy on Plasma MMP-3, MMP-9, and TIMP-1 Concentrations: A Systematic Review and Meta-Analysis of Randomized Placebo-Controlled Trials. Angiology 2017, 68, 850–862. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Agren, M.S.; Schnabel, R.; Christensen, L.H.; Mirastschijski, U. Tumor necrosis factor-alpha-accelerated degradation of type I collagen in human skin is associated with elevated matrix metalloproteinase (MMP)-1 and MMP-3 ex vivo. Eur. J. Cell Biol. 2015, 94, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kuang, X.R.; Lv, P.T.; Yan, X.X. Thymoquinone inhibits proliferation and invasion of human nonsmall-cell lung cancer cells via ERK pathway. Tumour Biol. 2015, 36, 259–269. [Google Scholar] [CrossRef]
- Goertzen, C.G.; Dragan, M.; Turley, E.; Babwah, A.V.; Bhattacharya, M. KISS1R signaling promotes invadopodia formation in human breast cancer cell via beta-arrestin2/ERK. Cell Signal 2016, 28, 165–176. [Google Scholar] [CrossRef]
- Belton, M.; Brilha, S.; Manavaki, R.; Mauri, F.; Nijran, K.; Hong, Y.T.; Patel, N.H.; Dembek, M.; Tezera, L.; Green, J.; et al. Hypoxia and tissue destruction in pulmonary TB. Thorax 2016, 71, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Brilha, S.; Sathyamoorthy, T.; Stuttaford, L.H.; Walker, N.F.; Wilkinson, R.J.; Singh, S.; Moores, R.C.; Elkington, P.T.; Friedland, J.S. Early Secretory Antigenic Target-6 Drives Matrix Metalloproteinase-10 Gene Expression and Secretion in Tuberculosis. Am. J. Respir. Cell Mol. Biol. 2017, 56, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Vendramini-Costa, D.B.; Carvalho, J.E. Molecular Link Mechanisms between Inflammation and Cancer. Curr. Pharm. Des. 2012, 18, 3831–3852. [Google Scholar] [CrossRef]
- Wu, K.I.; Schmid-Schonbein, G.W. Nuclear factor kappa B and matrix metalloproteinase induced receptor cleavage in the spontaneously hypertensive rat. Hypertension 2011, 57, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Chase, A.J.; Bond, M.; Crook, M.F.; Newby, A.C. Role of nuclear factor-kappa B activation in metalloproteinase-1,-3, and-9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Xu, X.B.; Chen, X.H.; Wei, G.; He, B.; Wang, J.D. The nuclear factor-B pathway is involved in matrix metalloproteinase-9 expression in RU486-induced endometrium breakdown in mice. Hum. Reprod. 2012, 27, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Jang, B.; Jung, H.; Choi, S.; Lee, Y.H.; Lee, S.T.; Oh, E.S. Syndecan-2 cytoplasmic domain up-regulates matrix metalloproteinase-7 expression via the protein kinase Cgamma-mediated FAK/ERK signaling pathway in colon cancer. J. Biol. Chem. 2017, 292, 16321–16332. [Google Scholar] [CrossRef] [Green Version]
- Malfait, A.M.; Liu, R.Q.; Ijiri, K.; Komiya, S.; Tortorella, M.D. Inhibition of ADAM-TS4 and ADAM-TS5 prevents aggrecan degradation in osteoarthritic cartilage. J. Biol. Ogical Chem. 2002, 277, 22201–22208. [Google Scholar] [CrossRef] [Green Version]
- Organ, S.L.; Tsao, M.S. An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R. Notch signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Bozkulak, E.C.; Weinmaster, G. Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Mol. Cell Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [Green Version]
- Groot, A.J.; Vooijs, M.A. The role of Adams in Notch signaling. Adv. Exp. Med. Biol. 2012, 727, 15–36. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, Z.; Yan, Y.; Qiang, X.; Zhou, C.; Li, R.; Chen, H.; Zhang, Y. Demethyleneberberine Protects against Hepatic Fibrosis in Mice by Modulating NF-kappaB Signaling. Int. J. Mol. Sci. 2016, 17, 1036. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Gao, H.; Ju, P.; Gao, M.Q.; Yuan, Y.P.; Chen, X.H.; Liu, K.L.; Han, Y.T.; Han, Z.W. Hispidulin inhibits hepatocellular carcinoma growth and metastasis through AMPK and ERK signaling mediated activation of PPARgamma. Biomed. Pharm. 2018, 103, 272–283. [Google Scholar] [CrossRef]
- Kolb, C.; Mauch, S.; Peter, H.H.; Krawinkel, U.; Sedlacek, R. The matrix metalloproteinase RASI-1 is expressed in synovial blood vessels of a rheumatoid arthritis patient. Immunol. Lett. 1997, 57, 83–88. [Google Scholar] [CrossRef]
- Kolb, S.A.; Lahrtz, F.; Paul, R.; Leppert, D.; Nadal, D.; Pfister, H.W.; Fontana, A. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in viral meningitis: Upregulation of MMP-9 and TIMP-1 in cerebrospinal fluid. J. Neuroimmunol. 1998, 84, 143–150. [Google Scholar] [CrossRef]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. (AMST) 2019, 2019, 9423907. [Google Scholar] [CrossRef] [Green Version]
- Al-Raawi, D.; Abu-El-Zahab, H.; El-Shinawi, M.; Mohamed, M.M. Membrane type-1 matrix metalloproteinase (MT1-MMP) correlates with the expression and activation of matrix metalloproteinase-2 (MMP-2) in inflammatory breast cancer. Int. J. Clin. Exp. Med. 2011, 4, 265–275. [Google Scholar]
- Mehner, C.; Hockla, A.; Miller, E.; Ran, S.; Radisky, D.C.; Radisky, E.S. Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 2014, 5, 2736–2749. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, C.; Ran, R.; Liu, G.; Yang, Y.; Zhao, W.; Xie, X.; Li, J. Matrix metalloproteinase family gene polymorphisms and lung cancer susceptibility: An updated meta-analysis. J. Thorac. Dis. 2020, 12, 349–362. [Google Scholar] [CrossRef]
- Justilien, V.; Regala, R.P.; Tseng, I.C.; Walsh, M.P.; Batra, J.; Radisky, E.S.; Murray, N.R.; Fields, A.P. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS ONE 2012, 7, e35040. [Google Scholar] [CrossRef]
- Radisky, D.C.; Przybylo, J.A. Matrix metalloproteinase-induced fibrosis and malignancy in breast and lung. Proc. Am. Thorac. Soc. 2008, 5, 316–322. [Google Scholar] [CrossRef]
- Stallings-Mann, M.L.; Waldmann, J.; Zhang, Y.; Miller, E.; Gauthier, M.L.; Visscher, D.W.; Downey, G.P.; Radisky, E.S.; Fields, A.P.; Radisky, D.C. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci. Transl. Med. 2012, 4, 142ra195. [Google Scholar] [CrossRef] [Green Version]
- El-Chaer, W.K.; Moraes, C.F.; Nobrega, O.T. Diagnosis and Prognosis of Prostate Cancer from Circulating Matrix Metalloproteinases and Inhibitors. J. Aging Res. 2018, 2018, 7681039. [Google Scholar] [CrossRef]
- Jiang, C.; Wang, J.; Dong, C.; Wei, W.; Li, J.; Li, X. Membranous type matrix metalloproteinase 16 induces human prostate cancer metastasis. Oncol. Lett. 2017, 14, 3096–3102. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhu, X. Association between matrix-metalloproteinase polymorphisms and prostate cancer risk: A meta-analysis and systematic review. Cancer Manag. Res. 2018, 10, 5247–5259. [Google Scholar] [CrossRef] [Green Version]
- Marshall, D.C.; Lyman, S.K.; McCauley, S.; Kovalenko, M.; Spangler, R.; Liu, C.; Lee, M.; O’Sullivan, C.; Barry-Hamilton, V.; Ghermazien, H.; et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix metalloproteinases in cancer: Their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Knapinska, A.M.; Fields, G.B. The Expanding Role of MT1-MMP in Cancer Progression. Pharmaceuticals 2019, 12, 77. [Google Scholar] [CrossRef] [Green Version]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar] [CrossRef] [Green Version]
- Pahwa, S.; Stawikowski, M.J.; Fields, G.B. Monitoring and Inhibiting MT1-MMP during Cancer Initiation and Progression. Cancers 2014, 6, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Wang, B.; Li, K.; Sun, H.; Hai, T.; Zhang, Y.; Kang, H. Upregulating MMP-1 in carcinoma-associated fibroblasts reduces the efficacy of Taxotere on breast cancer synergized by Collagen IV. Oncol. Lett. 2018, 16, 3537–3544. [Google Scholar] [CrossRef]
- Wang, Q.M.; Lv, L.; Tang, Y.; Zhang, L.; Wang, L.F. MMP-1 is overexpressed in triple-negative breast cancer tissues and the knockdown of MMP-1 expression inhibits tumor cell malignant behaviors in vitro. Oncol. Lett. 2019, 17, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Vanlaere, I.; Van Hauwermeiren, F.; Van Wonterghem, E.; Wilson, C.; Libert, C. Pro-inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunol. 2014, 7, 579–588. [Google Scholar] [CrossRef]
- Yamada, T.; Oshima, T.; Yoshihara, K.; Tamura, S.; Kanazawa, A.; Inagaki, D.; Yamamoto, N.; Sato, T.; Fujii, S.; Numata, K.; et al. Overexpression of MMP-13 gene in colorectal cancer with liver metastasis. Anticancer Res. 2010, 30, 2693–2699. [Google Scholar]
- Ghasemi, A.; Hashemy, S.I.; Aghaei, M.; Panjehpour, M. Leptin induces matrix metalloproteinase 7 expression to promote ovarian cancer cell invasion by activating ERK and JNK pathways. J. Cell Biochem. 2018, 119, 2333–2344. [Google Scholar] [CrossRef]
- Shao, S.H.; Li, Z.L.; Gao, W.; Yu, G.H.; Liu, D.X.; Pan, F. ADAM-12 as a Diagnostic Marker for the Proliferation, Migration and Invasion in Patients with Small Cell Lung Cancer. PLoS ONE 2014, 9, e85936. [Google Scholar] [CrossRef]
- Blacker, M.; Noe, M.C.; Carty, T.J.; Goodyer, C.G.; LeBlanc, A.C. Effect of tumor necrosis factor-alpha converting enzyme (TACE) and metalloprotease inhibitor on amyloid precursor protein metabolism in human neurons. J. Neurochem. 2002, 83, 1349–1357. [Google Scholar] [CrossRef]
- Caiazza, F.; McGowan, P.M.; Mullooly, M.; Murray, A.; Synnott, N.; O’Donovan, N.; Flanagan, L.; Tape, C.J.; Murphy, G.; Crown, J.; et al. Targeting ADAM-17 with an inhibitory monoclonal antibody has antitumour effects in triple-negative breast cancer cells. Br. J. Cancer 2015, 112, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Rocks, N.; Paulissen, G.; Quesada Calvo, F.; Polette, M.; Gueders, M.; Munaut, C.; Foidart, J.M.; Noel, A.; Birembaut, P.; Cataldo, D. Expression of a disintegrin and metalloprotease (ADAM and ADAMTS) enzymes in human non-small-cell lung carcinomas (NSCLC). Br. J. Cancer 2006, 94, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Le Pabic, H.; Bonnier, D.; Wewer, U.M.; Coutand, A.; Musso, O.; Baffet, G.; Clement, B.; Theret, N. ADAM12 in human liver cancers: TGF-beta-regulated expression in stellate cells is associated with matrix remodeling. Hepatology 2003, 37, 1056–1066. [Google Scholar] [CrossRef]
- Duhachek-Muggy, S.; Qi, Y.; Wise, R.; Alyahya, L.; Li, H.; Hodge, J.; Zolkiewska, A. Metalloprotease-disintegrin ADAM12 actively promotes the stem cell-like phenotype in claudin-low breast cancer. Mol. Cancer 2017, 16, 32. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Seubert, B.; Stahl, E.; Dietz, H.; Reuning, U.; Moreno-Leon, L.; Ilie, M.; Hofman, P.; Nagase, H.; Mari, B.; et al. Tissue inhibitor of metalloproteinases-1 induces a pro-tumourigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene 2015, 34, 3640–3650. [Google Scholar] [CrossRef]
- Gong, Y.; Scott, E.; Lu, R.; Xu, Y.; Oh, W.K.; Yu, Q. TIMP-1 promotes accumulation of cancer associated fibroblasts and cancer progression. PLoS ONE 2013, 8, e77366. [Google Scholar] [CrossRef] [Green Version]
- Roeb, E.; Bosserhoff, A.K.; Hamacher, S.; Jansen, B.; Dahmen, J.; Wagner, S.; Matern, S. Enhanced migration of tissue inhibitor of metalloproteinase overexpressing hepatoma cells is attributed to gelatinases: Relevance to intracellular signaling pathways. World J. Gastroenterol. 2005, 11, 1096–1104. [Google Scholar] [CrossRef]
- Nakatsukasa, H.; Ashida, K.; Higashi, T.; Ohguchi, S.; Tsuboi, S.; Hino, N.; Nouso, K.; Urabe, Y.; Kinugasa, N.; Yoshida, K.; et al. Cellular distribution of transcripts for tissue inhibitor of metalloproteinases 1 and 2 in human hepatocellular carcinomas. Hepatology 1996, 24, 82–88. [Google Scholar] [CrossRef]
- Giannelli, G.; Bergamini, C.; Marinosci, F.; Fransvea, E.; Quaranta, M.; Lupo, L.; Schiraldi, O.; Antonaci, S. Clinical role of MMP-2/TIMP-2 imbalance in hepatocellular carcinoma. Int. J. Cancer 2002, 97, 425–431. [Google Scholar] [CrossRef]
- Shirian, J.; Arkadash, V.; Cohen, I.; Sapir, T.; Radisky, E.S.; Papo, N.; Shifman, J.M. Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14. FEBS Lett. 2018, 592, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.H.; Ebrahem, Q.; Ali, M.; Cutler, A.; Bell, B.; Prayson, N.; Sears, J.; Knauper, V.; Murphy, G.; Anand-Apte, B. Tissue inhibitor of metalloproteinases-3 peptides inhibit angiogenesis and choroidal neovascularization in mice. PLoS ONE 2013, 8, e55667. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.H.; Ebrahem, Q.; Moore, N.; Murphy, G.; Claesson-Welsh, L.; Bond, M.; Baker, A.; Anand-Apte, B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): Inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003, 9, 407–415. [Google Scholar] [CrossRef]
- Qi, J.H.; Anand-Apte, B. Tissue inhibitor of metalloproteinase-3 (TIMP3) promotes endothelial apoptosis via a caspase-independent mechanism. Apoptosis 2015, 20, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Lipton, A.; Leitzel, K.; Chaudri-Ross, H.A.; Evans, D.B.; Ali, S.M.; Demers, L.; Hamer, P.; Brown-Shimer, S.; Pierce, K.; Gaur, V.; et al. Serum TIMP-1 and response to the aromatase inhibitor letrozole versus tamoxifen in metastatic breast cancer. J. Clin. Oncol. 2008, 26, 2653–2658. [Google Scholar] [CrossRef]
- Jenkinson, C.; Elliott, V.; Menon, U.; Apostolidou, S.; Fourkala, O.E.; Gentry-Maharaj, A.; Pereira, S.P.; Jacobs, I.; Cox, T.F.; Greenhalf, W.; et al. Evaluation in pre-diagnosis samples discounts ICAM-1 and TIMP-1 as biomarkers for earlier diagnosis of pancreatic cancer. J. Proteom. 2015, 113, 400–402. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Zurakowski, D.; Wischhusen, J.; Frauenhoffer, C.; Hooshmand, S.; Kulke, M.; Moses, M.A. Urinary TIMP-1 and MMP-2 levels detect the presence of pancreatic malignancies. Br. J. Cancer 2014, 111, 1772–1779. [Google Scholar] [CrossRef] [Green Version]
- Brzdak, P.; Nowak, D.; Wiera, G.; Mozrzymas, J.W. Multifaceted Roles of Metzincins in CNS Physiology and Pathology: From Synaptic Plasticity and Cognition to Neurodegenerative Disorders. Front. Cell. Neurosci. 2017, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or Foes: Matrix Metalloproteinases and Their Multifaceted Roles in Neurodegenerative Diseases. Mediat. Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, H.; Takuma, K.; Fukuzaki, E.; Ibi, D.; Someya, E.; Akazawa, K.H.; Alkam, T.; Tsunekawa, H.; Mouri, A.; Noda, Y.; et al. Matrix metalloprotease-9 inhibition improves amyloid beta-mediated cognitive impairment and neurotoxicity in mice. J. Pharmacol. Exp. Ther. 2009, 331, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Shackleton, B.; Ringland, C.; Abdullah, L.; Mullan, M.; Crawford, F.; Bachmeier, C. Influence of Matrix Metallopeptidase 9 on Beta-Amyloid Elimination Across the Blood-Brain Barrier. Mol. Neurobiol. 2019, 56, 8296–8305. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef]
- Berg, G.; Miksztowicz, V.; Schreier, L. Metalloproteinases in metabolic syndrome. Clin. Chim. Acta 2011, 412, 1731–1739. [Google Scholar] [CrossRef]
- Gasche, Y.; Soccal, P.M.; Kanemitsu, M.; Copin, J.C. Matrix metalloproteinases and diseases of the central nervous system with a special emphasis on ischemic brain. Front. Biosci. 2006, 11, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.A.; Lee, Y.; Marshall, J. Understanding the complexity of the matrix metalloproteinase system and its relevance to age-related diseases: Age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 2019, 100775. [Google Scholar] [CrossRef]
- Ren, Y.; Liu, W.; Jiang, H.; Jiang, Q.; Feng, J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J. Biol. Chem. 2005, 280, 34105–34112. [Google Scholar] [CrossRef] [Green Version]
- Stomrud, E.; Bjorkqvist, M.; Janciauskiene, S.; Minthon, L.; Hansson, O. Alterations of matrix metalloproteinases in the healthy elderly with increased risk of prodromal Alzheimer’s disease. Alzheimer Res. Ther. 2010, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharm. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Lim, E.W.; Aarsland, D.; Ffytche, D.; Taddei, R.N.; van Wamelen, D.J.; Wan, Y.M.; Tan, E.K.; Chaudhuri, K.R.; Kings Parcog group, M.D.S.N.S. Amyloid-beta and Parkinson’s disease. J. Neurol. 2019, 266, 2605–2619. [Google Scholar] [CrossRef]
- Musardo, S.; Marcello, E. Synaptic dysfunction in Alzheimer’s disease: From the role of amyloid beta-peptide to the alpha-secretase ADAM10. Eur. J. Pharmacol. 2017, 817, 30–37. [Google Scholar] [CrossRef]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–321. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Metalloproteinases and neurodegenerative diseases: Pathophysiological and therapeutic perspectives. Met. Med. 2015, 2015, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Cheng, C.L.; Mair, W.; Almeida, S.; Fong, H.; Biswas, M.H.U.; Zhang, Z.; Huang, Y.; Temple, S.; Coppola, G.; et al. Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Rep. 2016, 7, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Fragkouli, A.; Tsilibary, E.C.; Tzinia, A.K. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol. Dis. 2014, 70, 179–189. [Google Scholar] [CrossRef]
- Agrawal, S.M.; Lau, L.; Yong, V.W. MMPs in the central nervous system: Where the good guys go bad. Semin. Cell Dev. Biol. 2008, 19, 42–51. [Google Scholar] [CrossRef]
- Storck, S.E.; Pietrzik, C.U. Endothelial LRP1-A Potential Target for the Treatment of Alzheimer’s Disease. Pharm. Res. 2017, 34, 2637–2651. [Google Scholar] [CrossRef]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.Z.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix Metalloproteinases Are Modifiers of Huntingtin Proteolysis and Toxicity in Huntington’s Disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef] [Green Version]
- George, A.K.; Homme, R.P.; Majumder, A.; Tyagi, S.C.; Singh, M. Effect of MMP-9 gene knockout on retinal vascular form and function. Physiol. Genom. 2019, 51, 613–622. [Google Scholar] [CrossRef]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B.J. Genetics of dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in multiple sclerosis. Neurobiol. Mult. Scler. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Latronico, T.; Liuzzi, G.M. Metalloproteinases and their inhibitors as therapeutic targets for multiple sclerosis: Current evidence and future perspectives. Met. Med. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Iannetta, M.; Zingaropoli, M.A.; Latronico, T.; Pati, I.; Pontecorvo, S.; Prezioso, C.; Pietropaolo, V.; Cortese, A.; Frontoni, M.; D’Agostino, C.; et al. Dynamic changes of MMP-9 plasma levels correlate with JCV reactivation and immune activation in natalizumab-treated multiple sclerosis patients. Sci. Rep. 2019, 9, 9. [Google Scholar] [CrossRef]
- Saribas, A.S.; Ozdemir, A.; Lam, C.; Safak, M. JC virus-induced progressive multifocal leukoencephalopathy. Future Virol. 2010, 5, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieseier, B.C.; Kiefer, R.; Clements, J.M.; Miller, K.; Wells, G.M.A.; Schweitzer, T.; Gearing, A.J.H.; Hartung, H.P. Matrix metalloproteinase-9 and -7 are regulated in experimental autoimmune encephalomyelitis. Brain 1998, 121, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J. Cereb. Blood Flow Metab. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [Green Version]
- Polonikov, A.; Rymarova, L.; Klyosova, E.; Volkova, A.; Azarova, I.; Bushueva, O.; Bykanova, M.; Bocharova, I.; Zhabin, S.; Churnosov, M.; et al. Matrix metalloproteinases as target genes for gene regulatory networks driving molecular and cellular pathways related to a multistep pathogenesis of cerebrovascular disease. J. Cell. Biochem. 2019, 120, 16467–16482. [Google Scholar] [CrossRef] [Green Version]
- Nath, N.; Prasad, H.K.; Kumar, M. Cerebroprotective effects of hydrogen sulfide in homocysteine-induced neurovascular permeability: Involvement of oxidative stress, arginase, and matrix metalloproteinase-9. J. Cell. Physiol. 2019, 234, 3007–3019. [Google Scholar] [CrossRef]
- Zhong, C.K.; Wang, G.L.; Xu, T.; Zhu, Z.B.; Guo, D.X.; Zheng, X.W.; Wang, A.L.; Bu, X.Q.; Peng, H.; Chen, J.; et al. Tissue inhibitor metalloproteinase-1 and clinical outcomes after acute ischemic stroke. Neurology 2019, 93, E1675–E1685. [Google Scholar] [CrossRef]
- Sole, S.; Petegnief, V.; Gorina, R.; Chamorro, A.; Planas, A.M. Activation of matrix metalloproteinase-3 and agrin cleavage in cerebral ischemia/reperfusion. J. Neuropathol. Exp. Neurol. 2004, 63, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Che, B.; Zhong, C.; Ge, J.; Li, R.; Zhu, Z.; Bu, X.; Xu, T.; Ju, Z.; Liu, J.; Zhang, J.; et al. Serum Matrix Metalloproteinase-9 Is Associated With Depression After Acute Ischemic Stroke. Circ. J. 2019, 83, 2303–2311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and involvement of matrix metalloproteinases in vascular diseases. Front. Biosci. -Landmark 2016, 21, 89–118. [Google Scholar] [CrossRef] [Green Version]
- Guizani, I.; Zidi, W.; Zayani, Y.; Boudiche, S.; Hadj-Taieb, S.; Sanhaji, H.; Zaroui, A.; Mechmeche, R.; Mourali, M.S.; Feki, M.; et al. Matrix metalloproteinase-3 predicts clinical cardiovascular outcomes in patients with coronary artery disease: A 5 years cohort study. Mol. Biol. Rep. 2019, 46, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis—The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [CrossRef]
- Provenzano, M.; Andreucci, M.; Garofalo, C.; Faga, T.; Michael, A.; Ielapi, N.; Grande, R.; Sapienza, P.; de Franciscis, S.; Mastroroberto, P.; et al. The Association of Matrix Metalloproteinases with Chronic Kidney Disease and Peripheral Vascular Disease: A Light at the End of the Tunnel? Biomolecules 2020, 10, 154. [Google Scholar] [CrossRef] [Green Version]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. In Matrix Metalloproteinses and Tissue Remodeling in Health and Disease: Cardiovascular Remodeling; Khalil, R.A., Ed.; Elsevier Academic Press Inc.: San Diego, CA, USA, 2017; Volume 147, pp. 75–100. [Google Scholar]
- Hu, W.; Wei, R.; Wang, L.Y.; Lu, J.Q.; Liu, H.M.; Zhang, W. Correlations of MMP-1, MMP-3, and MMP-12 with the degree of atherosclerosis, plaque stability and cardiovascular and cerebrovascular events. Exp. Ther. Med. 2018, 15, 1994–1998. [Google Scholar] [CrossRef]
- Bouvet, C.; Gilbert, L.A.; Girardot, D.; deBlois, D.; Moreau, P. Different involvement of extracellular matrix components in small and large arteries during chronic NO synthase inhibition. Hypertension 2005, 45, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Pasterkamp, G.; Schoneveld, A.H.; Hijnen, D.J.; de Kleijn, D.P.V.; Teepen, H.; van der Wal, A.C.; Borst, C. Atherosclerotic arterial remodeling and the localization of macrophages and matrix metalloproteases 1, 2 and 9 in the human coronary artery. Atherosclerosis 2000, 150, 245–253. [Google Scholar] [CrossRef]
- Lee, H.S.; Noh, J.Y.; Shin, O.S.; Song, J.Y.; Cheong, H.J.; Kim, W.J. Matrix Metalloproteinase-13 in Atherosclerotic Plaque Is Increased by Influenza A Virus Infection. J. Infect. Dis. 2020, 221, 256–266. [Google Scholar] [CrossRef]
- Moreno-Ajona, D.; Irimia, P.; Rodriguez, J.A.; Garcia-Velloso, M.J.; Lopez-Fidalgo, J.; Fernandez-Alonso, L.; Grochowitz, L.; Munoz, R.; Dominguez, P.; Gallego-Cullere, J.; et al. Elevated circulating metalloproteinase 7 predicts recurrent cardiovascular events in patients with carotid stenosis: A prospective cohort study. BMC Cardiovasc. Disord. 2020, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Cuvelliez, M.; Vandewalle, V.; Brunin, M.; Beseme, O.; Hulot, A.; de Groote, P.; Amouyel, P.; Bauters, C.; Marot, G.; Pinet, F. Circulating proteomic signature of early death in heart failure patients with reduced ejection fraction. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Kozakova, M.; Morizzo, C.; Goncalves, I.; Natali, A.; Nilsson, J.; Palombo, C. Cardiovascular organ damage in type 2 diabetes mellitus: The role of lipids and inflammation. Cardiovasc. Diabetol. 2019, 18, 11. [Google Scholar] [CrossRef]
- Baum, O.; Bernd, J.; Becker, S.; Odriozola, A.; Zuber, B.; Tschanz, S.A.; Zakrzewicz, A.; Egginton, S.; Berkholz, J. Structural Microangiopathies in Skeletal Muscle Related to Systemic Vascular Pathologies in Humans. Front. Physiol. 2020, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Arreguin-Cano, J.A.; Ayerdi-Najera, B.; Tacuba-Saavedra, A.; Navarro-Tito, N.; Davalos-Martinez, A.; Emigdio-Vargas, A.; Barrera-Rodriguez, E.; Blanco-Garcia, N.; Gutierrez-Venegas, G.; Ventura-Molina, E.; et al. MMP-2 salivary activity in type 2 diabetes mellitus patients. Diabetol. Metab. Syndr. 2019, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masciantonio, M.G.; Lee, C.K.S.; Arpino, V.; Mehta, S.; Gill, S.E. The Balance Between Metalloproteinases and TIMPs: Critical Regulator of Microvascular Endothelial Cell Function in Health and Disease. Matrix Met. Tissue Remodel. Health Dis. Cardiovasc. Remodel. 2017, 147, 101–131. [Google Scholar] [CrossRef]
- Huang, M.; Sharma, S.; Zhu, L.X.; Keane, M.P.; Luo, J.; Zhang, L.; Burdick, M.D.; Lin, Y.Q.; Dohadwala, M.; Gardner, B.; et al. IL-7 inhibits fibroblast TGF-beta production and signaling in pulmonary fibrosis. J. Clin. Investig. 2002, 109, 931–937. [Google Scholar] [CrossRef]
- Yu, S.-h.; Liu, L.-j.; Lv, B.; Che, C.-l.; Fan, D.-p.; Wang, L.-f.; Zhang, Y.-m. Inhibition of bleomycin-induced pulmonary fibrosis by bone marrow-derived mesenchymal stem cells might be mediated by decreasing MMP9, TIMP-1, INF- and TGF. Cell Biochem. Funct. 2015, 33, 356–366. [Google Scholar] [CrossRef]
- Edwards, S.T.; Cruz, A.C.; Donnelly, S.; Dazin, P.F.; Schulman, E.S.; Jones, K.D.; Wolters, P.J.; Hoopes, C.; Dolganov, G.M.; Fang, K.C. c-Kit immunophenotyping and metal loproteinase expression profiles of mast cells in interstitial lung diseases. J. Pathol. 2005, 206, 279–290. [Google Scholar] [CrossRef]
- Nareznoi, D.; Konikov-Rozenman, J.; Petukhov, D.; Breuer, R.; Wallach-Dayan, S.B. Matrix Metalloproteinases Retain Soluble FasL-mediated Resistance to Cell Death in Fibrotic-Lung Myofibroblasts. Cells 2020, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, H.; Fujita, M.; Ikegame, S.; Ye, Q.; Inoshima, I.; Harada, E.; Kuwano, K.; Nakanishi, Y. The role of collagenases in experimental pulmonary fibrosis. Pulm. Pharmacol. Ther. 2008, 21, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Corbel, M.; Belleguic, C.; Boichot, E.; Lagente, V. Involvement of gelatinases (MMP-2 and MMP-9) in the development of airway inflammation and pulmonary fibrosis. Cell Biol. Toxicol. 2002, 18, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Summer, R.; Krishna, R.; Schriner, D.; Cuevas-Mora, K.; Sales, D.; Para, R.; Roman, J.; Nieweld, C.; Gochuico, B.R.; Romero, F. Matrix metalloproteinase activity in the lung is increased in Hermansky-Pudlak syndrome. Orphanet J. Rare Dis. 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Frevert, C.W.; Debley, J.S.; Reeves, S.R.; Parks, W.C.; Ziegler, S.F. Interplay of extracellular matrix and leukocytes in lung inflammation. Cell. Immunol. 2017, 312, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dancer, R.C.A.; Wood, A.M.; Thickett, D.R. SERIES “MATRIX METALLOPROTEINASES IN LUNG HEALTH AND DISEASE” Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 1461–1467. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.Y.; Kovkarova-Naumovski, E.; Jara, P.; Parwani, A.; Kass, D.; Ruiz, V.; Lopez-Otin, C.; Rosas, I.O.; Gibson, K.F.; Cabrera, S.; et al. Matrix Metalloproteinase-19 Is a Key Regulator of Lung Fibrosis in Mice and Humans. Am. J. Respir. Crit. Care Med. 2012, 186, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.J.; Huang, T.J.; Zhang, Q.Q.; Zhang, H.Y.; Guo, X.H.; Fan, H.Q.; Li, R.K.; Liu, L.X. Insulin-like growth factor binding protein related protein 1 knockdown attenuates hepatic fibrosis via the regulation of MMPs/TIMPs in mice. Hepatobiliary Pancreat. Dis. Int. 2019, 18, 38–47. [Google Scholar] [CrossRef]
- Duarte, S.; Saber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Balog, S.; Li, Y.; Ogawa, T.; Miki, T.; Saito, T.; French, S.W.; Asahina, K. Development of Capsular Fibrosis Beneath the Liver Surface in Humans and Mice. Hepatology 2020, 71, 291–305. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Zhang, J.; Lu, L.; Feng, M.; Wang, J. Dynamic features of liver fibrogenesis and fibrosis resolution in the absence of matrix metalloproteinase-9. Mol. Med. Rep. 2019, 20, 5239–5248. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, N.; Jacobs-Cacha, C.; Mora-Gutierrez, J.M.; Vergara, A.; Orbe, J.; Soler, M.J. Matrix Metalloproteinases in Diabetic Kidney Disease. J. Clin. Med. 2020, 9, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, Z.Y.; Yilmaz, A.; Pehlivanoglu, C.; Gedikbasi, A.; Yuldiz, M.; Dirican, A.; Bundak, R.; Darendeliler, F.; Emre, S.; Nayir, A. Urine Levels of Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases in Children with Type 1 Diabetes Mellitus. J. Clin. Res. Pediatric Endocrinol. 2019, 11, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Bienias, B.; Sikora, P. Urinary metalloproteinases and tissue inhibitors of metalloproteinases as potential early biomarkers for renal fibrosis in children with nephrotic syndrome. Medicine 2018, 97, 6. [Google Scholar] [CrossRef]
- Mora-Gutierrez, J.M.; Rodriguez, J.A.; Fernandez-Seara, M.A.; Orbe, J.; Escalada, F.J.; Soler, M.J.; Roblero, M.F.S.; Riera, M.; Paramo, J.A.; Garcia-Fernandez, N. MMP-10 is Increased in Early Stage Diabetic Kidney Disease and can be Reduced by Renin-Angiotensin System Blockade. Sci. Rep. 2020, 10, 12. [Google Scholar] [CrossRef]
- Ke, B.; Fan, C.Q.; Yang, L.P.; Fang, X.D. Matrix Metalloproteinases-7 and Kidney Fibrosis. Front. Physiol. 2017, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. Inhibition of MMPs and ADAM/ADAMTS. Biochem. Pharm. 2019, 165, 33–40. [Google Scholar] [CrossRef]
- Naqvi, M.A.; Li, H.; Gao, W.; Naqvi, S.Z.; Jamil, T.; Aimulajiang, K.; Xu, L.; Song, X.; Li, X.; Yan, R. Haemonchus contortus: siRNA mediated knockdown of matrix metalloproteinase 12A (MMP-12) results in reduction of infectivity. Parasit Vectors 2020, 13, 151. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.H.; Chua, S.; Chen, K.H.; Sun, C.K.; Li, Y.C.; Huang, C.R.; Luo, C.W.; Chai, H.T.; Lu, H.I.; Yip, H.K. Role of double knockdown of tPA and MMP-9 on regulating the left ventricular function and remodeling followed by transverse aortic constriction-induced hypertrophic cardiomyopathy in mice. Am. J. Transl. Res. 2018, 10, 2781–2795. [Google Scholar]
- Zhang, W.; Li, Y.; Yang, L.; Zhou, B.; Chen, K.L.; Meng, W.J.; Liu, Y.; Hu, J.K.; Sun, X.F.; Zhou, Z.G. Knockdown of MMP-7 inhibits cell proliferation and enhances sensitivity to 5-Fluorouracil and X-ray irradiation in colon cancer cells. Clin. Exp. Med. 2014, 14, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, Z.N.; Guo, Y.J.; Mei, Y.W. Favorable effects of MMP-9 knockdown in murine herpes simplex encephalitis using small interfering RNA. Neurol. Res. 2010, 32, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Juric, V.; O’Sullivan, C.; Stefanutti, E.; Kovalenko, M.; Greenstein, A.; Barry-Hamilton, V.; Mikaelian, I.; Degenhardt, J.; Yue, P.; Smith, V.; et al. MMP-9 inhibition promotes anti-tumor immunity through disruption of biochemical and physical barriers to T-cell trafficking to tumors. PLoS ONE 2018, 13, e0207255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Atteridge, C.L.; Wang, X.; Lundgren, A.D.; Wu, J.D. The membrane type matrix metalloproteinase MMP14 mediates constitutive shedding of MHC class I chain-related molecule A independent of A disintegrin and metalloproteinases. J. Immunol. 2010, 184, 3346–3350. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar] [CrossRef] [Green Version]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Devel, L.; Garcia, S.; Czarny, B.; Beau, F.; LaJeunesse, E.; Vera, L.; Georgiadis, D.; Stura, E.; Dive, V. Insights from selective non-phosphinic inhibitors of MMP-12 tailored to fit with an S1’ loop canonical conformation. J. Biol. Chem. 2010, 285, 35900–35909. [Google Scholar] [CrossRef] [Green Version]
- Devel, L.; Beau, F.; Amoura, M.; Vera, L.; Cassar-Lajeunesse, E.; Garcia, S.; Czarny, B.; Stura, E.A.; Dive, V. Simple pseudo-dipeptides with a P2’ glutamate: A novel inhibitor family of matrix metalloproteases and other metzincins. J. Biol. Chem. 2012, 287, 26647–26656. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Riedl, R. Inhibitory Antibodies Designed for Matrix Metalloproteinase Modulation. Molecules 2019, 24, 2265. [Google Scholar] [CrossRef] [Green Version]
- Nara, H.; Kaieda, A.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; et al. Discovery of Novel, Highly Potent, and Selective Matrix Metalloproteinase (MMP)-13 Inhibitors with a 1,2,4-Triazol-3-yl Moiety as a Zinc Binding Group Using a Structure-Based Design Approach. J. Med. Chem. 2017, 60, 608–626. [Google Scholar] [CrossRef] [PubMed]
- Hugenberg, V.; Wagner, S.; Kopka, K.; Schafers, M.; Schuit, R.C.; Windhorst, A.D.; Hermann, S. Radiolabeled Selective Matrix Metalloproteinase 13 (MMP-13) Inhibitors: (Radio)Syntheses and in Vitro and First in Vivo Evaluation. J. Med. Chem. 2017, 60, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertini, I.; Fragai, M.; Luchinat, C. Intra- and interdomain flexibility in matrix metalloproteinases: Functional aspects and drug design. Curr. Pharm. Des. 2009, 15, 3592–3605. [Google Scholar] [CrossRef] [PubMed]
- Sela-Passwell, N.; Rosenblum, G.; Shoham, T.; Sagi, I. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim. Biophys. Acta 2010, 1803, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Udi, Y.; Fragai, M.; Grossman, M.; Mitternacht, S.; Arad-Yellin, R.; Calderone, V.; Melikian, M.; Toccafondi, M.; Berezovsky, I.N.; Luchinat, C.; et al. Unraveling hidden regulatory sites in structurally homologous metalloproteases. J. Mol. Biol. 2013, 425, 2330–2346. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Li, J.; Kuscu, C.; Rizzo, R.C.; Deleon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011, 71, 4977–4988. [Google Scholar] [CrossRef] [Green Version]
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012, 72, 2339–2349. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.F.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndinguri, M.W.; Bhowmick, M.; Tokmina-Roszyk, D.; Robichaud, T.K.; Fields, G.B. Peptide-based selective inhibitors of matrix metalloproteinase-mediated activities. Molecules 2012, 17, 14230–14248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, J.M.; Bihan, D.; Slatter, D.A.; Hamaia, S.W.; Packman, L.C.; Knauper, V.; Visse, R.; Farndale, R.W. The recognition of collagen and triple-helical toolkit peptides by MMP-13: Sequence specificity for binding and cleavage. J. Biol. Chem. 2014, 289, 24091–24101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer-Fields, J.L.; Whitehead, J.K.; Li, S.; Hammer, R.P.; Brew, K.; Fields, G.B. Selective modulation of matrix metalloproteinase 9 (MMP-9) functions via exosite inhibition. J. Biol. Chem. 2008, 283, 20087–20095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, M.; Stawikowska, R.; Tokmina-Roszyk, D.; Fields, G.B. Matrix metalloproteinase inhibition by heterotrimeric triple-helical Peptide transition state analogues. Chembiochem 2015, 16, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Czarny, B.; Stura, E.A.; Devel, L.; Vera, L.; Cassar-Lajeunesse, E.; Beau, F.; Calderone, V.; Fragai, M.; Luchinat, C.; Dive, V. Molecular determinants of a selective matrix metalloprotease-12 inhibitor: Insights from crystallography and thermodynamic studies. J. Med. Chem. 2013, 56, 1149–1159. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Calderone, V.; Fragai, M.; Giachetti, A.; Loconte, M.; Luchinat, C.; Maletta, M.; Nativi, C.; Yeo, K.J. Exploring the subtleties of drug-receptor interactions: The case of matrix metalloproteinases. J. Am. Chem. Soc. 2007, 129, 2466–2475. [Google Scholar] [CrossRef]
- Boder, E.T.; Raeeszadeh-Sarmazdeh, M.; Price, J.V. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 2012, 526, 99–106. [Google Scholar] [CrossRef]
- Doerner, A.; Rhiel, L.; Zielonka, S.; Kolmar, H. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014, 588, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur. J. Biochem. 1995, 234, 759–765. [Google Scholar] [CrossRef]
- Martens, E.; Leyssen, A.; Van Aelst, I.; Fiten, P.; Piccard, H.; Hu, J.; Descamps, F.J.; Van den Steen, P.E.; Proost, P.; Van Damme, J.; et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim. Biophys. Acta 2007, 1770, 178–186. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Remacle, A.G.; Golubkov, V.S.; Ingvarsen, S.; Porse, A.; Behrendt, N.; Cieplak, P.; Strongin, A.Y. A monoclonal antibody interferes with TIMP-2 binding and incapacitates the MMP-2-activating function of multifunctional, pro-tumorigenic MMP-14/MT1-MMP. Oncogenesis 2013, 2, e80. [Google Scholar] [CrossRef] [Green Version]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingvarsen, S.; Porse, A.; Erpicum, C.; Maertens, L.; Jurgensen, H.J.; Madsen, D.H.; Melander, M.C.; Gardsvoll, H.; Hoyer-Hansen, G.; Noel, A.; et al. Targeting a single function of the multifunctional matrix metalloprotease MT1-MMP: Impact on lymphangiogenesis. J. Biol. Chem. 2013, 288, 10195–10204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Udi, Y.; Grossman, M.; Solomonov, I.; Dym, O.; Rozenberg, H.; Moreno, V.; Cuniasse, P.; Dive, V.; Arroyo, A.G.; Sagi, I. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure 2015, 23, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Van den Steen, P.E.; Houde, M.; Ilenchuk, T.T.; Opdenakker, G. Inhibitors of gelatinase B/matrix metalloproteinase-9 activity comparison of a peptidomimetic and polyhistidine with single-chain derivatives of a neutralizing monoclonal antibody. Biochem. Pharm. 2004, 67, 1001–1009. [Google Scholar] [CrossRef]
- Appleby, T.C.; Greenstein, A.E.; Hung, M.; Liclican, A.; Velasquez, M.; Villasenor, A.G.; Wang, R.; Wong, M.H.; Liu, X.; Papalia, G.A.; et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J. Biol. Chem. 2017, 292, 6810–6820. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Bhandari, B.R.; Fogel, R.; Onken, J.; Yen, E.; Zhao, X.; Jiang, Z.; Ge, D.; Xin, Y.; Ye, Z.; et al. Randomised clinical trial: A phase 1, dose-ranging study of the anti-matrix metalloproteinase-9 monoclonal antibody GS-5745 versus placebo for ulcerative colitis. Aliment. Pharm. Ther. 2016, 44, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Gossage, D.L.; Cieslarova, B.; Ap, S.; Zheng, H.; Xin, Y.; Lal, P.; Chen, G.; Smith, V.; Sundy, J.S. Phase 1b Study of the Safety, Pharmacokinetics, and Disease-related Outcomes of the Matrix Metalloproteinase-9 Inhibitor Andecaliximab in Patients With Rheumatoid Arthritis. Clin. Ther. 2018, 40, 156–165.e155. [Google Scholar] [CrossRef]
- Baer, P.N. Periostat: Low-dose doxycycline. A commentary. Periodontal Clin. Investig. 1998, 20, 5. [Google Scholar]
- Caton, J.G. Evaluation of Periostat for patient management. Compend. Contin. Educ. Dent. 1999, 20, 451–456, 458–460, 462, quiz 463. [Google Scholar]
- Kocer, S.S.; Walker, S.G.; Zerler, B.; Golub, L.M.; Simon, S.R. Metalloproteinase inhibitors, nonantimicrobial chemically modified tetracyclines, and ilomastat block Bacillus anthracis lethal factor activity in viable cells. Infect. Immun 2005, 73, 7548–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirt, I.; Bodurth, A.; Lohmann, R.; Rusthoven, J.; Belanger, K.; Young, V.; Wainman, N.; Stewar, W.; Eisenhauer, E.; National Cancer Institute of Canada Clinical Trials, G. Phase II study of marimastat (BB-2516) in malignant melanoma: A clinical and tumor biopsy study of the National Cancer Institute of Canada Clinical Trials Group. Investig. New Drugs 2002, 20, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Ratnikov, B.I.; Strongin, A.Y. Prinomastat, a hydroxamate inhibitor of matrix metalloproteinases, has a complex effect on migration of breast carcinoma cells. Int. J. Cancer 2003, 104, 533–541. [Google Scholar] [CrossRef]
- Ge, S.L.; Gong, W.H.; Zhang, C.X.; Zhang, L.; Han, P.H.; Zhang, S.Q.; Feng, J.B.; Zhou, D.C. Protective role of MMP-9 inhibitor batimastat in acute lung injury after cardiopulmonary bypass. Zhonghua Wai Ke Za Zhi 2010, 48, 57–61. [Google Scholar]
- Prontera, C.; Mariani, B.; Rossi, C.; Poggi, A.; Rotilio, D. Inhibition of gelatinase A (MMP-2) by batimastat and captopril reduces tumor growth and lung metastases in mice bearing Lewis lung carcinoma. Int. J. Cancer 1999, 81, 761–766. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Bhandari, B.R.; Randall, C.; Younes, Z.H.; Romanczyk, T.; Xin, Y.; Wendt, E.; Chai, H.; McKevitt, M.; Zhao, S.; et al. Andecaliximab [Anti-matrix Metalloproteinase-9] Induction Therapy for Ulcerative Colitis: A Randomised, Double-Blind, Placebo-Controlled, Phase 2/3 Study in Patients With Moderate to Severe Disease. J. Crohns Colitis 2018, 12, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; Siegel, C.A.; Friedenberg, K.A.; Younes, Z.H.; Seidler, U.; Bhandari, B.R.; Wang, K.; Wendt, E.; McKevitt, M.; Zhao, S.; et al. A Phase 2, Randomized, Placebo-Controlled Study Evaluating Matrix Metalloproteinase-9 Inhibitor, Andecaliximab, in Patients With Moderately to Severely Active Crohn’s Disease. J. Crohns Colitis 2018, 12, 1014–1020. [Google Scholar] [CrossRef]
- Lopez, T.; Nam, D.H.; Kaihara, E.; Mustafa, Z.; Ge, X. Identification of highly selective MMP-14 inhibitory Fabs by deep sequencing. Biotechnol. Bioeng. 2017. [Google Scholar] [CrossRef]
- Nam, D.H.; Fang, K.; Rodriguez, C.; Lopez, T.; Ge, X. Generation of inhibitory monoclonal antibodies targeting matrix metalloproteinase-14 by motif grafting and CDR optimization. Protein Eng. Des. Sel. 2017, 30, 113–118. [Google Scholar] [CrossRef]
- Pearson, J.R.; Zurita, F.; Tomas-Gallardo, L.; Diaz-Torres, A.; Diaz de la Loza Mdel, C.; Franze, K.; Martin-Bermudo, M.D.; Gonzalez-Reyes, A. ECM-Regulator timp Is Required for Stem Cell Niche Organization and Cyst Production in the Drosophila Ovary. PLoS Genet. 2016, 12, e1005763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, J.; Robinson, J.; Mehner, C.; Hockla, A.; Miller, E.; Radisky, D.C.; Radisky, E.S. PEGylation extends circulation half-life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases-1 (TIMP-1). PLoS ONE 2012, 7, e50028. [Google Scholar] [CrossRef] [Green Version]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.S.; Diaz, R.; Korets, L.; Hodgson, J.G.; Coussens, L.M. TIMP-1 alters susceptibility to carcinogenesis. Cancer Res. 2004, 64, 952–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedez, L.; McMarlin, A.J.; Kingma, D.W.; Bennett, T.A.; Stetler-Stevenson, M.; Stetler-Stevenson, W.G. Tissue inhibitor of metalloproteinase-1 alters the tumorigenicity of Burkitt’s lymphoma via divergent effects on tumor growth and angiogenesis. Am. J. Pathol. 2001, 158, 1207–1215. [Google Scholar] [CrossRef]
- Elezkurtaj, S.; Kopitz, C.; Baker, A.H.; Perez-Canto, A.; Arlt, M.J.; Khokha, R.; Gansbacher, B.; Anton, M.; Brand, K.; Kruger, A. Adenovirus-mediated overexpression of tissue inhibitor of metalloproteinases-1 in the liver: Efficient protection against T-cell lymphoma and colon carcinoma metastasis. J. Gene Med. 2004, 6, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Brand, K. Cancer gene therapy with tissue inhibitors of metalloproteinases (TIMPs). Curr. Gene Ther. 2002, 2, 255–271. [Google Scholar] [CrossRef]
- Meng, Q.; Malinovskii, V.; Huang, W.; Hu, Y.; Chung, L.; Nagase, H.; Bode, W.; Maskos, K.; Brew, K. Residue 2 of TIMP-1 is a major determinant of affinity and specificity for matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1’ residue of substrate. J. Biol. Chem. 1999, 274, 10184–10189. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Chen, Y.; Chung, L.; Nagase, H.; Brew, K. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP-1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J. Biol. Chem. 2003, 278, 9831–9834. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Rapti, M.; Knauper, V.; Murphy, G. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J. Biol. Chem. 2004, 279, 17562–17569. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Rapti, M.; Murphy, G. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J. Biol. Chem. 2003, 278, 40224–40230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.A.; Hutton, M.; Vogt, G.; Rapti, M.; Knauper, V.; Carr, M.D.; Murphy, G. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases-2 with matrix metalloproteinase-14. J. Biol. Chem. 2001, 276, 32966–32970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahudhanapati, H.; Zhang, Y.; Sidhu, S.S.; Brew, K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): Identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J. Biol. Chem. 2011, 286, 31761–31770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamze, A.B.; Wei, S.; Bahudhanapati, H.; Kota, S.; Acharya, K.R.; Brew, K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007, 16, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, O.; Shirian, J.; Grossman, M.; Lebendiker, M.; Sagi, I.; Shifman, J. Affinity- and specificity-enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS ONE 2014, 9, e93712. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Tworowski, D.; Dym, O.; Lee, M.H.; Levy, Y.; Murphy, G.; Sagi, I. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010, 49, 6184–6192. [Google Scholar] [CrossRef]
- Lee, M.H.; Atkinson, S.; Rapti, M.; Handsley, M.; Curry, V.; Edwards, D.; Murphy, G. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)-1 against native membrane type 1 matrix metalloproteinase (MT1-MMP) in a cell-based environment. Cancer Lett. 2010, 290, 114–122. [Google Scholar] [CrossRef]
- Arkadash, V.; Yosef, G.; Shirian, J.; Cohen, I.; Horev, Y.; Grossman, M.; Sagi, I.; Radisky, E.S.; Shifman, J.M.; Papo, N. Development of high-affinity and high-specificity inhibitors of metalloproteinase 14 through computational design and directed evolution. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [Green Version]
- Koslawsky, D.; Zaretsky, M.; Alcalay, R.; Mazor, O.; Aharoni, A.; Papo, N. A bi-specific inhibitor targeting IL-17A and MMP-9 reduces invasion and motility in MDA-MB-231 cells. Oncotarget 2018, 9, 28500–28513. [Google Scholar] [CrossRef]
- Yosef, G.; Arkadash, V.; Papo, N. Targeting the MMP-14/MMP-2/integrin alphavbeta3 axis with multispecific N-TIMP2-based antagonists for cancer therapy. J. Biol. Chem. 2018, 293, 13310–13326. [Google Scholar] [CrossRef] [Green Version]
- Chirco, R.; Liu, X.W.; Jung, K.K.; Kim, H.R. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 2006, 25, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Tissue Inhibitors of Metalloproteinases in Cell Signaling: Metalloproteinase-Independent Biological Activities. Sci. Signal. 2008, 1, re6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Lutolf, M.P.; Raeber, G.P.; Zisch, A.H.; Tirelli, N.; Hubbell, J.A. Cell-responsive synthetic hydrogels. Adv. Mater. 2003, 15, 888–892. [Google Scholar] [CrossRef]
- Schultz, K.M.; Anseth, K.S. Monitoring degradation of matrix metalloproteinases-cleavable PEG hydrogels via multiple particle tracking microrheology. Soft Matter 2013, 9, 1570–1579. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Sun, H.; Lei, W.; Tang, Y.; Hong, S.; Yang, H.; Tay, F.R.; Huang, C. MMP-8-Responsive Polyethylene Glycol Hydrogel for Intraoral Drug Delivery. J. Dent. Res. 2019, 98, 564–571. [Google Scholar] [CrossRef]
- Secret, E.; Crannell, K.E.; Kelly, S.J.; Villancio-Wolter, M.; Andrew, J.S. Matrix metalloproteinase-sensitive hydrogel microparticles for pulmonary drug delivery of small molecule drugs or proteins. J. Mater. Chem. B 2015, 3, 5629–5634. [Google Scholar] [CrossRef]
- Yan, X.; Gemeinhart, R.A. Cisplatin delivery from poly(acrylic acid-co-methyl methacrylate) microparticles. J. Control Release 2005, 106, 198–208. [Google Scholar] [CrossRef]
- Ribeiro, J.S.; Bordini, E.A.F.; Ferreira, J.A.; Mei, L.; Dubey, N.; Fenno, J.C.; Piva, E.; Lund, R.G.; Schwendeman, A.; Bottino, M.C. Injectable MMP-Responsive Nanotube-Modified Gelatin Hydrogel for Dental Infection Ablation. ACS Appl Mater. Interfaces 2020, 12, 16006–16017. [Google Scholar] [CrossRef]
- Yao, Q.; Kou, L.; Tu, Y.; Zhu, L. MMP-Responsive ‘Smart’ Drug Delivery and Tumor Targeting. Trends Pharm. Sci. 2018, 39, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Shargh, V.H.; Hondermarck, H.; Liang, M. Gelatin-albumin hybrid nanoparticles as matrix metalloproteinases-degradable delivery systems for breast cancer therapy. Nanomedicine (Lond.) 2017, 12, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.P.; Carlini, A.S.; Hu, D.; Barback, C.V.; Rush, A.M.; Hall, D.J.; Orr, G.; Gianneschi, N.C. Enzyme-directed assembly of nanoparticles in tumors monitored by in vivo whole animal imaging and ex vivo super-resolution fluorescence imaging. J. Am. Chem. Soc. 2013, 135, 18710–18713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.H.; Luo, G.F.; Qiu, W.X.; Lei, Q.; Liu, L.H.; Zheng, D.W.; Hong, S.; Cheng, S.X.; Zhang, X.Z. Tumor-Triggered Drug Release with Tumor-Targeted Accumulation and Elevated Drug Retention To Overcome Multidrug Resistance. Chem. Mater. 2016, 28, 6742–6752. [Google Scholar] [CrossRef]
- Kinoh, H.; Inoue, M.; Washizawa, K.; Yamamoto, T.; Fujikawa, S.; Tokusumi, Y.; Iida, A.; Nagai, Y.; Hasegawa, M. Generation of a recombinant Sendai virus that is selectively activated and lyses human tumor cells expressing matrix metalloproteinases. Gene Ther. 2004, 11, 1137–1145. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMP Inhibitor | Type | Clinical Trial | Specificity | Reference |
|---|---|---|---|---|
| Periostat (doxycycline hydraxate) | Small molecule | FDA approved | Broad spectrum | [222,223] |
| IImostat; GM6001(Hydroxamate derivative) | Small molecule | Phase I, II | MMP-1, -2, -9 | [224] |
| Marimastat (BB-2516) | Small molecule | Phase II | Broad spectrum | [225] |
| Prinomastat (AG-3340) | Small molecule | Phase III | MMP-1, -2, -9 | [226] |
| Batimastat (BB-94) | peptide | Phase I | Broad spectrum | [227,228] |
| GA-5745/andecaliximab | mAb | Phase I, II, III | MMP-9 | [221,229,230] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells 2020, 9, 1313. https://doi.org/10.3390/cells9051313
Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells. 2020; 9(5):1313. https://doi.org/10.3390/cells9051313
Chicago/Turabian StyleRaeeszadeh-Sarmazdeh, Maryam, Linh D. Do, and Brianne G. Hritz. 2020. "Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics" Cells 9, no. 5: 1313. https://doi.org/10.3390/cells9051313
APA StyleRaeeszadeh-Sarmazdeh, M., Do, L. D., & Hritz, B. G. (2020). Metalloproteinases and Their Inhibitors: Potential for the Development of New Therapeutics. Cells, 9(5), 1313. https://doi.org/10.3390/cells9051313