The Survey of Cells Responsible for Heterotopic Ossification Development in Skeletal Muscles—Human and Mouse Models

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Heterotopic Ossification as a Clinical Issue
2.1. Traumatic HO
2.2. Surgery-Induced HO
2.3. Neurogenic HO
2.4. Genetic HO
2.5. Diagnostic Imaging
2.6. Biomarkers
2.7. Prophylaxis
2.8. Treatment
3. Heterotopic Ossification Precursor Cells
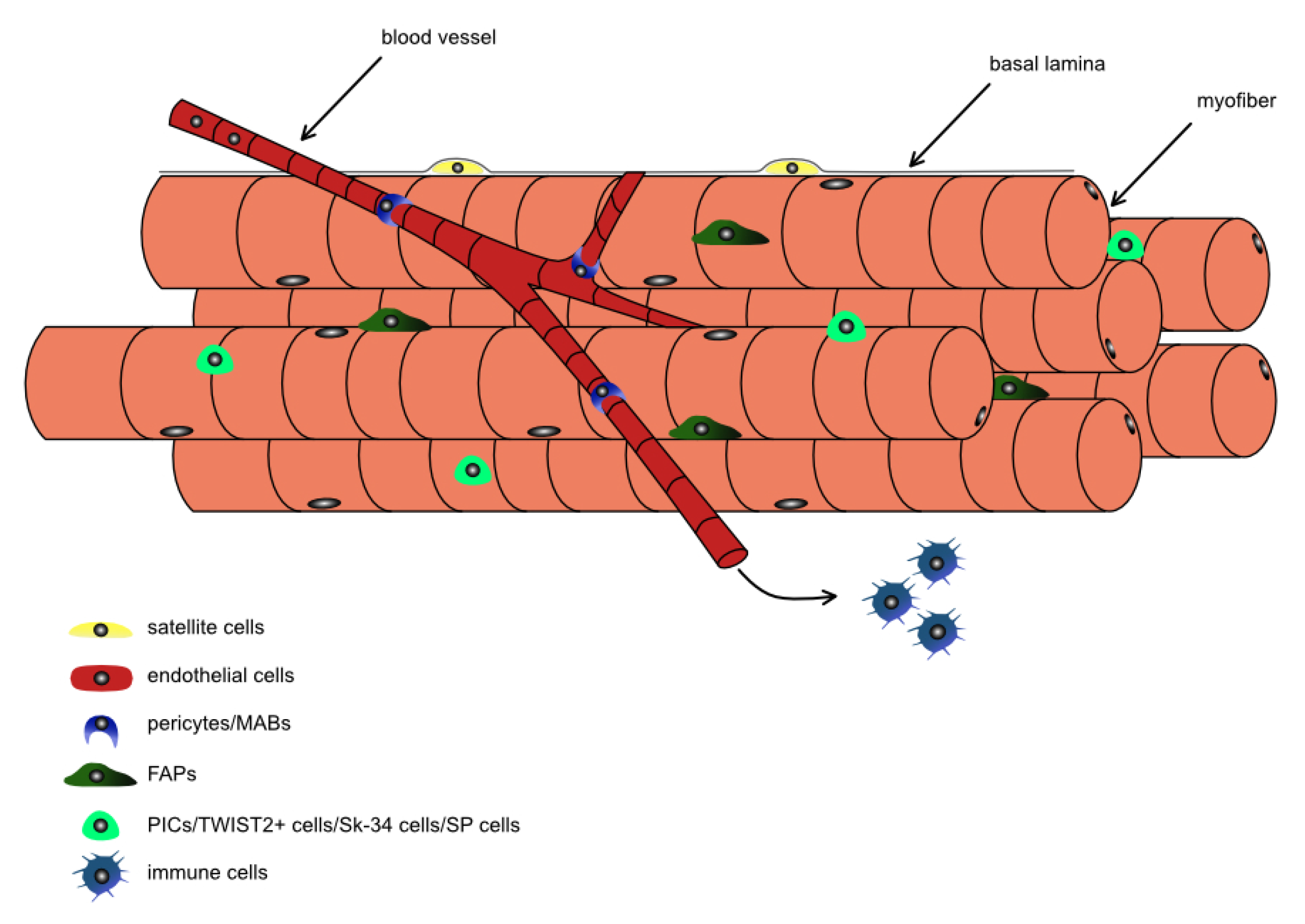
3.1. Stem and Progenitor Cells in Skeletal Muscles
3.2. The Osteogenic Potential of Stem and Progenitor Cells Residing in Skeletal Muscle—In Vitro Studies
3.3. The Cells Directly Participating in Heterotropic Ossification Formation In Vivo
4. Possible Signaling Mechanisms of Ectopic Osteogenesis in Skeletal Muscles
5. Future Therapeutic Options
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACVR1/ALK2 | Activin A receptor type 1/activin-like kinase 2 |
| ALP | alkaline phosphatase |
| BAF60c | 60 KDa BRG-1/Brm-Associated Factor Subunit C |
| BAP | bone-specific isoform of alkaline phosphatase |
| BMP | bone morphogenetic protein |
| CD | cluster of differentiation |
| CDC6 | cell division cycle 6 protein |
| COL-1 | collagen alpha 1 |
| COX-2 | cyclooxygenase-2 |
| CRP | C-reactive protein |
| CTDA | cervical total disc arthroplasty |
| CTX-1 | type I collagen cross-linked C-telopeptide |
| Dlx5 | Distal-Less Homeobox 5 |
| DVT | deep venous thrombosis |
| ECM | extracellular matrix |
| ENO3 | enolase 3 |
| ERK | extracellular signal-regulated kinase |
| FAP | fibro-adipogenic progenitors |
| FOP | fibrodysplasia ossificans progressive |
| FSP1 | fibroblast-specific protein 1 |
| GLAST | glial high affinity glutamate transporter |
| GLI1 | glioma-associated oncogene 1 |
| GNAS | guanine nucleotide-binding protein, subunit alpha |
| HDAC | histone deacetylase |
| HGF | hepatocyte growth factor |
| HIF1α | hypoxia-inducible factor 1α |
| HLA | human leukocyte antigen |
| HO | heterotopic ossification |
| ID-1 | DNA-Binding Protein Inhibitor ID-1 |
| IFN γ | interferon γ |
| IFOPA | international FOP Association |
| IGF1 | insulin-like growth factor 1 |
| IL | Interleukin |
| iPSC | induced pluripotent stem cell |
| LTA | lipoteichoic acid |
| MAPK | mitogen-activated protein kinases |
| MCK | muscle creatine kinase |
| MCM2 | minichromosome maintenance complex component 2 |
| MCP-1 | monocyte chemoattractant protein-1 |
| MHC | myosin heavy chains |
| miRNA | microRNA |
| MIS | micro-invasive surgery |
| MRF | myogenic regulatory factor |
| MRI | magnetic resonance imaging |
| MRSA | methicillin-resistant Staphylococcus aureus |
| MSC | mesenchymal stromal cell |
| mTOR | mammalian target of rapamycin |
| NF-κB | Nuclear factor-κB |
| NG2 | neural-glial antigen 2 |
| NHO | neurogenic heterotopic ossification |
| NSAID | nonsteroidal anti-inflammatory drug |
| NT-3 | neurotrophin-3 |
| OC/BGLAP | osteocalcin/Bone Gamma-Carboxyglutamate Protein |
| OMD | Osteomodulin |
| OSX/Sp7 | Osterix |
| Pax7 | paired box transcription factor 7 |
| PDGFRα | platelet derived growth factor receptor α |
| PGE2 | prostaglandin E2 |
| PIC | PW1 interstitial cell |
| POH | progressive osseous heteroplasia |
| Prx1 | peroxiredoxin Prx1 |
| ROM | range of motion |
| RT | Radiotherapy |
| RUNX2 | runt-related transcription factor 2 |
| SC | satellite cell |
| SCA1 | stem cell antigen 1 |
| SCI | spinal cord injury |
| Scx | Scleraxis |
| Sox6 | SRY-Box Transcription Factor 6 |
| SP cell | side population cell |
| SPECT | single-photon emission computed tomography |
| SWI/SNF | SWItch/Sucrose NonFermentable |
| TAB1 | TAK1 binding protein |
| TBI | traumatic brain injury |
| TBX18 | T-box transcription factor 18 |
| TCF1 | T cell factor 1 |
| TGFβ | transforming growth factor β |
| THR | total hip replacement |
| TIE2 | angiopoietin receptor |
| TLR | toll-like receptor |
| TNF-α | tumor necrosis factor-α |
| TRAF6 | TNF Receptor Associated Factor 6 |
| TWIST2 | Twist Basic Helix-Loop-Helix Transcription Factor 2/DERMO1 |
| VE-cadherin | vascular endothelial cadherin (Cdh15) |
| VEGF | vascular endothelial growth factor |
| VTE | venous thromboembolism |
| WNT | wingless/integrated |
References
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3, e10172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauth, A.; Giles, E.; Potter, B.K.; Nesti, L.J.; O’brien, F.P.; Bosse, M.J.; Anglen, J.O.; Mehta, S.; Ahn, J.; Miclau, T.; et al. Heterotopic ossification in orthopaedic trauma. J. Orthop. Trauma 2012, 26, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kuijk, A.A.; Geurts, A.C.H.; van Kuppevelt, H.J.M. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord. 2002, 40, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Łęgosz, P.; Drela, K.; Pulik, Ł.; Sarzyńska, S.; Małdyk, P. Challenges of heterotopic ossification-Molecular background and current treatment strategies. Clin. Exp. Pharm. Physiol. 2018, 45, 1229–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees-Shepard, J.B.; Goldhamer, D.J. Stem cells and heterotopic ossification: Lessons from animal models. Bone 2018, 109, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Convente, M.R.; Wang, H.; Pignolo, R.J.; Kaplan, F.S.; Shore, E.M. The immunological contribution to heterotopic ossification disorders. Curr. Osteoporos. Rep. 2015, 13, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Levesque, J.P.; Sims, N.A.; Pettit, A.R.; Alexander, K.A.; Tseng, H.W.; Torossian, F.; Genet, F.; Lataillade, J.J.; Le Bousse-Kerdiles, M.C. Macrophages Driving Heterotopic Ossification: Convergence of Genetically-Driven and Trauma-Driven Mechanisms. J. Bone Miner. Res. 2018, 33, 365–366. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, M.; Huber, A.K.; Hwang, C.; Carson, W.F.; Menon, R.; Li, J.; Vasquez, K.; Pagani, C.; Patel, N.; Li, S.; et al. Regulation of heterotopic ossification by monocytes in a mouse model of aberrant wound healing. Nat. Commun. 2020, 11, 722. [Google Scholar] [CrossRef]
- Kan, C.; Yang, J.; Na, D.; Xu, Y.; Yang, B.; Zhao, H.; Lu, H.; Li, Y.; Zhang, K.; McGuire, T.L.; et al. Inhibition of immune checkpoints prevents injury-induced heterotopic ossification. Bone Res. 2019, 7, 33. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Chu, J.; Ao, X.; Jiang, T.; Bin, Y.; Huang, M.; Zhang, Z. Macrophage-derived neurotrophin-3 promotes heterotopic ossification in rats. Lab. Investig. 2020. [Google Scholar] [CrossRef]
- Bossche, L.V.; Vanderstraeten, G. Heterotopic ossification: A review. J. Rehabil. Med. 2005, 37, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, K.L.; Hebela, N.; Keenan, M.A.; Pignolo, R.J. Histopathology of periarticular non-hereditary heterotopic ossification. Bone 2018, 109, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, J.L.; Crues, J.V.; Reimers, C.D. Muscle Imaging in Health and Disease; Springer: New York, NY, USA, 1996; ISBN 978-1-4612-2314-6. [Google Scholar]
- Ohlmeier, M.; Krenn, V.; Thiesen, D.M.; Sandiford, N.A.; Gehrke, T.; Citak, M. Heterotopic Ossification in Orthopaedic and Trauma surgery: A Histopathological Ossification Score. Sci. Rep. 2019, 9, 18401. [Google Scholar] [CrossRef]
- Barfield, W.R.; Holmes, R.E.; Hartsock, L.A. Heterotopic Ossification in Trauma. Orthop. Clin. N. Am. 2017, 48, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, N.; Stapley, S.; Grover, L. Post-Traumatic Heterotopic Ossification: An Old Problem in Need of New Solutions. J. Orthop. Res. 2018, 36, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-C.; Yang, J.-Y.; Chuang, S.-S.; Huang, C.-Y.; Yang, S.-Y. Heterotopic ossification in burns: Our experience and literature reviews. Burns 2009, 35, 857–862. [Google Scholar] [CrossRef]
- Thefenne, L.; de Brier, G.; Leclerc, T.; Jourdan, C.; Nicolas, C.; Truffaut, S.; Lapeyre, E.; Genet, F. Two new risk factors for heterotopic ossification development after severe burns. PLoS ONE 2017, 12, e0182303. [Google Scholar] [CrossRef]
- Hong, C.C.; Nashi, N.; Hey, H.W.; Chee, Y.H.; Murphy, D. Clinically relevant heterotopic ossification after elbow fracture surgery: A risk factors study. Orthop. Traumatol. Surg. Res. 2015, 101, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Firoozabadi, R.; O’Mara, T.J.; Swenson, A.; Agel, J.; Beck, J.D.; Routt, M. Risk Factors for the Development of Heterotopic Ossification After Acetabular Fracture Fixation. Clin. Orthop. Relat. Res. 2014, 472, 3383–3388. [Google Scholar] [CrossRef] [Green Version]
- Dey, D.; Wheatley, B.M.; Cholok, D.; Agarwal, S.; Yu, P.B.; Levi, B.; Davis, T.A. The traumatic bone: Trauma-induced heterotopic ossification. Transl. Res. 2017, 186, 95–111. [Google Scholar] [CrossRef]
- Orchard, G.R.; Paratz, J.D.; Blot, S.; Roberts, J.A. Risk Factors in Hospitalized Patients With Burn Injuries for Developing Heterotopic Ossification—A Retrospective Analysis. J. Burn Care Res. 2015, 36, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Bemenderfer, T.B.; Davis, W.H.; Anderson, R.B.; Wing, K.; Escudero, M.I.; Waly, F.; Penner, M. Heterotopic Ossification in Total Ankle Arthroplasty: Case Series and Systematic Review. J. Foot Ankle Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, F.; Chen, W.; Zhang, Q.; Liu, S.; Zhang, Y. Incidence and risk factors for heterotopic ossification after total hip arthroplasty: A meta-analysis. Arch. Orthop. Trauma Surg. 2015, 135, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, X.; Bai, W.; Shen, X.; Yuan, W. Prevalence of heterotopic ossification after cervical total disc arthroplasty: A meta-analysis. Eur. Spine J. 2012, 21, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-B.; Cho, Y.-J.; Park, J.-K.; Song, E.-K.; Yoon, T.-R.; Seon, J.-K. Heterotopic Ossification After Primary Total Ankle Arthroplasty. JBJS 2011, 93, 751–758. [Google Scholar] [CrossRef]
- Łęgosz, P.; Sarzyńska, S.; Pulik, Ł.; Stępiński, P.; Niewczas, P.; Kotela, A.; Małdyk, P. Heterotopic ossification and clinical results after total hip arthroplasty using the anterior minimally invasive and anterolateral approaches. Arch. Med. Sci. 2018. [Google Scholar] [CrossRef]
- Anthonissen, J.; Ossendorf, C.; Hock, J.L.; Steffen, C.T.; Goetz, H.; Hofmann, A.; Rommens, P.M. The role of muscular trauma in the development of heterotopic ossification after hip surgery: An animal-model study in rats. Injury 2016, 47, 613–616. [Google Scholar] [CrossRef]
- Yi, S.; Shin, D.A.; Kim, K.N.; Choi, G.; Shin, H.C.; Kim, K.S.; Yoon, D.H. The predisposing factors for the heterotopic ossification after cervical artificial disc replacement. Spine J. 2013, 13, 1048–1054. [Google Scholar] [CrossRef]
- Kocic, M.; Lazovic, M.; Mitkovic, M.; Djokic, B. Clinical significance of the heterotopic ossification after total hip arthroplasty. Orthopedics 2010, 33, 16. [Google Scholar] [CrossRef]
- Sundseth, J.; Jacobsen, E.A.; Kolstad, F.; Sletteberg, R.O.; Nygaard, O.P.; Johnsen, L.G.; Pripp, A.H.; Andresen, H.; Fredriksli, O.A.; Myrseth, E.; et al. Heterotopic ossification and clinical outcome in nonconstrained cervical arthroplasty 2 years after surgery: The Norwegian Cervical Arthroplasty Trial (NORCAT). Eur. Spine J. 2016, 25, 2271–2278. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, E.F.; Sundaram, M. Heterotopic ossification: A review. Skelet. Radiol. 2005, 34, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Nalbantoglu, M.; Tuncer, O.G.; Acik, M.E.; Matur, Z.; Altunrende, B.; Ozgonenel, E.; Ozgonenel, L. Neurogenic heterotopic ossification in Guillain-Barre syndrome: A rare case report. J. Musculoskelet. Neuronal Interact. 2020, 20, 160–164. [Google Scholar] [PubMed]
- Zhang, Y.; Zhan, Y.; Kou, Y.; Yin, X.; Wang, Y.; Zhang, D. Identification of biological pathways and genes associated with neurogenic heterotopic ossification by text mining. PeerJ 2020, 8, e8276. [Google Scholar] [CrossRef] [PubMed]
- Pek, C.H.; Lim, M.C.; Yong, R.; Wong, H.P. Neurogenic heterotopic ossification after a stroke: Diagnostic and radiological challenges. Singap. Med. J. 2014, 55, e119–e122. [Google Scholar] [CrossRef] [Green Version]
- Reznik, J.; Biros, E.; Marshall, R.; Jelbart, M.; Milanese, S.; Gordon, S.; Galea, M. Prevalence and risk-factors of neurogenic heterotopic ossification in traumatic spinal cord and traumatic brain injured patients admitted to specialised units in Australia. J. Musculoskelet. Neuronal Interact. 2014, 14, 19–28. [Google Scholar]
- Citak, M.; Suero, E.M.; Backhaus, M.; Aach, M.; Godry, H.; Meindl, R.; Schildhauer, T.A. Risk Factors for Heterotopic Ossification in Patients With Spinal Cord Injury: A Case-Control Study of 264 Patients. Spine 2012, 37, 1953–1957. [Google Scholar] [CrossRef]
- Sullivan, M.P.; Torres, S.J.; Mehta, S.; Ahn, J. Heterotopic ossification after central nervous system trauma: A current review. Bone Jt. Res. 2013, 2, 51–57. [Google Scholar] [CrossRef]
- Citak, M.; Grasmücke, D.; Suero, E.M.; Cruciger, O.; Meindl, R.; Schildhauer, T.A.; Aach, M. The roles of serum alkaline and bone alkaline phosphatase levels in predicting heterotopic ossification following spinal cord injury. Spinal Cord. 2016, 54, 368–370. [Google Scholar] [CrossRef] [Green Version]
- Dizdar, D.; Tiftik, T.; Kara, M.; Tunç, H.; Ersöz, M.; Akkuş, S. Risk factors for developing heterotopic ossification in patients with traumatic brain injury. Brain Inj. 2013, 27, 807–811. [Google Scholar] [CrossRef]
- Ohlmeier, M.; Suero, E.M.; Aach, M.; Meindl, R.; Schildhauer, T.A.; Citak, M. Muscle localization of heterotopic ossification following spinal cord injury. Spine J. 2017, 17, 1519–1522. [Google Scholar] [CrossRef]
- Suero, E.M.; Meindl, R.; Schildhauer, T.A.; Citak, M. Clinical Prediction Rule for Heterotopic Ossification of the Hip in Patients with Spinal Cord Injury. Spine 2018, 43, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Johns, J.S.; Cifu, D.X.; Keyser-Marcus, L.; Jolles, P.R.; Fratkin, M.J. Impact of Clinically Significant Heterotopic Ossification on Functional Outcome after Traumatic Brain Injury. J. Head Trauma Rehabil. 1999, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Adegbite, N.S.; Xu, M.; Kaplan, F.S.; Shore, E.M.; Pignolo, R.J. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am. J. Med. Genet. Part A 2008, 146, 1788–1796. [Google Scholar] [CrossRef] [Green Version]
- Valer, J.A.; Sanchez-de-Diego, C.; Pimenta-Lopes, C.; Rosa, J.L.; Ventura, F. ACVR1 Function in Health and Disease. Cells 2019, 8, 1366. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Ramaswamy, G.; Fong, J.T.; Shore, E.M.; Kaplan, F.S. Progressive osseous heteroplasia: Diagnosis, treatment, and prognosis. Appl. Clin. Genet. 2015, 8, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Cheung, K.; Kile, S.; Fitzpatrick, M.A.; De Cunto, C.; Al Mukaddam, M.; Hsiao, E.C.; Baujat, G.; Delai, P.; Eekhoff, E.M.W.; et al. Self-reported baseline phenotypes from the International Fibrodysplasia Ossificans Progressiva (FOP) Association Global Registry. Bone 2020. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia Ossificans Progressiva: Clinical and Genetic Aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, F.S.; Le Merrer, M.; Glaser, D.L.; Pignolo, R.J.; Goldsby, R.E.; Kitterman, J.A.; Groppe, J.; Shore, E.M. Fibrodysplasia ossificans progressiva. Best Pr. Res. Clin. Rheumatol. 2008, 22, 191–205. [Google Scholar] [CrossRef]
- Towler, O.W.; Shore, E.M.; Kaplan, F.S. Skeletal malformations and developmental arthropathy in individuals who have fibrodysplasia ossificans progressiva. Bone 2020, 130, 115116. [Google Scholar] [CrossRef]
- Mujtaba, B.; Taher, A.; Fiala, M.J.; Nassar, S.; Madewell, J.E.; Hanafy, A.K.; Aslam, R. Heterotopic ossification: Radiological and pathological review. Radiol. Oncol. 2019, 53, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, M.A.; Dannoon, S.; Elgazzar, A.H. The added value of SPECT-CT in the detection of heterotopic ossification on bone scintigraphy. Skelet. Radiol. 2020, 49, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.C.; Passarelli, M.C.; Dario, V.; Lebani, B.R.; Monteiro, P.H.S.; Ramos, C.D. The use of spect/ct in the evaluation of heterotopic ossification in para/tetraplegics. Acta Ortopédica Bras. 2014, 22, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botman, E.; Raijmakers, P.G.H.M.; Yaqub, M.; Teunissen, B.; Netelenbos, C.; Lubbers, W.; Schwarte, L.A.; Micha, D.; Bravenboer, N.; Schoenmaker, T.; et al. Evolution of heterotopic bone in fibrodysplasia ossificans progressiva: An [18F]NaF PET/CT study. Bone 2019, 124, 1–6. [Google Scholar] [CrossRef]
- Rosteius, T.; Suero, E.; Grasmücke, D.; Aach, M.; Gisevius, A.; Ohlmeier, M.; Meindl, R.; Schildhauer, T.; Citak, M. The sensitivity of ultrasound screening examination in detecting heterotopic ossification following spinal cord injury. Spinal Cord. 2017, 55, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Shehab, D.; Elgazzar, A.H.; Collier, B.D. Heterotopic ossification. J. Nucl. Med. 2002, 43, 346–353. [Google Scholar]
- Łęgosz, P.; Pulik, Ł.; Stępiński, P.; Janowicz, J.; Wirkowska, A.; Kotela, A.; Sarzyńska, S.; Małdyk, P. The Use of Type I Collagen Cross-Linked C-Telopeptide (CTX-1) as a Biomarker Associated with the Formation of Periprosthetic Ossifications Following Total Hip JoInt. Arthroplasty. Ann. Clin. Lab. Sci. 2018, 48, 183–190. [Google Scholar]
- Sung Hsieh, H.H.; Chung, M.T.; Allen, R.M.; Ranganathan, K.; Habbouche, J.; Cholok, D.; Butts, J.; Kaura, A.; Tiruvannamalai-Annamalai, R.; Breuler, C.; et al. Evaluation of Salivary Cytokines for Diagnosis of both Trauma-Induced and Genetic Heterotopic Ossification. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, J.A.; Potter, B.K.; Polfer, E.M.; Safford, S.D.; Elster, E.A. Do Inflammatory Markers Portend Heterotopic Ossification and Wound Failure in Combat Wounds? Clin. Orthop. Relat. Res. 2014, 472, 2845–2854. [Google Scholar] [CrossRef] [Green Version]
- Edsberg, L.E.; Crowgey, E.L.; Osborn, P.M.; Wyffels, J.T. A survey of proteomic biomarkers for heterotopic ossification in blood serum. J. Orthop. Surg. Res. 2017, 12, 69. [Google Scholar] [CrossRef]
- Sun, Y.; Cai, J.; Yu, S.; Chen, S.; Li, F.; Fan, C. MiR-630 Inhibits Endothelial-Mesenchymal Transition by Targeting Slug in Traumatic Heterotopic Ossification. Sci. Rep. 2016, 6, 22729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, C.; Lv, Z.; Zhang, C.; Jiao, Y. Regulatory effect of miR-421 on humeral fracture and heterotopic ossification in elderly patients. Exp. Ther. Med. 2019, 17, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Liu, S.; Yu, B.; Zhu, J.; Ruan, H.; Tang, T.; Fan, C. miR-203 inhibits the traumatic heterotopic ossification by targeting Runx2. Cell Death Dis. 2016, 7, e2436. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, D.; Pan, X.; Zhang, X.; Mi, J.; Rui, Y. Celecoxib cannot inhibit the progression of initiated traumatic heterotopic ossification. J. Shoulder Elb. Surg. 2019, 28, 2379–2385. [Google Scholar] [CrossRef] [PubMed]
- Joice, M.; Vasileiadis, G.I.; Amanatullah, D.F. Non-steroidal anti-inflammatory drugs for heterotopic ossification prophylaxis after total hip arthroplasty. Bone Jt. J. 2018, 100, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Zheng, Q.; Li, H.; Qian, S.; Zhang, B.; Pan, Z. Selective COX-2 inhibitor versus nonselective COX-1 and COX-2 inhibitor in the prevention of heterotopic ossification after total hip arthroplasty: A meta-analysis of randomised trials. Int. Orthop. 2011, 35, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Kan, S.L.; Yang, B.; Ning, G.Z.; Chen, L.X.; Li, Y.L.; Gao, S.J.; Chen, X.Y.; Sun, J.C.; Feng, S.Q. Nonsteroidal Anti-inflammatory Drugs as Prophylaxis for Heterotopic Ossification after Total Hip Arthroplasty: A Systematic Review and Meta-Analysis. Medicine 2015, 94, e828. [Google Scholar] [CrossRef]
- Winkler, S.; Wagner, F.; Weber, M.; Matussek, J.; Craiovan, B.; Heers, G.; Springorum, H.R.; Grifka, J.; Renkawitz, T. Current therapeutic strategies of heterotopic ossification—A survey amongst orthopaedic and trauma departments in Germany. BMC Musculoskelet. Disord. 2015, 16, 313. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.C.; Rodgers, A.; Clark, T.; Gray, H.; Reid, I.R.; Dunn, L.; MacMahon, S.W. A systematic survey of 13 randomized trials of non-steroidal anti-inflammatory drugs for the prevention of heterotopic bone formation after major hip surgery. Acta Orthop. Scand. 2000, 71, 122–128. [Google Scholar] [CrossRef]
- Strauss, J.B.; Chen, S.S.; Shah, A.P.; Coon, A.B.; Dickler, A. Cost of radiotherapy versus NSAID administration for prevention of heterotopic ossification after total hip arthroplasty. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 1460–1464. [Google Scholar] [CrossRef]
- Vavken, P.; Dorotka, R. Economic evaluation of NSAID and radiation to prevent heterotopic ossification after hip surgery. Arch. Orthop. Trauma Surg. 2011, 131, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Milakovic, M.; Popovic, M.; Raman, S.; Tsao, M.; Lam, H.; Chow, E. Radiotherapy for the prophylaxis of heterotopic ossification: A systematic review and meta-analysis of randomized controlled trials. Radiother. Oncol. 2015, 116, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Gao, H.; Huang, S.; Wang, G.; Jiang, X.; Li, J.; Shou, Z. NSAIDs combined with radiotherapy to prevent heterotopic ossification after total hip arthroplasty. Zhongguo Gu Shang China J. Orthop. Traumatol. 2018, 31, 538–542. [Google Scholar]
- Pakos, E.E.; Papadopoulos, D.V.; Gelalis, I.D.; Tsantes, A.G.; Gkiatas, I.; Kosmas, D.; Tsekeris, P.G.; Xenakis, T.A. Is prophylaxis for heterotopic ossification with radiation therapy after THR associated with early loosening or carcinogenesis? Hip. Int. 2019, 1120700019842724. [Google Scholar] [CrossRef] [PubMed]
- Seavey, J.G.; Wheatley, B.M.; Pavey, G.J.; Tomasino, A.M.; Hanson, M.A.; Sanders, E.M.; Dey, D.; Moss, K.L.; Potter, B.K.; Forsberg, J.A. Early local delivery of vancomycin suppresses ectopic bone formation in a rat model of trauma-induced heterotopic ossification. J. Orthop. Res. 2017, 35, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Vasileiadis, G.I.; Sakellariou, V.I.; Kelekis, A.; Galanos, A.; Soucacos, P.N.; Papagelopoulos, P.J.; Babis, G.C. Prevention of heterotopic ossification in cases of hypertrophic osteoarthritis submitted to total hip arthroplasty. Etidronate or Indomethacin? J. Musculoskelet. Neuronal Interact. 2010, 10, 159–165. [Google Scholar]
- Haykal, T.; Kheiri, B.; Zayed, Y.; Barbarawi, M.; Miran, M.S.; Chahine, A.; Katato, K.; Bachuwa, G. Aspirin for venous thromboembolism prophylaxis after hip or knee arthroplasty: An updated meta-analysis of randomized controlled trials. J. Orthop. 2019, 16, 294–302. [Google Scholar] [CrossRef]
- Brouwer, K.M.; Lindenhovius, A.L.; de Witte, P.B.; Jupiter, J.B.; Ring, D. Resection of heterotopic ossification of the elbow: A comparison of ankylosis and partial restriction. J. Hand Surg. 2010, 35, 1115–1119. [Google Scholar] [CrossRef]
- Almangour, W.; Schnitzler, A.; Salga, M.; Debaud, C.; Denormandie, P.; Genet, F. Recurrence of heterotopic ossification after removal in patients with traumatic brain injury: A systematic review. Ann. Phys. Rehabil. Med. 2016, 59, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Genet, F.; Marmorat, J.L.; Lautridou, C.; Schnitzler, A.; Mailhan, L.; Denormandie, P. Impact of late surgical intervention on heterotopic ossification of the hip after traumatic neurological injury. J. Bone Jt. Surg. Br. Vol. 2009, 91, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.H.; Lim, T.K.; Lee, H.I.; Park, M.J. Surgical treatment of elbow stiffness caused by post-traumatic heterotopic ossification. J. Shoulder Elb. Surg. 2013, 22, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Jayasundara, J.A.; Punchihewa, G.L.; de Alwis, D.S.; Renuka, M.D. Short-term outcome after resection of neurogenic heterotopic ossification around the hips and elbow following encephalitis. Singap. Med. J. 2012, 53, e97–e100. [Google Scholar]
- Pontell, M.E.; Sparber, L.S.; Chamberlain, R.S. Corrective and reconstructive surgery in patients with postburn heterotopic ossification and bony ankylosis: An evidence-based approach. J. Burn Care Res. 2015, 36, 57–69. [Google Scholar] [CrossRef]
- Rubayi, S.; Gabbay, J.; Kruger, E.; Ruhge, K. The Modified Girdlestone Procedure With Muscle Flap for Management of Pressure Ulcers and Heterotopic Ossification of the Hip Region in Spinal Injury Patients: A 15-Year Review With Long-term Follow-up. Ann. Plast. Surg. 2016, 77, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Pansard, E.; Schnitzler, A.; Lautridou, C.; Judet, T.; Denormandie, P.; Genet, F. Heterotopic ossification of the shoulder after central nervous system lesion: Indications for surgery and results. J. Shoulder Elb. Surg. 2013, 22, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Mitsionis, G.I.; Lykissas, M.G.; Kalos, N.; Paschos, N.; Beris, A.E.; Georgoulis, A.D.; Xenakis, T.A. Functional outcome after excision of heterotopic ossification about the knee in ICU patients. Int. Orthop. 2009, 33, 1619–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akman, S.; Sonmez, M.M.; Erturer, R.E.; Seckin, M.F.; Kara, A.; Ozturk, I. The results of surgical treatment for posttraumatic heterotopic ossification and ankylosis of the elbow. Acta Orthop. Traumatol. Turc. 2010, 44, 206–211. [Google Scholar] [CrossRef]
- Denormandie, P.; de l’Escalopier, N.; Gatin, L.; Grelier, A.; Genêt, F. Resection of neurogenic heterotopic ossification (NHO) of the hip. Orthop. Traumatol. Surg. Res. 2018, 104, S121–S127. [Google Scholar] [CrossRef]
- Łęgosz, P.; Stępiński, P.; Pulik, Ł.; Kotela, A.; Małdyk, P. Total hip replacement vs femoral neck osteotomy in the treatment of heterotopic ossifications, neurogenic in the IV degree by scale Brooker–comparison of treatment results. Chir. Narz. Ruchu Ortop. Pol. 2017, 82, 28–40. [Google Scholar]
- Forcina, L.; Miano, C.; Pelosi, L.; Musaro, A. An Overview about the Biology of Skeletal Muscle Satellite Cells. Curr. Genom. 2019, 20, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Brzoska, E.; Ciemerych, M.A.; Przewozniak, M.; Zimowska, M. Regulation of muscle stem cells activation: The role of growth factors and extracellular matrix. Vitam. Horm. 2011, 87, 239–276. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Zammit, P.S. Satellite cells are essential for skeletal muscle regeneration: The cell on the edge returns centre stage. Development 2012, 139, 2845–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Gagan, J.; Dutta, A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol. Cell Biol. 2011, 31, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. Embo J. 2012, 31, 301–316. [Google Scholar] [CrossRef]
- Chazaud, B. Inflammation during skeletal muscle regeneration and tissue remodeling: Application to exercise-induced muscle damage management. Immunol. Cell Biol. 2016, 94, 140–145. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y.; Zhao, L.; Zeng, Z.; Xiao, W.; Chen, P. Macrophage depletion impairs skeletal muscle regeneration: The roles of regulatory factors for muscle regeneration. Cell Biol. Int. 2017, 41, 228–238. [Google Scholar] [CrossRef]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Galvin, C.D.; Hardiman, O.; Nolan, C.M. IGF-1 receptor mediates differentiation of primary cultures of mouse skeletal myoblasts. Mol. Cell Endocrinol. 2003, 200, 19–29. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S. Macrophage-Derived IGF-1 Is a Potent Coordinator of Myogenesis and Inflammation in Regenerating Muscle. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1134–1135. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.E.; Jin, B.; Li, Y.P. TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am. J. Physiol. Cell Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef] [PubMed]
- Latroche, C.; Weiss-Gayet, M.; Chazaud, B. Investigating the Vascular Niche: Three-Dimensional Co-culture of Human Skeletal Muscle Stem Cells and Endothelial Cells. Methods Mol. Biol. 2019, 2002, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Abou-Khalil, R.; Mounier, R.; Chazaud, B. Regulation of myogenic stem cell behavior by vessel cells: The “menage a trois” of satellite cells, periendothelial cells and endothelial cells. Cell Cycle 2010, 9, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christov, C.; Chretien, F.; Abou-Khalil, R.; Bassez, G.; Vallet, G.; Authier, F.J.; Bassaglia, Y.; Shinin, V.; Tajbakhsh, S.; Chazaud, B.; et al. Muscle satellite cells and endothelial cells: Close neighbors and privileged partners. Mol. Biol. Cell 2007, 18, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, D.; Judson, R.N.; Low, M.; Lee, S.; Zhang, E.; Hopkins, C.; Xu, P.; Lenzi, A.; Rossi, F.M.; Lemos, D.R. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 2016, 17, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Mathew, S.J.; Hansen, J.M.; Merrell, A.J.; Murphy, M.M.; Lawson, J.A.; Hutcheson, D.A.; Hansen, M.S.; Angus-Hill, M.; Kardon, G. Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 2011, 138, 371–384. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wosczyna, M.N.; Konishi, C.T.; Perez Carbajal, E.E.; Wang, T.T.; Walsh, R.A.; Gan, Q.; Wagner, M.W.; Rando, T.A. Mesenchymal Stromal Cells Are Required for Regeneration and Homeostatic Maintenance of Skeletal Muscle. Cell Rep. 2019, 27, 2029–2035.e5. [Google Scholar] [CrossRef] [Green Version]
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Musaro, A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells 2019, 8, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierzejewski, B.; Archacka, K.; Grabowska, I.; Florkowska, A.; Ciemerych, M.A.; Brzoska, E. Human and mouse skeletal muscle stem and progenitor cells in health and disease. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A.; Delbono, O. Pericytes are Essential for Skeletal Muscle Formation. Stem Cell Rev. Rep. 2015, 11, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Mintz, A.; Delbono, O. Type-1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 305, C1098–C1113. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Galvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, J.; Yao, Y. Pericytes in Skeletal Muscle. Adv. Exp. Med. Biol. 2019, 1122, 59–72. [Google Scholar] [CrossRef]
- Persichini, T.; Funari, A.; Colasanti, M.; Sacchetti, B. Clonogenic, myogenic progenitors expressing MCAM/CD146 are incorporated as adventitial reticular cells in the microvascular compartment of human post-natal skeletal muscle. PLoS ONE 2017, 12, e0188844. [Google Scholar] [CrossRef] [Green Version]
- Kostallari, E.; Baba-Amer, Y.; Alonso-Martin, S.; Ngoh, P.; Relaix, F.; Lafuste, P.; Gherardi, R.K. Pericytes in the myovascular niche promote post-natal myofiber growth and satellite cell quiescence. Development 2015, 142, 1242–1253. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.J.; Pannerec, A.; Cadot, B.; Parlakian, A.; Besson, V.; Gomes, E.R.; Marazzi, G.; Sassoon, D.A. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 2010, 12, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, T.; Akatsuka, A.; Ando, K.; Nakamura, Y.; Matsuzawa, H.; Hotta, T.; Roy, R.R.; Edgerton, V.R. Identification of myogenic-endothelial progenitor cells in the interstitial spaces of skeletal muscle. J. Cell Biol. 2002, 157, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Garry, G.A.; Li, S.; Bezprozvannaya, S.; Sanchez-Ortiz, E.; Chen, B.; Shelton, J.M.; Jaichander, P.; Bassel-Duby, R.; Olson, E.N. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat. Cell Biol. 2017, 19, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gussoni, E.; Soneoka, Y.; Strickland, C.D.; Buzney, E.A.; Khan, M.K.; Flint, A.F.; Kunkel, L.M.; Mulligan, R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature 1999, 401, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Sampaolesi, M.; Pisati, F.; Meregalli, M.; D’Antona, G.; Tonlorenzi, R.; Porretti, L.; Gavina, M.; Mamchaoui, K.; et al. Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J. Clin. Investig. 2004, 114, 182–195. [Google Scholar] [CrossRef]
- Asakura, A.; Komaki, M.; Rudnicki, M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68, 245–253. [Google Scholar] [CrossRef]
- Castiglioni, A.; Hettmer, S.; Lynes, M.D.; Rao, T.N.; Tchessalova, D.; Sinha, I.; Lee, B.T.; Tseng, Y.H.; Wagers, A.J. Isolation of progenitors that exhibit myogenic/osteogenic bipotency in vitro by fluorescence-activated cell sorting from human fetal muscle. Stem Cell Rep. 2014, 2, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes-Camboa, N.; Cattaneo, P.; Sun, Y.; Moore-Morris, T.; Gu, Y.; Dalton, N.D.; Rockenstein, E.; Masliah, E.; Peterson, K.L.; Stallcup, W.B.; et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell 2017, 20, 345–359.e5. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, B.; Funari, A.; Remoli, C.; Giannicola, G.; Kogler, G.; Liedtke, S.; Cossu, G.; Serafini, M.; Sampaolesi, M.; Tagliafico, E.; et al. No Identical “Mesenchymal Stem Cells” at Different Times and Sites: Human Committed Progenitors of Distinct Origin and Differentiation Potential Are Incorporated as Adventitial Cells in Microvessels. Stem Cell Rep. 2016, 6, 897–913. [Google Scholar] [CrossRef] [Green Version]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef] [Green Version]
- Pannerec, A.; Formicola, L.; Besson, V.; Marazzi, G.; Sassoon, D.A. Defining skeletal muscle resident progenitors and their cell fate potentials. Development 2013, 140, 2879–2891. [Google Scholar] [CrossRef] [Green Version]
- Tamaki, T.; Akatsuka, A.; Yoshimura, S.; Roy, R.R.; Edgerton, V.R. New fiber formation in the interstitial spaces of rat skeletal muscle during postnatal growth. J. Histochem. Cytochem. 2002, 50, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, T.; Uchiyama, Y.; Okada, Y.; Ishikawa, T.; Sato, M.; Akatsuka, A.; Asahara, T. Functional recovery of damaged skeletal muscle through synchronized vasculogenesis, myogenesis, and neurogenesis by muscle-derived stem cells. Circulation 2005, 112, 2857–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negroni, E.; Riederer, I.; Chaouch, S.; Belicchi, M.; Razini, P.; Di Santo, J.; Torrente, Y.; Butler-Browne, G.S.; Mouly, V. In vivo myogenic potential of human CD133+ muscle-derived stem cells: A quantitative study. Moleculus 2009, 17, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Torrente, Y.; Belicchi, M.; Marchesi, C.; D’Antona, G.; Cogiamanian, F.; Pisati, F.; Gavina, M.; Giordano, R.; Tonlorenzi, R.; Fagiolari, G.; et al. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transpl. 2007, 16, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Kaji, D.A.; Tan, Z.; Johnson, G.L.; Huang, W.; Vasquez, K.; Lehoczky, J.A.; Levi, B.; Cheah, K.S.E.; Huang, A.H. Cellular Plasticity in Musculoskeletal Development, Regeneration, and Disease. J. Orthop. Res. 2020, 38, 708–718. [Google Scholar] [CrossRef]
- Lounev, V.Y.; Ramachandran, R.; Wosczyna, M.N.; Yamamoto, M.; Maidment, A.D.; Shore, E.M.; Glaser, D.L.; Goldhamer, D.J.; Kaplan, F.S. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Jt. Surg. Am. 2009, 91, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef]
- Kan, L.; Liu, Y.; McGuire, T.L.; Berger, D.M.; Awatramani, R.B.; Dymecki, S.M.; Kessler, J.A. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells 2009, 27, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.; Marini, S.; Huber, A.K.; Stepien, D.M.; Sorkin, M.; Loder, S.; Pagani, C.A.; Li, J.; Visser, N.D.; Vasquez, K.; et al. Mesenchymal VEGFA induces aberrant differentiation in heterotopic ossification. Bone Res. 2019, 7, 36. [Google Scholar] [CrossRef]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Ikemoto-Uezumi, M.; Nakatani, M.; Morita, M.; Yamaguchi, A.; Yamada, H.; Nishino, I.; Hamada, Y.; et al. Identification and characterization of PDGFRalpha+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014, 5, e1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regard, J.B.; Malhotra, D.; Gvozdenovic-Jeremic, J.; Josey, M.; Chen, M.; Weinstein, L.S.; Lu, J.; Shore, E.M.; Kaplan, F.S.; Yang, Y. Activation of Hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013, 19, 1505–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Loder, S.; Brownley, C.; Cholok, D.; Mangiavini, L.; Li, J.; Breuler, C.; Sung, H.H.; Li, S.; Ranganathan, K.; et al. Inhibition of Hif1alpha prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl. Acad. Sci. USA 2016, 113, E338–E347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Kan, C.; Chen, L.; Hu, Y.; Ding, N.; Li, Y.; McGuire, T.L.; Lu, H.; Kessler, J.A.; Kan, L. Gli1-labeled adult mesenchymal stem/progenitor cells and hedgehog signaling contribute to endochondral heterotopic ossification. Bone 2018, 109, 71–79. [Google Scholar] [CrossRef]
- Kan, L.; Peng, C.Y.; McGuire, T.L.; Kessler, J.A. Glast-expressing progenitor cells contribute to heterotopic ossification. Bone 2013, 53, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Loder, S.J.; Cholok, D.; Peterson, J.; Li, J.; Breuler, C.; Cameron Brownley, R.; Hsin Sung, H.; Chung, M.T.; Kamiya, N.; et al. Scleraxis-Lineage Cells Contribute to Ectopic Bone Formation in Muscle and Tendon. Stem Cells 2017, 35, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Komori, T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem. Cell Biol. 2018, 149, 313–323. [Google Scholar] [CrossRef]
- Montecino, M.; Stein, G.; Stein, J.; Zaidi, K.; Aguilar, R. Multiple levels of epigenetic control for bone biology and pathology. Bone 2015, 81, 733–738. [Google Scholar] [CrossRef] [Green Version]
- Uchida, K.; Yayama, T.; Cai, H.X.; Nakajima, H.; Sugita, D.; Guerrero, A.R.; Kobayashi, S.; Yoshida, A.; Chen, K.B.; Baba, H. Ossification process involving the human thoracic ligamentum flavum: Role of transcription factors. Arthritis Res. Ther. 2011, 13, R144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Shen, Q.; Xue, T.; Yu, C. Heterotopic ossification induced by Achilles tenotomy via endochondral bone formation: Expression of bone and cartilage related genes. Bone 2010, 46, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of Runx2 function by TGF-beta through recruitment of class II histone deacetylases by Smad3. Embo J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Hong, S.H.; Bae, S.C. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 2002, 21, 7156–7163. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Chen, G.; Li, Y.P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-beta/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005. [Google Scholar] [CrossRef] [Green Version]
- Grenier, G.; Leblanc, E.; Faucheux, N.; Lauzier, D.; Kloen, P.; Hamdy, R.C. BMP-9 expression in human traumatic heterotopic ossification: A case report. Skelet. Muscle 2013, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Ikeya, M.; Hino, K.; Horigome, K.; Fukuta, M.; Watanabe, M.; Nagata, S.; Yamamoto, T.; Otsuka, T.; Toguchida, J. New Protocol to Optimize iPS Cells for Genome Analysis of Fibrodysplasia Ossificans Progressiva. Stem Cells 2015, 33, 1730–1742. [Google Scholar] [CrossRef]
- Medici, D.; Olsen, B.R. The role of endothelial-mesenchymal transition in heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Loder, S.; Li, J.; Brownley, C.; Peterson, J.R.; Oluwatobi, E.; Drake, J.; Cholok, D.; Ranganathan, K.; Sung, H.H.; et al. Diminished Chondrogenesis and Enhanced Osteoclastogenesis in Leptin-Deficient Diabetic Mice (ob/ob) Impair Pathologic, Trauma-Induced Heterotopic Ossification. Stem Cells Dev. 2015, 24, 2864–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Ren, B.; Shi, F.; Hua, P.; Lin, H. BMP and mTOR signaling in heterotopic ossification: Does their crosstalk provide therapeutic opportunities? J. Cell Biochem. 2019, 120, 12108–12122. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Ecsit-ement on the crossroads of Toll and BMP signal transduction. Genes Dev. 2003, 17, 2855–2859. [Google Scholar] [CrossRef] [Green Version]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem 2005, 280, 33132–33140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J. Bone Miner. Res. 2014, 29, 1960–1969. [Google Scholar] [CrossRef]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Wang, X.; Lin, H. The hypoxic microenvironment: A driving force for heterotopic ossification progression. Cell Commun. Signal. 2020, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.R.; De La Rosa, S.; Sun, H.; Eboda, O.; Cilwa, K.E.; Donneys, A.; Morris, M.; Buchman, S.R.; Cederna, P.S.; Krebsbach, P.H.; et al. Burn injury enhances bone formation in heterotopic ossification model. Ann. Surg. 2014, 259, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Croes, M.; Kruyt, M.C.; Boot, W.; Pouran, B.; Braham, M.V.; Pakpahan, S.A.; Weinans, H.; Vogely, H.C.; Fluit, A.C.; Dhert, W.J.; et al. The role of bacterial stimuli in inflammation-driven bone formation. Eur. Cells Mater. 2019, 37, 402–419. [Google Scholar] [CrossRef]
- Wang, H.; Behrens, E.M.; Pignolo, R.J.; Kaplan, F.S. ECSIT links TLR and BMP signaling in FOP connective tissue progenitor cells. Bone 2018, 109, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wi, S.M.; Moon, G.; Kim, J.; Kim, S.T.; Shim, J.H.; Chun, E.; Lee, K.Y. TAK1-ECSIT-TRAF6 complex plays a key role in the TLR4 signal to activate NF-kappaB. J. Biol. Chem 2014, 289, 35205–35214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-gamma agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Hino, K.; Zhao, C.; Horigome, K.; Nishio, M.; Okanishi, Y.; Nagata, S.; Komura, S.; Yamada, Y.; Toguchida, J.; Ohta, A. An mTOR signaling modulator suppressed heterotopic ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Rep. 2018, 11, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.R.; De La Rosa, S.; Eboda, O.; Cilwa, K.E.; Agarwal, S.; Buchman, S.R.; Cederna, P.S.; Xi, C.; Morris, M.D.; Herndon, D.N.; et al. Treatment of heterotopic ossification through remote ATP hydrolysis. Sci. Transl. Med. 2014, 6, 255ra132. [Google Scholar] [CrossRef] [Green Version]
- Hannallah, D.; Peng, H.; Young, B.; Usas, A.; Gearhart, B.; Huard, J. Retroviral delivery of Noggin inhibits the formation of heterotopic ossification induced by BMP-4, demineralized bone matrix, and trauma in an animal model. J. Bone Jt. Surg. Am. Vol. 2004, 86, 80–91. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Pignolo, R.J.; Al Mukaddam, M.M.; Shore, E.M. Hard targets for a second skeleton: Therapeutic horizons for fibrodysplasia ossificans progressiva (FOP). Expert Opin. Orphan Drugs 2017, 5, 291–294. [Google Scholar] [CrossRef]
- Xue, T.; Mao, Z.; Lin, L.; Hou, Y.; Wei, X.; Fu, X.; Zhang, J.; Yu, C. Non-virus-mediated transfer of siRNAs against Runx2 and Smad4 inhibit heterotopic ossification in rats. Gene Ther. 2010, 17, 370–379. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulik, Ł.; Mierzejewski, B.; Ciemerych, M.A.; Brzóska, E.; Łęgosz, P. The Survey of Cells Responsible for Heterotopic Ossification Development in Skeletal Muscles—Human and Mouse Models. Cells 2020, 9, 1324. https://doi.org/10.3390/cells9061324
Pulik Ł, Mierzejewski B, Ciemerych MA, Brzóska E, Łęgosz P. The Survey of Cells Responsible for Heterotopic Ossification Development in Skeletal Muscles—Human and Mouse Models. Cells. 2020; 9(6):1324. https://doi.org/10.3390/cells9061324
Chicago/Turabian StylePulik, Łukasz, Bartosz Mierzejewski, Maria A. Ciemerych, Edyta Brzóska, and Paweł Łęgosz. 2020. "The Survey of Cells Responsible for Heterotopic Ossification Development in Skeletal Muscles—Human and Mouse Models" Cells 9, no. 6: 1324. https://doi.org/10.3390/cells9061324
APA StylePulik, Ł., Mierzejewski, B., Ciemerych, M. A., Brzóska, E., & Łęgosz, P. (2020). The Survey of Cells Responsible for Heterotopic Ossification Development in Skeletal Muscles—Human and Mouse Models. Cells, 9(6), 1324. https://doi.org/10.3390/cells9061324