Comparison of Multiscale Imaging Methods for Brain Research

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Immunolabeling of Mouse Brain Sections
2.3. Culturing, Transfection and Immunostaining of Primary Rat Hippocampal neurons
2.4. Confocal Microscopy
2.5. STED Microscopy
2.6. Adaptive Optics (AO) z-STED Microscopy
2.7. Widefield Fluorescence and Structured Illumination Optical Sectioning (SIOS) Microscopy
2.8. Spinning Disc Confocal Microscopy (SPDM)
2.9. Airyscan Microscopy
2.10. HyVolution Imaging
2.11. Stereo Microscopy
2.12. Slide Scanner Microscopy
2.13. Lattice-SIM Microscopy
2.14. Deconvolution
3. Results
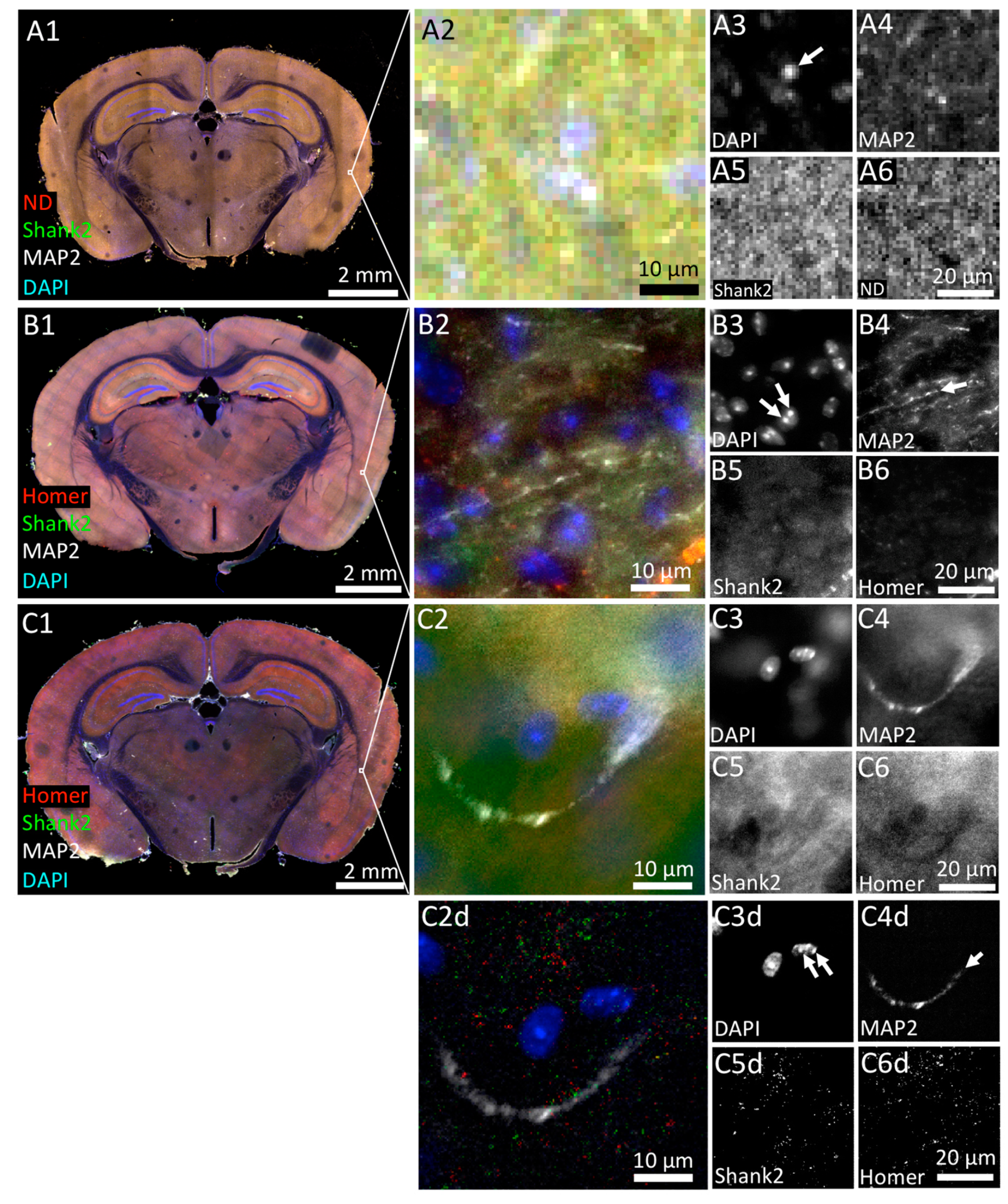
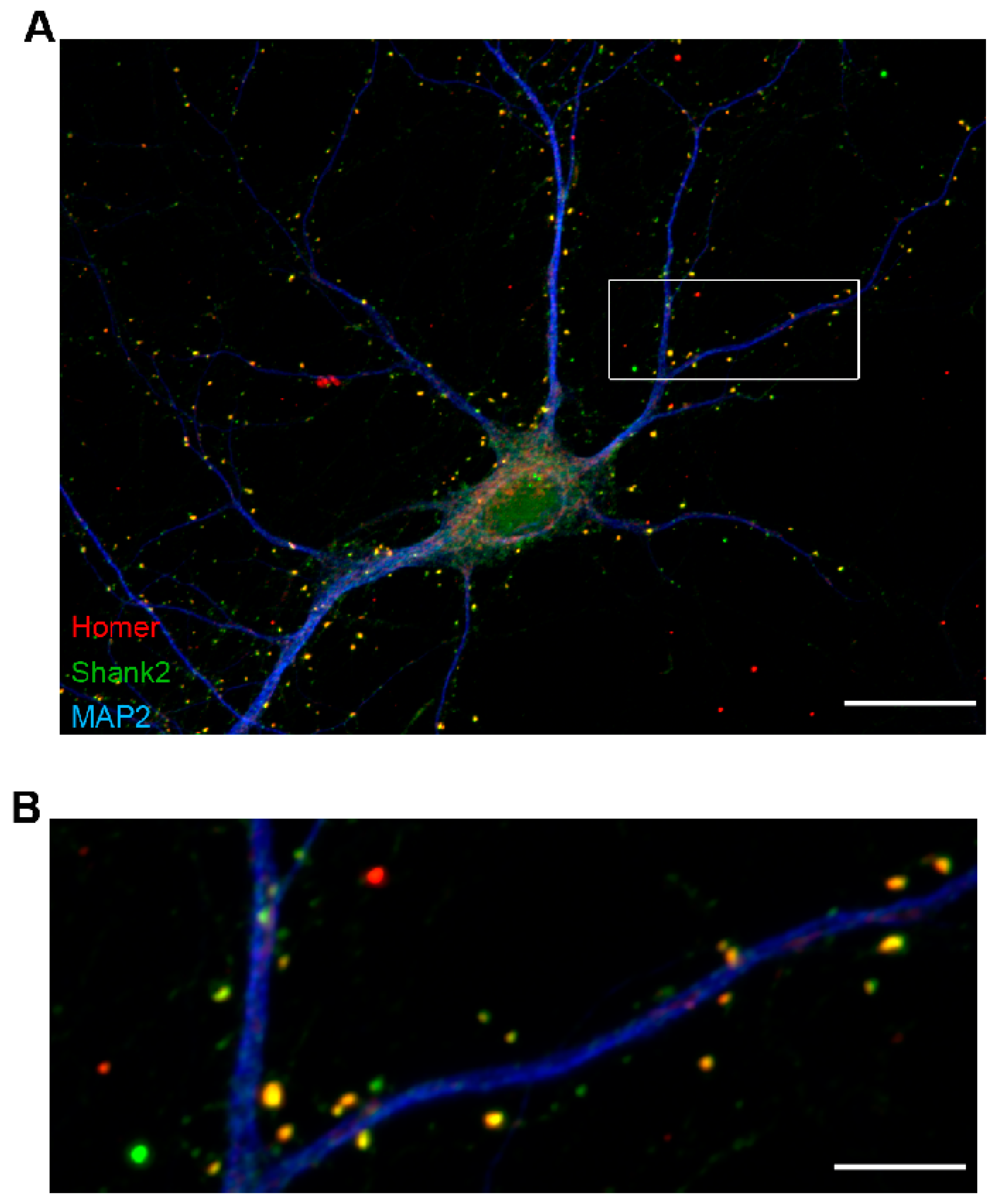
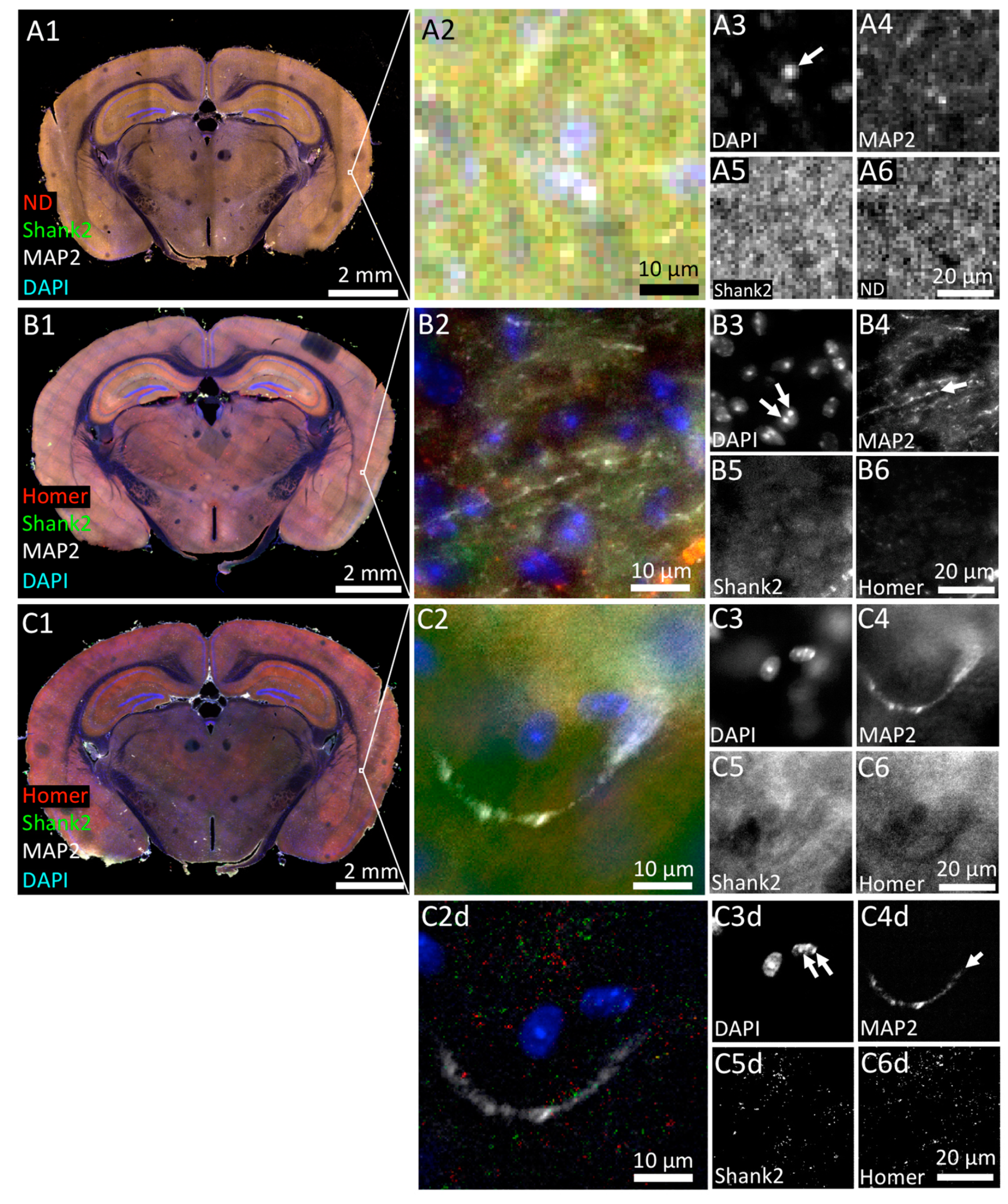
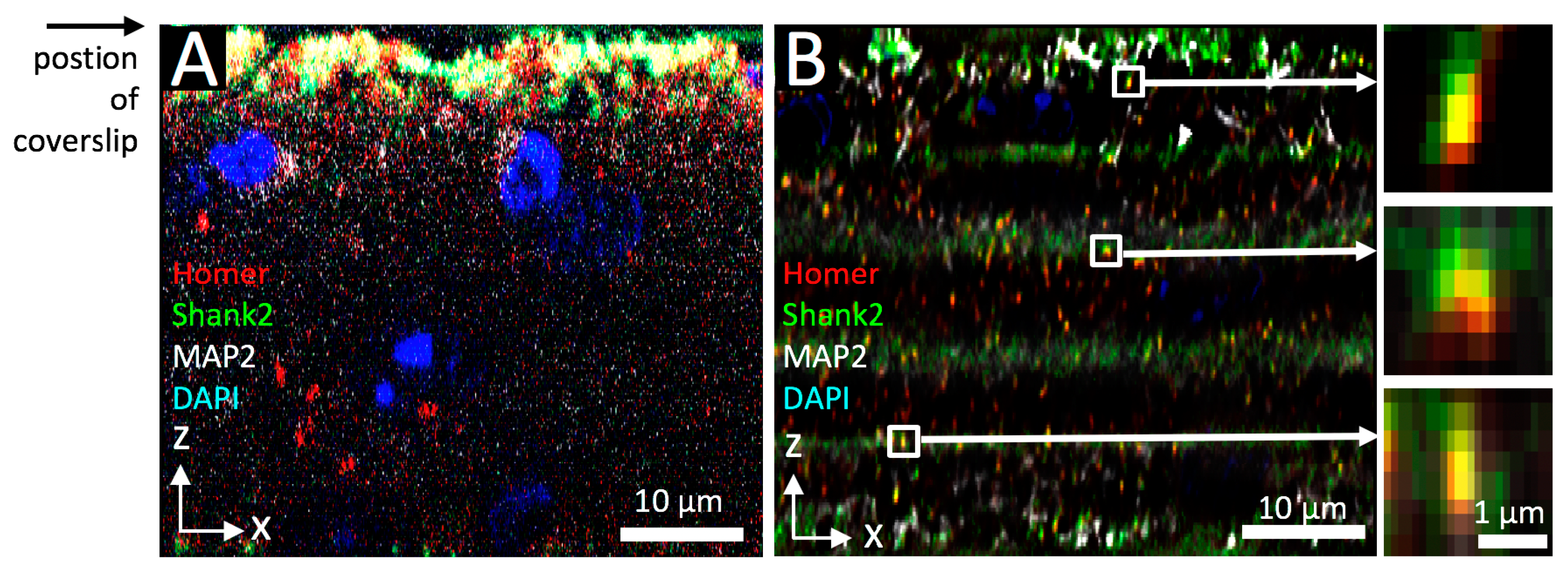
3.1. Tissue Imaging with Widefield Microscopy
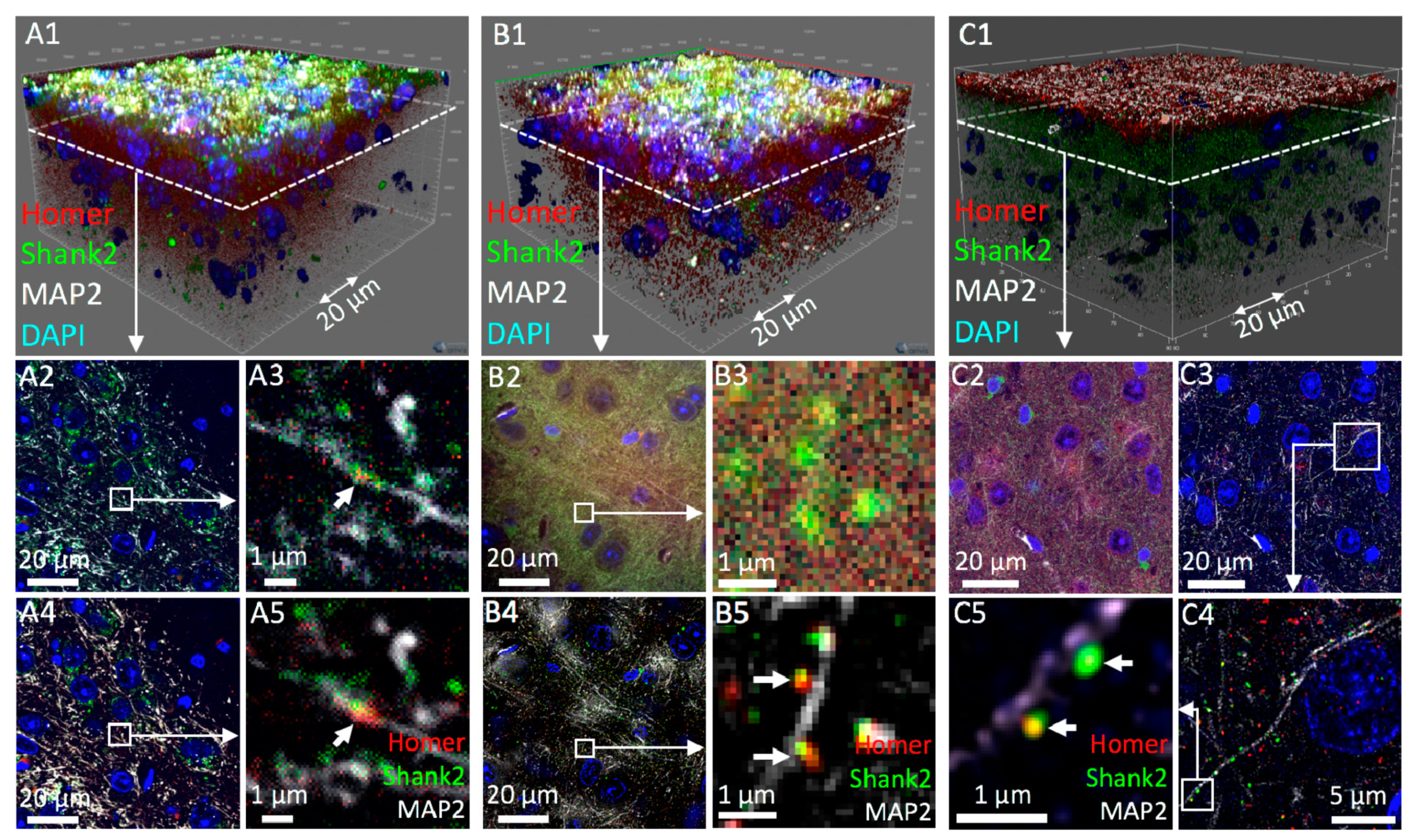
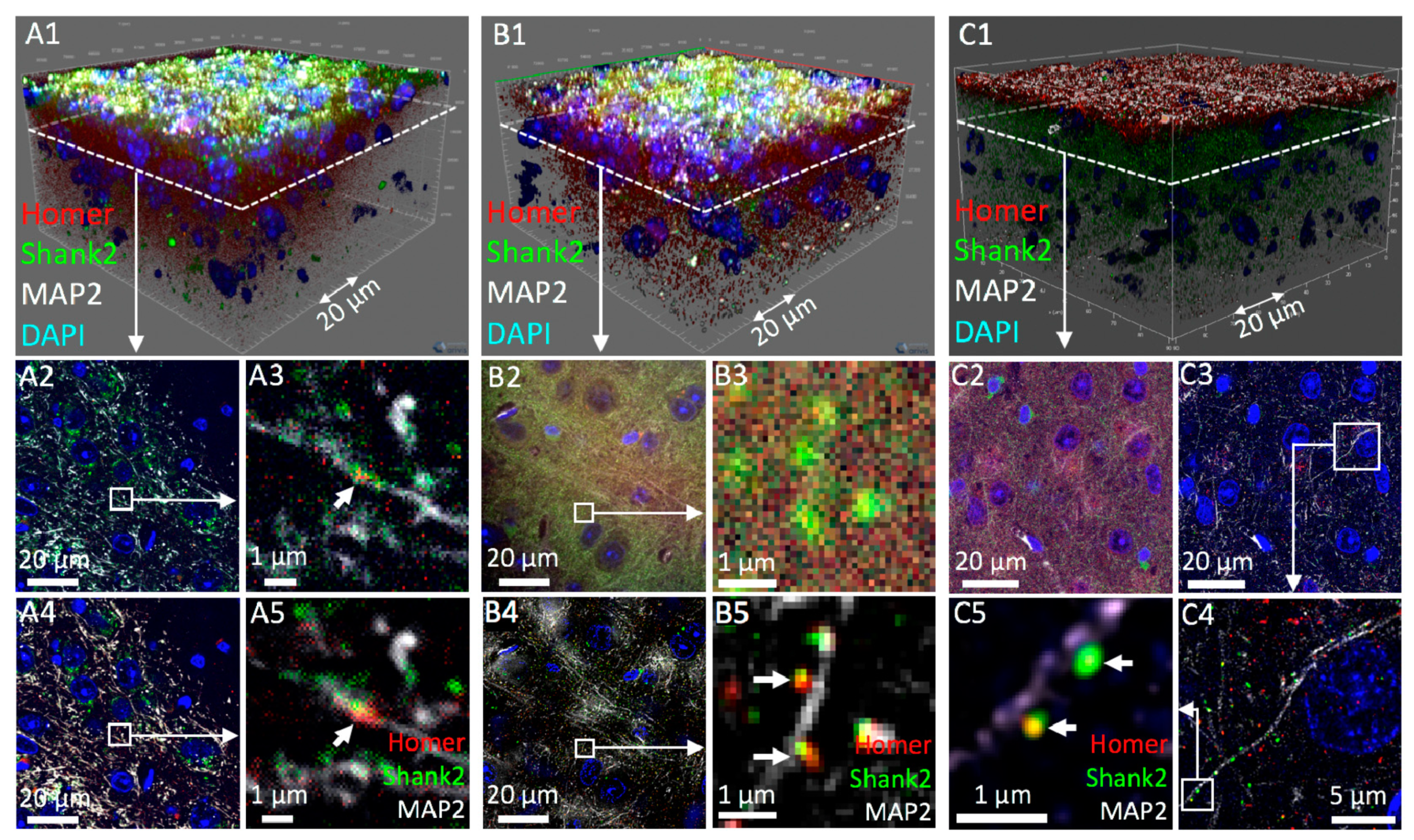
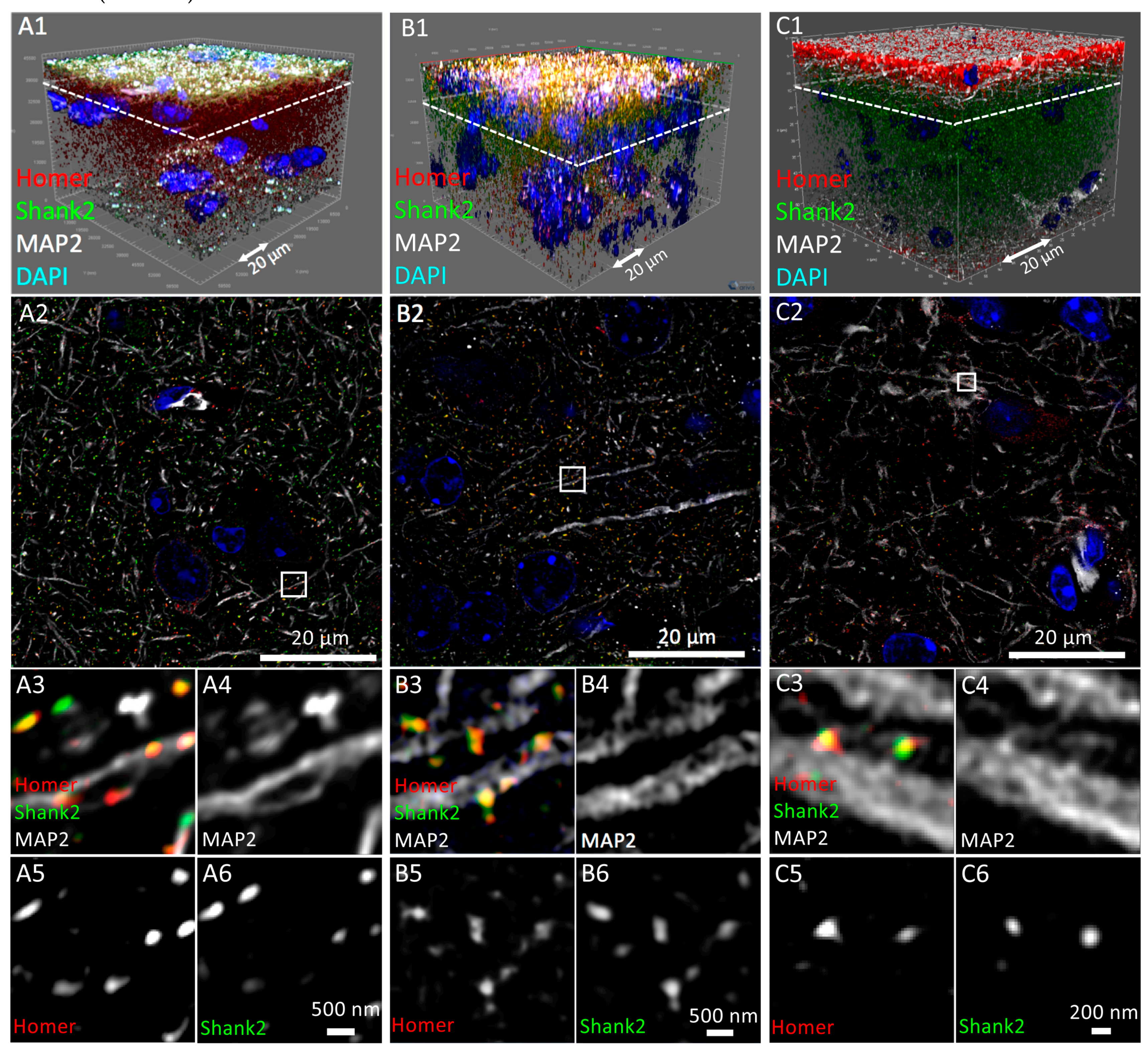
3.2. Confocal 3D Imaging of Brain Tissue
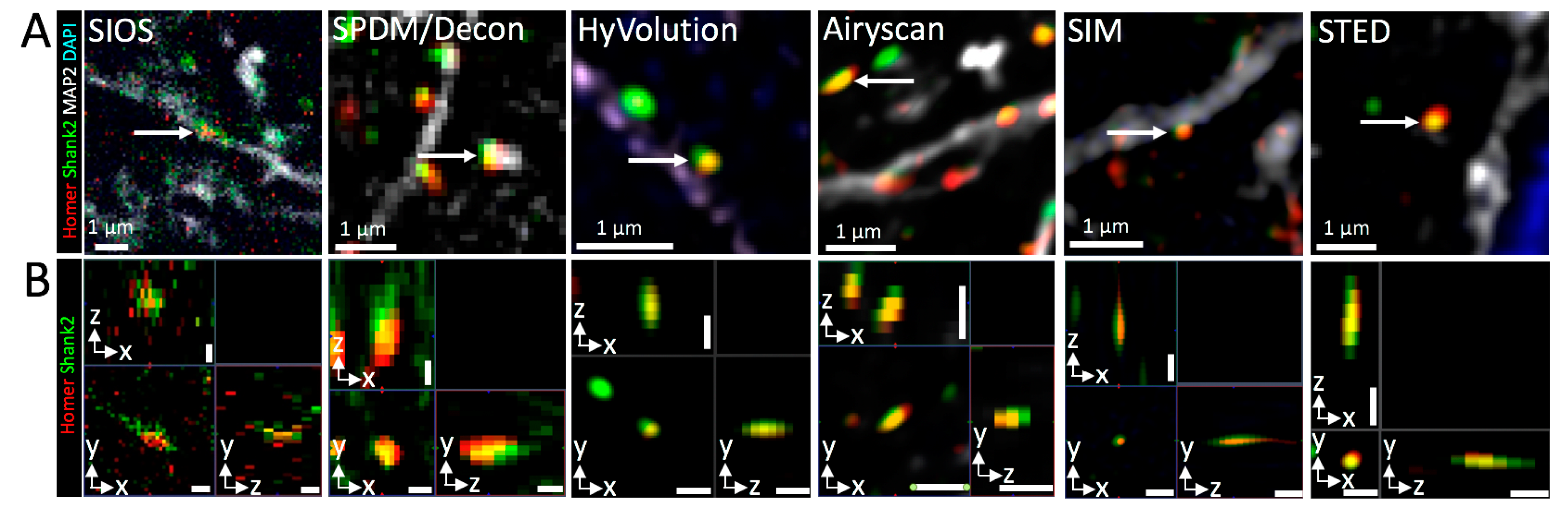
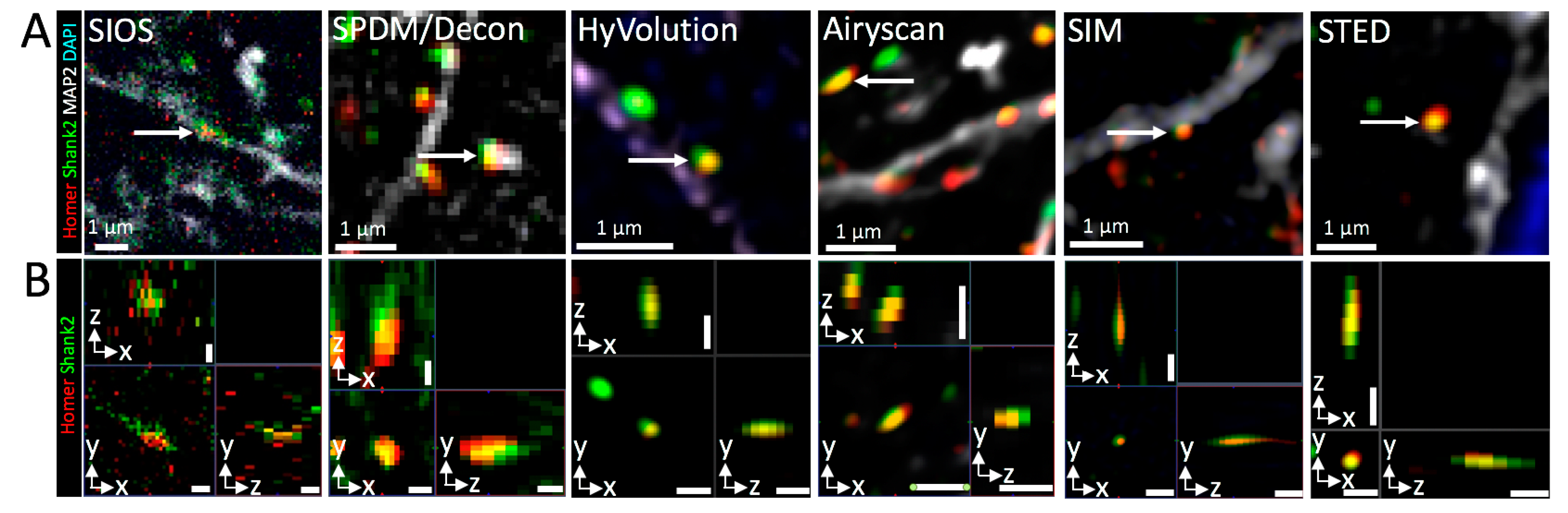
3.3. Three-Dimensional Super-Resolution Imaging of Mouse Brain Tissue
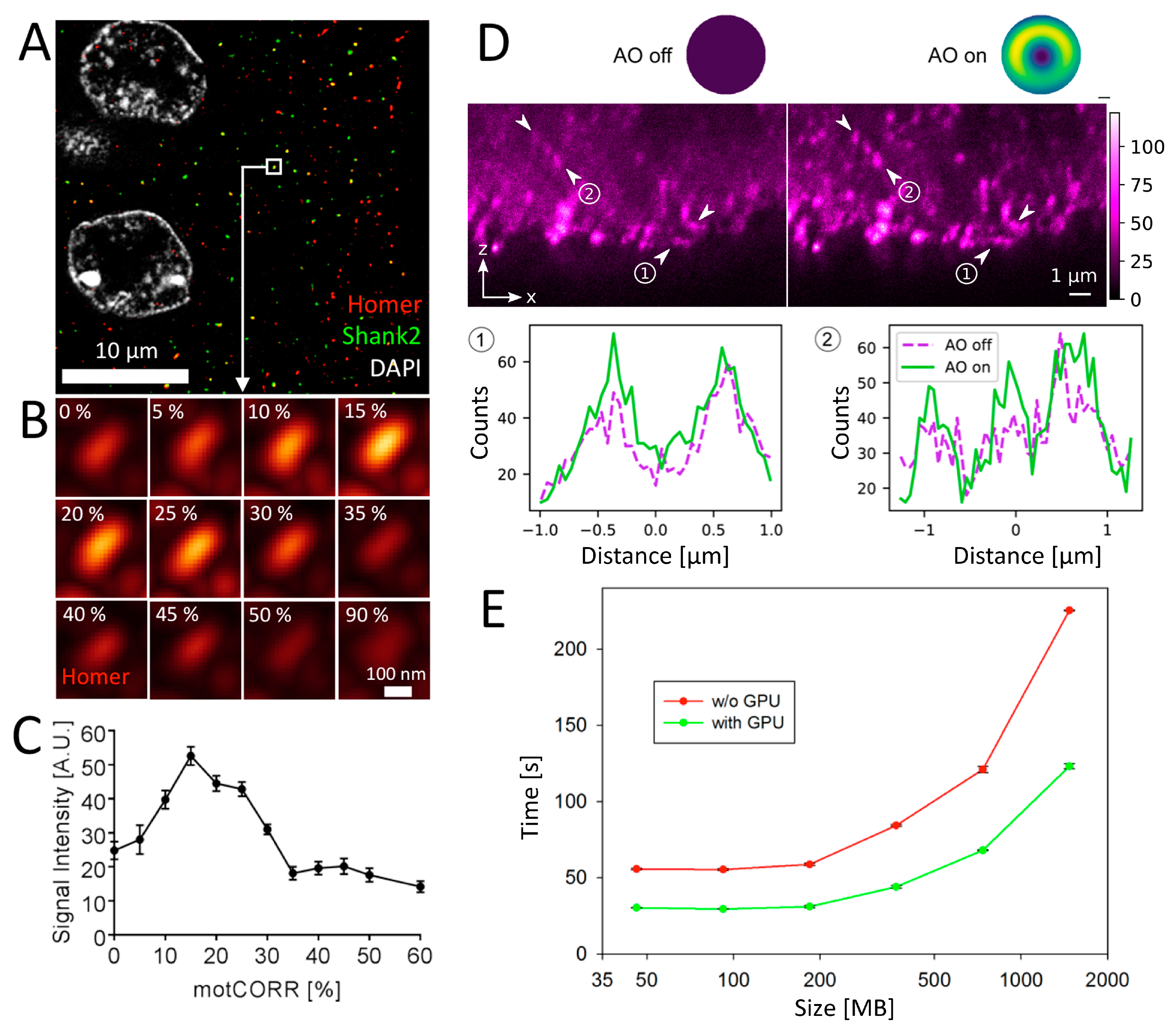
3.4. Approaches to Increase Performance in 3D Tissue Imaging
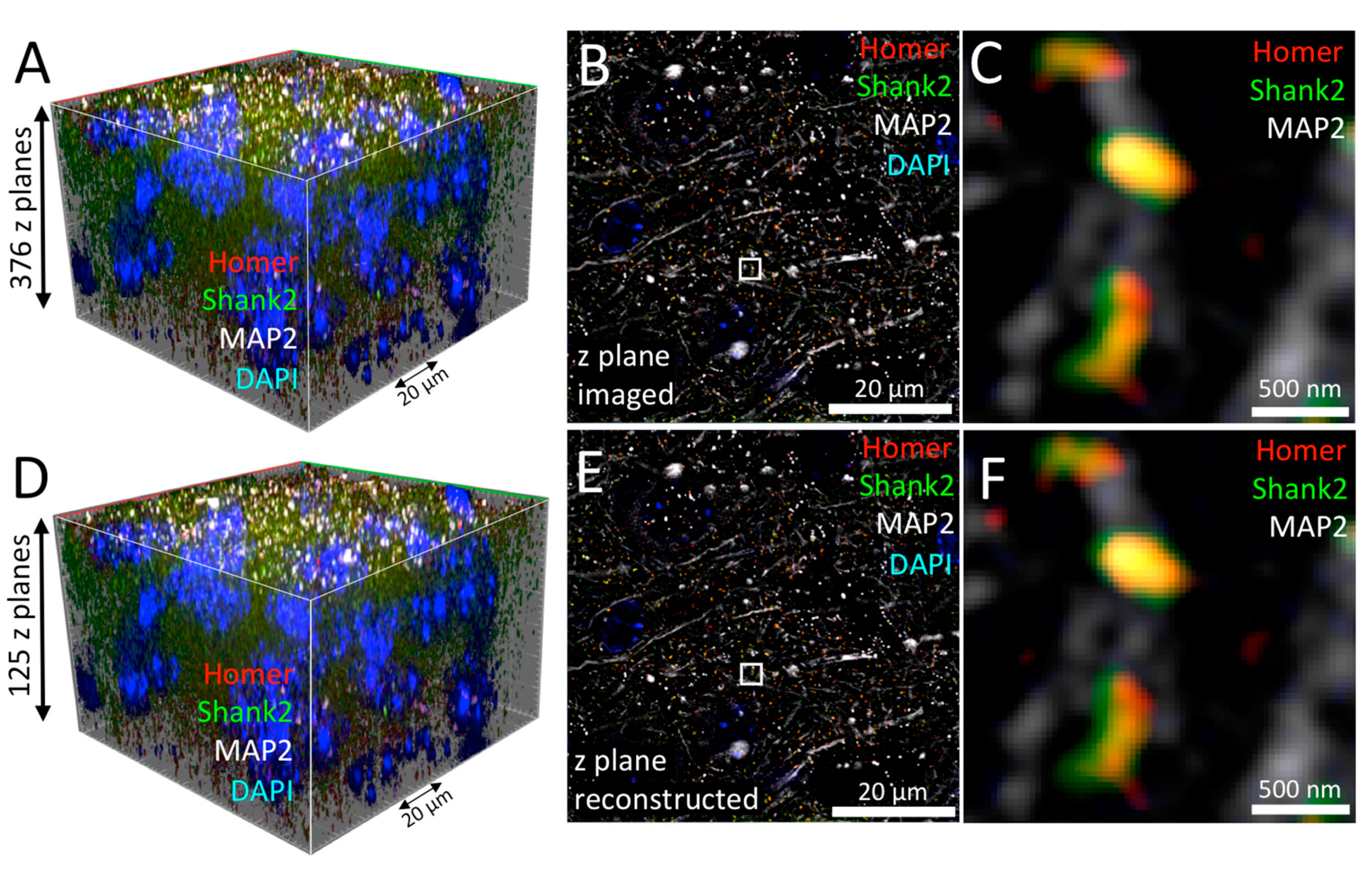
3.4.1. Leap Mode Virtual Reconstruction in Lattice-SIM Microscopy
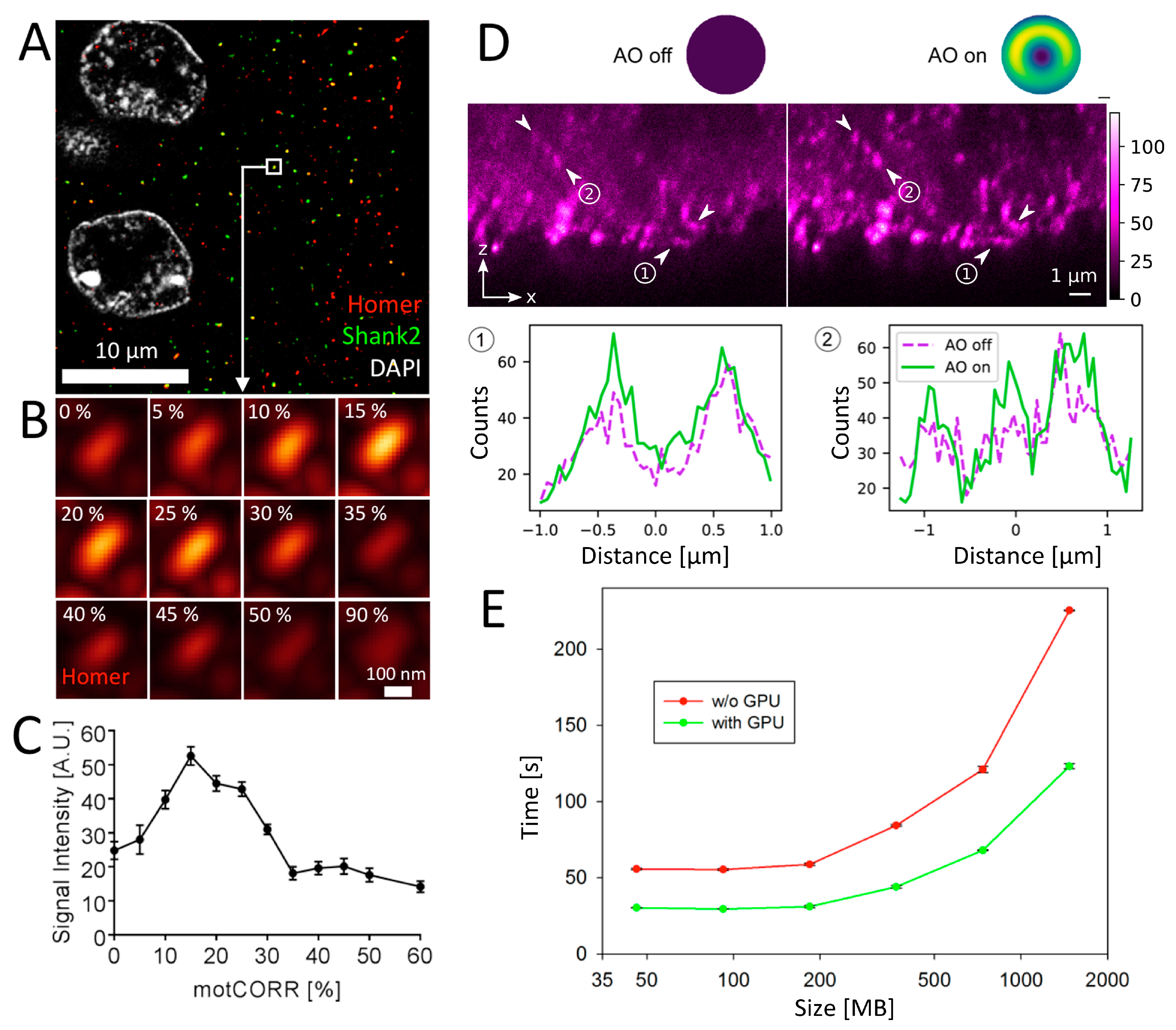
3.4.2. Motorized Correction Collar Objectives
3.4.3. Adaptive Optics (AO)
3.4.4. CUDA-GPU Accelerated Image Processing
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Mattay, V.S.; Fera, F.; Tessitore, A.; Hariri, A.R.; Berman, K.F.; Das, S.; Meyer-Lindenberg, A.; Goldberg, T.E.; Callicott, J.H.; Weinberger, D.R. Neurophysiological correlates of age-related changes in working memory capacity. Neurosci. Lett. 2006, 392, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Ouda, L.; Profant, O.; Syka, J. Age-related changes in the central auditory system. Cell Tissue Res. 2015, 361, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Sibille, E. Molecular aging of the brain, neuroplasticity, and vulnerability to depression and other brain-related disorders. Dialogues Clin. Neurosci. 2013, 15, 53–65. [Google Scholar] [PubMed]
- Lister, J.P.; Barnes, C.A. Neurobiological changes in the hippocampus during normative aging. Arch. Neurol. 2009, 66, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, D.L.; Weaver, C.M.; Luebke, J.I.; Hof, P.R. Dendritic spine changes associated with normal aging. Neuroscience 2013, 251, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Rozycka, A.; Liguz-Lecznar, M. The space where aging acts: focus on the GABAergic synapse. Aging Cell 2017, 16, 634–643. [Google Scholar] [CrossRef]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Tatti, R.; Haley, M.S.; Swanson, O.K.; Tselha, T.; Maffei, A. Neurophysiology and regulation of the balance between excitation and inhibition in neocortical circuits. Biol. Psychiatry 2017, 81, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Azpurua, J.; Eaton, B.A. Neuronal epigenetics and the aging synapse. Front. Cell Neurosci. 2015, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Dickstein, D.L.; Kabaso, D.; Rocher, A.B.; Luebke, J.I.; Wearne, S.L.; Hof, P.R. Changes in the structural complexity of the aged brain. Aging Cell 2007, 6, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, V.A.; Sabatini, B.L. Anatomical and physiological plasticity of dendritic spines. Annu Rev. Neurosci. 2007, 30, 79–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, V.A.; Firestein, B.L. The dendritic tree and brain disorders. Mol. Cell Neurosci. 2012, 50, 10–20. [Google Scholar] [CrossRef]
- Taoufik, E.; Kouroupi, G.; Zygogianni, O.; Matsas, R. Synaptic dysfunction in neurodegenerative and neurodevelopmental diseases: an overview of induced pluripotent stem-cell-based disease models. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Dresbach, T.; Qualmann, B.; Kessels, M.M.; Garner, C.C.; Gundelfinger, E.D. The presynaptic cytomatrix of brain synapses. Cell Mol. Life Sci. 2001, 58, 94–116. [Google Scholar] [CrossRef]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef]
- Maidorn, M.; Rizzoli, S.O.; Opazo, F. Tools and limitations to study the molecular composition of synapses by fluorescence microscopy. Biochem. J. 2016, 473, 3385–3399. [Google Scholar] [CrossRef]
- Chen, X.; Winters, C.; Crocker, V.; Lazarou, M.; Sousa, A.A.; Leapman, R.D.; Reese, T.S. Identification of PSD-95 in the postsynaptic density uUsing MiniSOG and EM tomography. Front. Neuroanat. 2018, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Harris, K.M.; Weinberg, R.J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Follain, G.; Mercier, L.; Osmani, N.; Harlepp, S.; Goetz, J.G. Seeing is believing—Multi-scale spatio-temporal imaging towards in vivo cell biology. J. Cell Sci. 2017, 130, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tønnesen, J.; Nägerl, U.V. Superresolution imaging for neuroscience. Exp. Neurol. 2013, 242, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Heller, J.P.; Rusakov, D.A. The nanoworld of the tripartite synapse: insights from super-resolution microscopy. Front. Cell Neurosci. 2017, 11, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, Y.; Nishimune, H. Super-resolution microscopy for analyzing neuromuscular junctions and synapses. Neurosci. Lett. 2020, 715, 134644. [Google Scholar] [CrossRef] [PubMed]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72–84. [Google Scholar] [CrossRef]
- Power, R.M.; Huisken, J. Putting advanced microscopy in the hands of biologists. Nat. Methods 2019, 16, 1069–1073. [Google Scholar] [CrossRef]
- Gao, R.; Asano, S.M.; Upadhyayula, S.; Pisarev, I.; Milkie, D.E.; Liu, T.L.; Singh, V.; Graves, A.; Huynh, G.H.; Zhao, Y.; et al. Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 2019, 363. [Google Scholar] [CrossRef]
- Haag, N.; Schwintzer, L.; Ahuja, R.; Koch, N.; Grimm, J.; Heuer, H.; Qualmann, B.; Kessels, M.M. The actin nucleator Cobl is crucial for Purkinje cell development and works in close conjunction with the F-actin binding protein Abp1. J. Neurosci. 2012, 32, 17842–17856. [Google Scholar] [CrossRef] [Green Version]
- Schwintzer, L.; Koch, N.; Ahuja, R.; Grimm, J.; Kessels, M.M.; Qualmann, B. The functions of the actin nucleator Cobl in cellular morphogenesis critically depend on syndapin I. EMBO J. 2011, 30, 3147–3159. [Google Scholar] [CrossRef]
- Izadi, M.; Schlobinski, D.; Lahr, M.; Schwintzer, L.; Qualmann, B.; Kessels, M.M. Cobl-like promotes actin filament formation and dendritic branching using only a single WH2 domain. J. Cell Biol. 2018, 217, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Koch, N.; Koch, D.; Krueger, S.; Tröger, J.; Sabanov, V.; Ahmed, T.; McMillan, L.E.; Wolf, D.; Montag, D.; Kessels, M.M.; et al. Syndapin I loss-of-function in mice leads to schizophrenia-like symptoms. Cereb. Cortex 2020. [Google Scholar] [CrossRef] [PubMed]
- Kessels, M.M.; Engqvist-Goldstein, A.E.; Drubin, D.G.; Qualmann, B. Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J. Cell Biol. 2001, 153, 351–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinyol, R.; Haeckel, A.; Ritter, A.; Qualmann, B.; Kessels, M.M. Regulation of N-WASP and the Arp2/3 complex by Abp1 controls neuronal morphology. PLoS ONE 2007, 2, e400. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Okada, Y.; Nakagawa, S. Super-resolution imaging of nuclear bodies by STED microscopy. Methods Mol. Biol. 2015, 1262, 21–35. [Google Scholar] [CrossRef]
- Barbotin, A.; Galiani, S.; Urbančič, I.; Eggeling, C.; Booth, M.J. Adaptive optics allows STED-FCS measurements in the cytoplasm of living cells. Opt. Express 2019, 27, 23378–23395. [Google Scholar] [CrossRef]
- Heine, J.; Wurm, C.A.; Keller-Findeisen, J.; Schonle, A.; Harke, B.; Reuss, M.; Winter, F.R.; Donnert, G. Three dimensional live-cell STED microscopy at increased depth using a water immersion objective. Rev. Sci. Instrum. 2018, 89, 053701. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.J.; Burke, D.; Bewersdorf, J.; Booth, M.J. Adaptive optics enables 3D STED microscopy in aberrating specimens. Opt. Express 2012, 20, 20998–21009. [Google Scholar] [CrossRef] [Green Version]
- Antonello, J.; Hao, X.; Allgeyer, E.S.; Bewersdorf, J.; Rittscher, J.; Booth, M.J. Sensorless adaptive optics for isoSTED nanoscopy. In Adaptive Optics and Wavefront Control for Biological Systems IV; Bifano, T.G., Kubby, J., Gigan, S., Eds.; SPIE—International Society for Optics and Photonics: Bellingham, WA, USA, 2018; Volume 10502, p. 1050206. [Google Scholar]
- Borlinghaus, R.T.; Kappel, C. HyVolution—the smart path to confocal super-resolution. Nat. Methods 2016, 13, i–iii. [Google Scholar] [CrossRef]
- Schrader, M.; Hell, S.W.; van der Voort, H.T.M. Potential of confocal microscopes to resolve in the 50–100 nm range. Appl. Phys. Lett. 1996, 69, 3644–3646. [Google Scholar] [CrossRef] [Green Version]
- Lam, F.; Cladière, D.; Guillaume, C.; Wassmann, K.; Bolte, S. Super-resolution for everybody: An image processing workflow to obtain high-resolution images with a standard confocal microscope. Methods 2017, 115, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. Communication in the presence of noise. In Proceedings of the IRE; IEEE: Piscataway, NJ, USA, 1949; Volume 37, pp. 10–21. [Google Scholar]
- Nothnagle, P.E.; Chambers, W.; Davidson, M. Introduction to Stereomicrosopy. 2020. Available online: https://www.microscopyu.com/techniques/stereomicroscopy/introduction-to-stereomicroscopy (accessed on 25 May 2017).
- Diaspro, A.; Bianchini, P.; Cella, F.C.; Usai, C. Optical fluorescence microscopy. In Encyclopedia of Biophysics; Robert, G.C.K., Ed.; Springer Heidelberg: New York, NY, USA, 2013. [Google Scholar]
- Pawley, J. (Ed.) Handbook of Biological Confocal Microscopy 3; Springer: Boston, MA, USA, 2006. [Google Scholar]
- Moser, G.C.; Müller, H. Cell cycle dependent changes of chromosomes in mouse fibroblasts. Eur. J. Cell Biol. 1979, 19, 116–119. [Google Scholar]
- McNally, J.G.; Karpova, T.; Cooper, J.; Conchello, J.A. Three-dimensional imaging by deconvolution microscopy. Methods 1999, 19, 373–385. [Google Scholar] [CrossRef]
- Jonkman, J.; Brown, C.M. Any way you slice it—A comparison of confocal microscopy techniques. J. Biomol. Tech. 2015, 26, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Bayguinov, P.O.; Oakley, D.M.; Shih, C.C.; Geanon, D.J.; Joens, M.S.; Fitzpatrick, J.A.J. Modern laser scanning confocal microscopy. Curr. Protoc. Cytom. 2018, 85, e39. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Smith, I.; Parker, I.; Bootman, M.D. Fluorescence microscopy. Cold Spring Harb. Protoc. 2014, 2014, pdb top071795. [Google Scholar] [CrossRef] [Green Version]
- Jonkman, J.; Brown, C.M.; Cole, R.W. Quantitative confocal microscopy: beyond a pretty picture. Methods Cell Biol. 2014, 123, 113–134. [Google Scholar] [CrossRef]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Shroff, H. Faster, sharper, and deeper: structured illumination microscopy for biological imaging. Nat. Methods 2018, 15, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Smith, N.I.; Fujita, K. Introduction to super-resolution microscopy. Microscopy 2014, 63, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.B.; Enderlein, J. Image scanning microscopy. Phys. Rev. Lett. 2010, 104, 198101. [Google Scholar] [CrossRef]
- Huff, J.; Bergter, A.; Birkenbeil, J.; Kleppe, I.; Engelmann, R.; Krzic, U. The new 2D Superresolution mode for ZEISS Airyscan. Nat. Methods 2017, 14, 1223. [Google Scholar] [CrossRef]
- York, A.G.; Parekh, S.H.; Dalle Nogare, D.; Fischer, R.S.; Temprine, K.; Mione, M.; Chitnis, A.B.; Combs, C.A.; Shroff, H. Resolution doubling in live, multicellular organisms via multifocal structured illumination microscopy. Nat. Methods 2012, 9, 749–754. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.Y.; Lee, E.S. Enhancing the isotropy of lateral resolution in coherent structured illumination microscopy. Biomed. Opt. Express 2014, 5, 1895–1912. [Google Scholar] [CrossRef] [Green Version]
- Eggeling, C.; Willig, K.I.; Sahl, S.J.; Hell, S.W. Lens-based fluorescence nanoscopy. Q Rev. Biophys. 2015, 48, 178–243. [Google Scholar] [CrossRef]
- Castello, M.; Diaspro, A.; Vicidomini, G. Multi-images deconvolution improves signal-to-noise ratio on gated stimulated emission depletion microscopy. Appl. Phys. Lett. 2014, 105, 234106. [Google Scholar] [CrossRef]
- Van der Voort, H.T.M. Deconvolution of Nanoscopic Imaging (Chapter 5). In Super-Resolution Imaging in Biomedicine; CRC Press: Cleveland, OH, USA, 2016. [Google Scholar]
- Urban, N.T.; Willig, K.I.; Hell, S.W.; Nägerl, U.V. STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys. J. 2011, 101, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Jost, A.; Tolstik, E.; Feldmann, P.; Wicker, K.; Sentenac, A.; Heintzmann, R. Optical sectioning and high resolution in single-slice structured illumination microscopy by thick slice blind-SIM reconstruction. PLoS ONE 2015, 10, e0132174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouquet, C.; Gilles, J.F.; Heck, N.; Dos Santos, M.; Schwartzmann, R.; Cannaya, V.; Morel, M.P.; Davidson, R.S.; Trembleau, A.; Bolte, S. Improving axial resolution in confocal microscopy with new high refractive index mounting media. PLoS ONE 2015, 10, e0121096. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W.; van Royen, M.E.; van Cappellen, W.A.; Houtsmuller, A.B.; Verhaegen, M.; Schitter, G. Automated spherical aberration correction in scanning confocal microscopy. Rev. Sci. Instrum. 2014, 85, 123706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, N. Adaptive optical fluorescence microscopy. Nat. Methods 2017, 14, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J. Adaptive optics in microscopy. Philos. Trans. A Math. Phys. Eng. Sci. 2007, 365, 2829–2843. [Google Scholar] [CrossRef]
- Schmid, B.; Huisken, J. Real-time multi-view deconvolution. Bioinformatics 2015, 31, 3398–3400. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.U.; Moon, S.; Song, H.; Kwon, H.S.; Kim, D.Y. Masked illumination scheme for a galvanometer scanning high-speed confocal fluorescence microscope. Scanning 2011, 33, 455–462. [Google Scholar] [CrossRef]
- Heine, J.; Reuss, M.; Harke, B.; D’Este, E.; Sahl, S.J.; Hell, S.W. Adaptive-illumination STED nanoscopy. Proc. Natl. Acad. Sci. USA 2017, 114, 9797–9802. [Google Scholar] [CrossRef] [Green Version]
- Keller, P.J.; Pampaloni, F.; Stelzer, E.H. Life sciences require the third dimension. Curr. Opin. Cell Biol. 2006, 18, 117–124. [Google Scholar] [CrossRef]
- Power, R.M.; Huisken, J. A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 2017, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Legant, W.R.; Shao, L.; Grimm, J.B.; Brown, T.A.; Milkie, D.E.; Avants, B.B.; Lavis, L.D.; Betzig, E. High-density three-dimensional localization microscopy across large volumes. Nat. Methods 2016, 13, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.C.; Tang, W.C.; Low, C.S.L.; Liu, Y.T.; Wu, J.S.; Lee, P.Y.; Chen, L.Q.; Lin, Y.L.; Kanchanawong, P.; Gao, L.; et al. Rapid high resolution 3D imaging of expanded biological specimens with lattice light sheet microscopy. Methods 2019. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.K.; Petrov, P.N.; Moerner, W.E. Light sheet approaches for improved precision in 3D localization-based super-resolution imaging in mammalian cells [Invited]. Opt. Express 2018, 26, 13122–13147. [Google Scholar] [CrossRef]
- Chakraborty, T.; Driscoll, M.K.; Jeffery, E.; Murphy, M.M.; Roudot, P.; Chang, B.J.; Vora, S.; Wong, W.M.; Nielson, C.D.; Zhang, H.; et al. Light-sheet microscopy of cleared tissues with isotropic, subcellular resolution. Nat. Methods 2019, 16, 1109–1113. [Google Scholar] [CrossRef]
- Balzarotti, F.; Eilers, Y.; Gwosch, K.C.; Gynnå, A.H.; Westphal, V.; Stefani, F.D.; Elf, J.; Hell, S.W. Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes. Science 2017, 355, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Azuma, T.; Kei, T. Super-resolution spinning-disk confocal microscopy using optical photon reassignment. Opt. Express 2015, 23, 15003–15011. [Google Scholar] [CrossRef]
- Oni—The Nanoimager Home Page. Available online: https://oni.bio/nanoimager (accessed on 28 May 2020).
- Facility Line Home Page. Available online: https://www.abberior-instruments.com/products/facility-line/ (accessed on 28 May 2020).
- Cabriel, C.; Bourg, N.; Dupuis, G.; Lévêque-Fort, S. Aberration-accounting calibration for 3D single-molecule localization microscopy. Opt. Lett. 2018, 43, 174–177. [Google Scholar] [CrossRef] [Green Version]
- Nanofleye Home Page. Available online: https://www.nanofleye.com/ (accessed on 28 May 2020).
- BioAxial Codim Imaging Home Page. Available online: http://www.bioaxial.com/codim-imaging/ (accessed on 28 May 2020).
- De Luca, G.M.; Breedijk, R.M.; Brandt, R.A.; Zeelenberg, C.H.; de Jong, B.E.; Timmermans, W.; Azar, L.N.; Hoebe, R.A.; Stallinga, S.; Manders, E.M. Re-scan confocal microscopy: scanning twice for better resolution. Biomed. Opt. Express 2013, 4, 2644–2656. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microscope | Stereo | Slide Scanner | Convent. Widefield | SIOS (Apotome) | SPDM | Confocal (Hyvolution) | Airyscan | Lattice-SIM | Lattice-SIM Leap | 2D-STED | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Objective | 2.3× | 20× | 20x | 63× Oil | 63× Oil | 63× Oil | 63× Oil | 63× Oil | 63× Oil | 100× Oil | |
| Sample Size [µm] | x | 1 × 104 | 1 × 104 | 1 × 104 | 90 | 90 | 90 | 65 | 65 | 65 | 65 |
| y | 0.8 × 104 | 0.8 × 104 | 0.8 × 104 | 90 | 90 | 90 | 65 | 65 | 65 | 65 | |
| z | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | |
| Optical Sections | 8 | 16 | 80 | 210 | 197 | 448 | 273 | 376 | 125 | 276 | |
| Voxel Size [nm] | x | n.a. | n.a. | n.a. | 93 | 102 | 35 | 40 | 31 | 31 | 31 |
| y | n.a. | n.a. | n.a. | 93 | 102 | 35 | 40 | 31 | 31 | 31 | |
| z | n.a. | n.a. | n.a. | 240 | 240 | 123 | 110 | 110 | 110 | 183 | |
| Theor. Resolution [nm] | x | n.a. | n.a. | n.a. | 220 | 220 | 140 | 120 | 100 | 100 | 40 |
| y | n.a. | n.a. | n.a. | 220 | 220 | 140 | 120 | 100 | 100 | 40 | |
| z | n.a. | n.a. | n.a. | 600 | 600 | 380 | 350 | 300 | 300 | 600 | |
| File Size | 2.7 GB | 16.6 GB | 840 GB *3 | 11 GB | 1.8 GB | 12 GB | 69 GB | 42 GB | 14 GB | 21 GB | |
| Acquisition Time | 40 min | 1 h 15 min | 10 h 52 min *3 | 18 min 25 s | 5 min 38 s | 3 h 42 min | 1 h 38 min | 12 min 44 s | 4 min 17 s | 5 h 26 min | |
| Processing Time *1 | 2 min 9 s | 0 min *2 | 51 h 12 min *3 | 2 min 30 s | n.a. | n.a. | 54 min | 9 min 13 s | 24 min 40 s | n.a. | |
| Deconvol-ution Time | n.a. | n.a. | n.a. | 8 min 16 s | 5 min 24 s | 0 min *2 | 16 min 3 s | n.a. | n.a. | 3 h 20 min | |
| Total Time | 42 min 9 s | 1 h 15 min | 62 h 2 min *3 | 20 min 55 s *4 26 min 41 s *5 | 11 min 2 s | 3 h 42 min | 2 h 48 min | 21 min 57 s | 28 min 57 s | 8 h 46 min | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tröger, J.; Hoischen, C.; Perner, B.; Monajembashi, S.; Barbotin, A.; Löschberger, A.; Eggeling, C.; Kessels, M.M.; Qualmann, B.; Hemmerich, P. Comparison of Multiscale Imaging Methods for Brain Research. Cells 2020, 9, 1377. https://doi.org/10.3390/cells9061377
Tröger J, Hoischen C, Perner B, Monajembashi S, Barbotin A, Löschberger A, Eggeling C, Kessels MM, Qualmann B, Hemmerich P. Comparison of Multiscale Imaging Methods for Brain Research. Cells. 2020; 9(6):1377. https://doi.org/10.3390/cells9061377
Chicago/Turabian StyleTröger, Jessica, Christian Hoischen, Birgit Perner, Shamci Monajembashi, Aurélien Barbotin, Anna Löschberger, Christian Eggeling, Michael M. Kessels, Britta Qualmann, and Peter Hemmerich. 2020. "Comparison of Multiscale Imaging Methods for Brain Research" Cells 9, no. 6: 1377. https://doi.org/10.3390/cells9061377
APA StyleTröger, J., Hoischen, C., Perner, B., Monajembashi, S., Barbotin, A., Löschberger, A., Eggeling, C., Kessels, M. M., Qualmann, B., & Hemmerich, P. (2020). Comparison of Multiscale Imaging Methods for Brain Research. Cells, 9(6), 1377. https://doi.org/10.3390/cells9061377