CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes



Abstract
1. Introduction
2. Materials and Methods
2.1. Bioethics
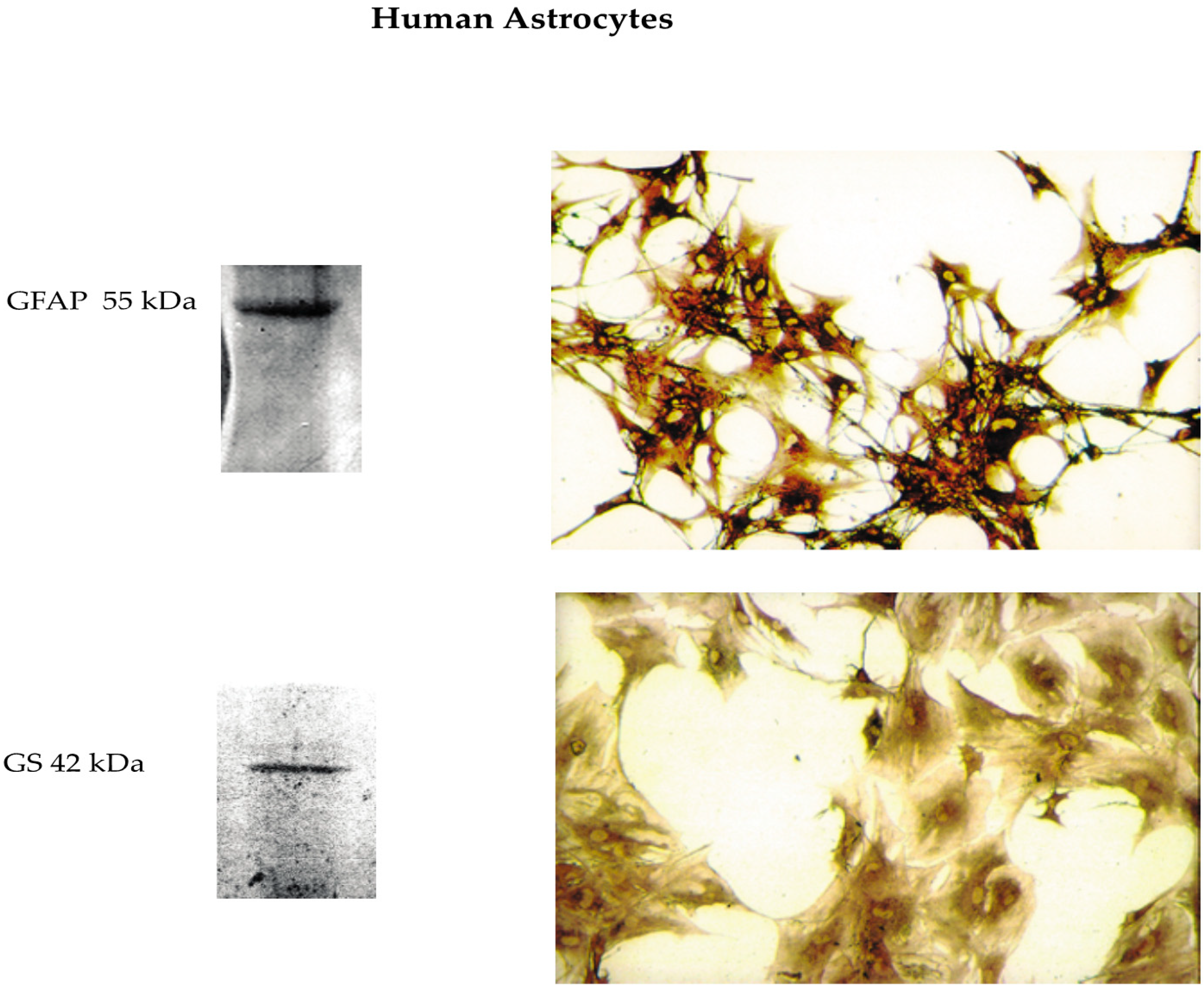
2.2. Isolation and Culture of Phenotypically Locked-in Nontumorigenic Adult Human Astrocytes (NAHAs)
2.3. Aβ Peptides
2.4. Experimental Protocol
2.5. Immunocytochemistry
2.6. Antibody Array
2.7. Enzyme-Linked Immunosorbent Assays (ELISAs) of IL-6, MCP-2, RANTES, and Soluble ICAM-1 Released into NAHAs-Conditioned Growth Media
2.8. Western Immunoblotting
2.9. Statistical Analysis
3. Results
3.1. Antibody Array-Detected Changes in Neuroinflammatory Agents Released from Untreated and fAβ25–35 ± NPS 2143-Treated NAHAs
- (i)
- a significant increase elicited by fAβ25–35 treatment, which fAβ25–35 + NPS 2143 cotreatment brought back to control levels by 96 h as was typical of IL-6, MCP-2, and soluble (s)-ICAM-1 fragment, whereas it only partially reduced RANTES secreted levels (Figure 2);
- (ii)
- a significant increase brought about by fAβ25–35 exposure that did not change when adding NPS 2143 to fAβ25–35, as was proper of IL-1β, IL-3, and IL-8 at both 48 h and 96 h, whereas the surge of IL-16 at 48-h was only transient (v. Figure S1); and
- (iii)
- no change vs. control levels after the addition of either fAβ25–35 by itself or fAβ25–35 + NPS 2143, as exemplified by MCP-1 and TIMP-2 (v. Figure S1).
3.2. Calcilytic NPS 2143 Effectively Hinders the Secretion of IL-6, MCP-2, RANTES, and s-ICAM-1 from NAHAs
3.2.1. IL-6 Secretion into NAHA-Conditioned Growth Media
3.2.2. s-ICAM-1 Shedding into NAHA-Conditioned Growth Media
3.2.3. RANTES Secretion into NAHA-Conditioned Growth Media
3.2.4. MCP-2 Secretion into NAHA-Conditioned Growth Media
3.3. Changes in Cytokine/Chemokine Levels in NAHAs Protein Lysates Evoked by with fAβ25–35 ± NPS 2143 Treatments
3.3.1. IL-6 Expression in NAHA Protein Lysates
3.3.2. ICAM-1 Holoprotein Expression in NAHA Protein Lysates
3.3.3. RANTES Expression in NAHAs Protein Lysates
3.3.4. MCP-2 Expression in NAHA Protein Lysates
4. Discussion
4.1. IL-6
4.2. ICAM-1 Holoprotein/s-ICAM-1
4.3. RANTES
4.4. MCP-2
4.5. Other Proinflammatory Agents Not Affected by Antagonizing Aβ•CaSR Signaling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate immunity and neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138 Pt 10, 2814–2833. [Google Scholar] [CrossRef]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Gondo, T.; Hoshii, Y.; Takahashi, M.; Yamada, M.; Ishihara, T. Confocal observation of senile plaques in Alzheimer’s disease: Senile plaque morphology and relationship between senile plaques and astrocytes. Pathol. Int. 1998, 48, 332–340. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Ramirez, G. Microglia-astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Johansen, N.; Penn, O.; et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 2019, 573, 61–68. [Google Scholar] [CrossRef]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of synapse number by glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Goodall, E.F.; Wang, C.; Simpson, J.E.; Baker, D.J.; Drew, D.R.; Heath, P.R.; Saffrey, M.J.; Romero, I.A.; Wharton, S.B. Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 2018, 44, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-Sensing Receptors of Human Astrocyte-Neuron Teams: Amyloid-β-Driven Mediators and Therapeutic Targets of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 353–364. [Google Scholar] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptors of Human Neural Cells Play Crucial Roles in Alzheimer’s Disease. Front. Physiol. 2016, 7, 134. [Google Scholar] [CrossRef]
- Dal Prà, I.; Armato, U.; Chiarini, A. Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282. [Google Scholar] [CrossRef]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25–35 in human cortical astrocytes and neurons¾Therapeutic relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277. [Google Scholar] [CrossRef]
- Nemeth, E.F.; Goodman, W.G. Calcimimetic and calcilytic drugs: Feats, flops, and futures. Calcif. Tissue Int. 2016, 98, 341–358. [Google Scholar] [CrossRef]
- Zhang, C.; Miller, C.L.; Brown, E.M.; Yang, J.J. The calcium sensing receptor: From calcium sensing to signaling. Sci. China Life Sci. 2015, 58, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, B.; Chattopadhyay, N.; Brown, E.M. Signaling through the extracellular calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 2012, 740, 103–142. [Google Scholar]
- Bandyopadhyay, S.; Tfelt-Hansen, J.; Chattopadhyay, N. Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci. Res. 2010, 88, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Evliyaoglu, C.; Heese, O.; Carroll, R.; Sanders, J.; Black, P.; Brown, E.M. Regulation of secretion of PTHrP by Ca2+-sensing receptor in human astrocytes, astrocytomas, and meningiomas. Am. J. Physiol. Cell. Physiol. 2000, 279, C691–C699. [Google Scholar] [CrossRef] [PubMed]
- Dal Prà, I.; Chiarini, A.; Nemeth, E.F.; Armato, U.; Whitfield, J.F. Roles of Ca2+ and the Ca2+-sensing receptor (CaSR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocytes. J. Cell. Biochem. 2005, 96, 428–438. [Google Scholar]
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, X.; He, J.; Liu, J.; Yang, S.; Dong, H. Important roles of the Ca2+-sensing receptor in vascular health and disease. Life Sci. 2018, 209, 217–227. [Google Scholar] [CrossRef]
- Conigrave, A.D.; Hampson, D.R. Broad-spectrum L-amino acid sensing by class 3 G-protein-coupled receptors. Trends Endocrinol. Metab. 2006, 17, 398–407. [Google Scholar] [CrossRef]
- Noh, J.S.; Pak, H.J.; Shin, Y.J.; Riew, T.R.; Park, J.H.; Moon, Y.W.; Lee, M.Y. Differential expression of the calcium-sensing receptor in the ischemic and border zones after transient focal cerebral ischemia in rats. J. Chem. Neuroanat. 2015, 66–67, 40–51. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Ye, C.; Yamaguchi, T.; Nakai, M.; Kifor, O.; Vassilev, P.M.; Nishimura, R.N.; Brown, E.M. The extracellular calcium-sensing receptor is expressed in rat microglia and modulates an outward K+ channel. J. Neurochem. 1999, 72, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Kemp, P.J. The calcium-sensing receptor beyond extracellular calcium homeostasis: Conception, development, adult physiology, and disease. Annu. Rev. Physyiol. 2012, 74, 271–297. [Google Scholar] [CrossRef] [PubMed]
- Ruat, M.; Traiffort, E. Roles of the calcium sensing receptor in the central nervous system. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Dal Prà, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromolecular Med. 2014, 16, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ho, H.; Kim, N.; Liu, J.; Yenari, M.A.; Wenhan, C. Calcium-sensing receptor (CaSR): A novel target for ischemic neuroprotection. Ann. Clin. Transl. Neurol. 2014, 1, 851–866. [Google Scholar] [CrossRef]
- Bai, S.; Mao, M.; Tian, L.; Yu, Y.; Zeng, J.; Ouyang, K.; Yu, L.; Li, L.; Wang, D.; Deng, X.; et al. Calcium sensing receptor mediated the excessive generation of β-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem. Biophys. Res. Commun. 2015, 459, 568–573. [Google Scholar] [CrossRef]
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased Calcium-Sensing Receptor Immunoreactivity in the Hippocampus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Klein, G.L.; Castro, S.M.; Garofalo, R.P. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56. [Google Scholar] [CrossRef]
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D., Jr.; Thompson, M.A.P.; Lowe, A.P.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.C.; et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 2015, 7, 284ra60. [Google Scholar] [CrossRef]
- Mattar, P.; Bravo-Sagua, R.; Tobar, N.; Fuentes, C.; Troncoso, R.; Breitwieser, G.; Lavandero, S.; Cifuentes, M. Autophagy mediates calcium-sensing receptor-induced TNFα production in human preadipocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3585–3594. [Google Scholar] [CrossRef] [PubMed]
- Iamartino, L.; Elajnaf, T.; Kallay, E.; Schepelmann, M. Calcium-sensing receptor in colorectal inflammation and cancer: Current insights and future perspectives. World J. Gastroenterol. 2018, 24, 4119–4131. [Google Scholar] [CrossRef] [PubMed]
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Méjean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 Prevents Calcium-Induced Progression of Early-Stage Prostate Tumors by Counteracting TRPC6 and Calcium Sensing Receptor Upregulation. Cancer Res. 2017, 77, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, H.A.; Kwon, O.K.; Park, J.W.; Lee, G.; Lee, H.J.; Lee, S.J.; Oh, S.R.; Ahn, K.S. NPS 2143, a selective calcium-sensing receptor antagonist inhibits lipopolysaccharide-induced pulmonary inflammation. Mol. Immunol. 2017, 90, 150–157. [Google Scholar] [CrossRef]
- Hu, B.; Tong, F.; Xu, L.; Shen, Z.; Yan, L.; Xu, G.; Shen, R. Role of Calcium Sensing Receptor in Streptozotocin-Induced Diabetic Rats Exposed to Renal Ischemia Reperfusion Injury. Kidney Blood Press. Res. 2018, 43, 276–286. [Google Scholar] [CrossRef]
- Lau, L.T.; Yu, A.C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef]
- Benveniste, E.N. Cytokine actions in the central nervous system. Cytokine Growth Factor Rev. 1998, 9, 259–275. [Google Scholar] [CrossRef]
- Pratt, B.M.; McPherson, J.M. TGF-beta in the central nervous system: Potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor Rev. 1997, 8, 267–292. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef]
- Ambrosini, E.; Remoli, M.E.; Giacomini, E.; Rosicarelli, B.; Serafini, B.; Lande, R.; Aloisi, F.; Coccia, E.M. Astrocytes produce dendritic cell-attracting chemokines in vitro and in multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2005, 64, 706–715. [Google Scholar] [CrossRef]
- Strack, A.; Asensio, V.C.; Campbell, I.L.; Schlüter, D.; Deckert, M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-gamma. Acta Neuropathol. 2002, 103, 458–468. [Google Scholar] [CrossRef]
- Sokolova, A.; Hill, M.D.; Rahimi, F.; Warden, L.A.; Halliday, G.M.; Shepherd, C.E. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2009, 19, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Dal Pra, I.; Menapace, L.; Pacchiana, R.; Whitfield, J.F.; Armato, U. Soluble amyloid beta-peptide and myelin basic protein strongly stimulate, alone and in synergism with joint proinflammatory cytokines, the expression of functional nitric oxide synthase-2 in normal adult human astrocytes. Int. J. Mol. Med. 2005, 16, 801–807. [Google Scholar]
- Dal Prà, I.; Armato, U.; Chiarini, A. Specific interactions of calcium-sensing receptors (CaSRs) with soluble amyloid-β peptides—A study using cultured normofunctioning adult human astrocytes. In Proceedings of the 2nd International Symposium on the Calcium-sensing Receptor, San Diego, CA, USA, 3–4 March 2015; pp. 90–91. [Google Scholar]
- Gulyaeva, N.V.; Stepanichev, M.Y. Abeta(25–35) as proxyholder for amyloidogenic peptides: In vivo evidence. Exp. Neurol. 2010, 222, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, A.; Otth, C.; Mujica, L.; Concha, I.I.; Maccioni, R.B. Interleukin-3 prevents neuronal death induced by amyloid peptide. BMC Neurosci. 2007, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M.; Dell’Ombra, N.; Zambito, A.M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Chitotriosidase and inflammatory mediator levels in Alzheimer’s disease and cerebrovascular dementia. Eur. J. Neurosci. 2006, 23, 2648–2656. [Google Scholar] [CrossRef]
- Ashutosh; Kou, W.; Cotter, R.; Borgmann, K.; Wu, L.; Persidsky, R.; Sakhuja, N.; Ghorpade, A. CXCL8 protects human neurons from amyloid-beta-induced neurotoxicity: Relevance to Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2011, 412, 565–571. [Google Scholar]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar]
- Weber, M.; Uguccioni, M.; Ochensberger, B.; Baggiolini, M.; Clark-Lewis, I.; Dahinden, C.A. Monocyte chemotactic protein MCP-2 activates human basophil and eosinophil leukocytes similar to MCP-3. J. Immunol. 1995, 154, 4166–4172. [Google Scholar]
- Kim, M.O.; Suh, H.S.; Brosnan, C.F.; Lee, S.C. Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-beta. J. Neurochem. 2004, 90, 297–308. [Google Scholar] [CrossRef]
- Lee, E.; Kim, H. The anti-inflammatory role of tissue inhibitor of metalloproteinase-2 in lipopolysaccharide-stimulated microglia. J. Neuroinflammation 2014, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Benveniste, E.N. Adhesion molecule expression and regulation on cells of the central nervous system. J. Neuroimmunol. 1999, 98, 77–88. [Google Scholar] [CrossRef]
- Zhou, Z.; Wu, Q.; Lu, Y.; Zhang, X.; Lv, S.; Shao, J.; Zhou, Y.; Chen, J.; Hou, L.; Huang, C.; et al. Crosstalk between soluble PDGF-BB and PDGFR b promotes astrocytic activation and synaptic recovery in the hippocampus after subarachnoid hemorrhage. FASEB J. 2019, 33, 9588–9601. [Google Scholar] [CrossRef] [PubMed]
- Croitoru-Lamoury, J.; Guillemin, G.J.; Boussin, F.D.; Mognetti, B.; Gigout, L.I.; Chéret, A.; Vaslin, B.; Le Grand, R.; Brew, B.J.; Dormont, D. Expression of chemokines and their receptors in human and simian astrocytes: Evidence for a central role of TNF alpha and IFN gamma in CXCR4 and CCR5 modulation. Glia 2003, 41, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Schmitz, M.L.; Weber, A.; Roxlau, T.; Gaestel, M.; Kracht, M. Signal integration, crosstalk mechanisms and networks in the function of inflammatory cytokines. Biochim. Biophys. Acta 2011, 1813, 2165–2175. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiyota, T.; Horiba, M.; Buescher, J.L.; Walsh, S.M.; Gendelman, H.E.; Ikezu, T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am. J. Pathol. 2007, 170, 680–692. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2011, 8, 150. [Google Scholar] [CrossRef]
- Tsakiri, N.; Kimber, I.; Rothwell, N.J.; Pinteaux, E. Mechanisms of interleukin-6 synthesis and release induced by interleukin-1 and cell depolarisation in neurones. Mol. Cell. Neurosci. 2008, 37, 110–118. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Zhou, X.; Hendy, G.N. The proinflammatory cytokine, interleukin-6, upregulates calcium-sensing receptor gene transcription via Stat 1/3 and Sp 1/3. J. Biol. Chem. 2008, 283, 13586–13600. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Glaser, N.; Griffen, S.; Wang-Polagruto, J.; Miguelino, E.; Jialal, I. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes 2006, 55, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Gruol, D.L.; Vo, K.; Bray, J.G. Increased astrocyte expression of IL-6 or CCL2 in transgenic mice alters levels of hippocampal and cerebellar proteins. Front. Cell. Neurosci. 2014, 8, 234. [Google Scholar] [CrossRef]
- Haim, L.B.; Ceyzériat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 Pathway Is a Common Inducer of Astrocyte Reactivity in Alzheimer’s and Huntington’s Diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef]
- Brugg, B.; Dubreuil, Y.L.; Huber, G.; Wollman, E.E.; Delhaye-Bouchaud, N.; Mariani, J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc. Natl. Acad. Sci. USA 1995, 92, 3032–3035. [Google Scholar] [CrossRef]
- Wennström, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [PubMed]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural. Plast. 2016, 2016, 6427537. [Google Scholar] [CrossRef] [PubMed]
- Staunton, D.E.; Marlin, S.D.; Stratowa, C.; Dustin, M.L.; Springer, T.A. Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell 1988, 52, 925–933. [Google Scholar] [CrossRef]
- Ramos, T.N.; Bullard, D.C.; Barnum, S.R. ICAM-1: Isoforms and phenotypes. J. Immunol. 2014, 192, 4469–4474. [Google Scholar] [CrossRef] [PubMed]
- Müller, N. The Role of Intercellular Adhesion Molecule-1 in the Pathogenesis of Psychiatric Disorders. Front. Pharmacol. 2019, 10, 1251. [Google Scholar] [CrossRef] [PubMed]
- Tsakadze, N.L.; Sithu, S.D.; Sen, U.; English, W.R.; Murphy, G.; D’Souza, S.E. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM- 1). J. Biol. Chem. 2006, 281, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef]
- Schmal, H.; Czermak, B.J.; Lentsch, A.B.; Bless, N.M.; Beck-Schimmer, B.; Friedl, H.P.; Ward, P.A. Soluble ICAM-1 activates lung macrophages and enhances lung injury. J. Immunol. 1998, 161, 3685–3693. [Google Scholar]
- Witkowska, A.M.; Borawska, M.H. Soluble intercellular adhesion molecule-1 (s-ICAM-1): An overview. Eur. Cytokine Netw. 2004, 15, 91–98. [Google Scholar]
- Hua, S. Targeting sites of inflammation: Intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Front. Pharmacol. 2013, 4, 127. [Google Scholar] [CrossRef]
- Luo, Y.; Berman, M.A.; Zhai, Q.; Fischer, F.R.; Abromson-Leeman, S.R.; Zhang, Y.; Kuziel, W.A.; Gerard, C.; Dorf, M.E. RANTES stimulates inflammatory cascades and receptor modulation in murine astrocytes. Glia 2002, 39, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Michalopoulou, M.; Nikolaou, C.; Cambouri, C.; Rombos, A.; Dimitrakopoulos, A.; Vassilopoulos, D. The role of soluble intercellular adhesion molecules in neurodegenerative disorders. J. Neurol. Sci. 2005, 228, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Hidalgo, J.J.; Nithuairisg, S.; Stockmeier, C.; Rajkowska, G. Distribution of ICAM-1 immunoreactivity during aging in the human orbitofrontal cortex. Brain Behav. Immun. 2007, 21, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.M.; Otte-Höller, I.; Westphal, J.R.; Wesseling, P.; Ruiter, D.J.; de Waal, R.M. Accumulation of intercellular adhesion molecule-1 in senile plaques in brain tissue of patients with Alzheimer’s disease. Am. J. Pathol. 1994, 144, 104–116. [Google Scholar] [PubMed]
- Akiyama, H.; Kawamata, T.; Yamada, T.; Tooyama, I.; Ishii, T.; McGeer, P.L. Expression of intercellular adhesion molecule (ICAM)-1 by a subset of astrocytes in Alzheimer disease and some other degenerative neurological disorders. Acta Neuropathol. 1993, 85, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.F.; Tang, T.M.; Adler, C.H.; Caviness, J.N.; Sabbagh, M.N.; Serrano, G.E.; Sue, L.I.; Beach, T.G. Changes in CD200 and intercellular adhesion molecule-1 (ICAM-1) levels in brains of Lewy body disorder cases are associated with amounts of Alzheimer’s pathology not alpha-synuclein pathology. Neurobiol. Aging 2017, 54, 175–186. [Google Scholar] [CrossRef]
- Rivieccio, M.A.; John, G.R.; Song, X.; Suh, H.S.; Zhao, Y.; Lee, S.C.; Brosnan, C.F. The cytokine IL-1beta activates IFN response factor 3 in human fetal astrocytes in culture. J. Immunol. 2005, 174, 3719–3726. [Google Scholar] [CrossRef]
- Lin, M.S.; Hung, K.S.; Chiu, W.T.; Sun, Y.Y.; Tsai, S.H.; Lin, J.W.; Lee, Y.H. Curcumin enhances neuronal survival in N-methyl-d-aspartic acid toxicity by inducing RANTES expression in astrocytes via PI-3K and MAPK signaling pathways. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 931–938. [Google Scholar] [CrossRef]
- Chiarini, A.; Dal Pra, I.; Marconi, M.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-sensing receptor (CaSR) in human brain’s pathophysiology: Roles in late-onset Alzheimer’s disease (LOAD). Curr. Pharm. Biotechnol. 2009, 10, 317–326. [Google Scholar] [CrossRef]
- Chou, S.Y.; Weng, J.Y.; Lai, H.L.; Liao, F.; Sun, S.H.; Tu, P.H.; Dickson, D.W.; Chern, Y. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J. Neurosci. 2008, 28, 3277–3290. [Google Scholar] [CrossRef]
- Stanley, A.C.; Lacy, P. Pathways for cytokine secretion. Physiology (Bethesda) 2010, 25, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. RANTES upregulation in the Alzheimer’s disease brain: A possible neuroprotective role. Neurobiol Aging 2010, 31, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, Y.; Zhai, Q.; Ma, L.; Dorf, M.E. Negative role of cAMP-dependent protein kinase A in RANTES-mediated transcription of proinflammatory mediators through Raf. FASEB J. 2003, 17, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Tripathy, D.; Grammas, P. RANTES release contributes to the protective action of PACAP38 against sodium nitroprusside in cortical neurons. Neuropeptides 2009, 43, 315–320. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galimberti, D.; Schoonenboom, N.; Scheltens, P.; Fenoglio, C.; Bouwman, F.; Venturelli, E.; Guidi, I.; Blankenstein, M.A.; Bresolin, N.; Scarpini, E. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch. Neurol. 2006, 63, 538–543. [Google Scholar] [CrossRef]
- Stuart, M.J.; Singhal, G.; Baune, B.T. Systematic Review of the Neurobiological Relevance of Chemokines to Psychiatric Disorders. Front. Cell. Neurosci. 2015, 9, 357. [Google Scholar] [CrossRef]
- Hayes, L.N.; Severance, E.G.; Leek, J.T.; Gressitt, K.L.; Rohleder, C.; Coughlin, J.M.; Leweke, F.M.; Yolken, R.H.; Sawa, A. Inflammatory molecular signature associated with infectious agents in psychosis. Schizophr. Bull. 2014, 40, 963–972. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokines | |||
|---|---|---|---|
| Name | Gene | UniProtKB ID | Expression |
| Interleukin-1α | IL1A | P01583 | nd |
| Interleukin-1β | IL1B | P01584 | + |
| Interleukin-2 | IL2 | P60568 | nd |
| Interleukin-3 | IL3 | P08700 | + |
| Interleukin-4 | IL4 | P05112 | nd |
| Interleukin-6 | IL6 | P05231 | + |
| Interleukin-7 | IL7 | P13232 | nd |
| Interleukin-10 | IL10 | P22301 | nd |
| Interleukin-11 | IL11 | P20809 | nd |
| Interleukin-12 subunit-α | IL12A | P29459 | nd |
| Interleukin-12 subunit-β | IL12B | P29460 | nd |
| Interleukin-13 | IL13 | P35225 | nd |
| Interleukin-15 | IL15 | P40933 | nd |
| Interleukin-16 | IL16 | Q14005 | + |
| Interleukin-17A | IL17 | Q16552 | nd |
| Interferon-γ | IFNG | P01579 | nd |
| Granulocyte colony-stimulating factor | CSF3 | P09919 | nd |
| Granulocyte-macrophage colony-stimulating factor | CSF2 | P04141 | nd |
| Lymphotoxin-α | TNFB | P01374 | nd |
| Macrophage colony-stimulating factor-1 | CSF1 | P09603 | nd |
| Transforming growth factor-β1 | TGFB1 | P01137 | nd |
| Tumor necrosis factor-α | TNFA | P01375 | nd |
| Cytokine Soluble Receptors | |||
| Interleukin-6 receptor subunit-α | IL6R | P08887 | nd |
| TNF receptor superfamily member-1α | TNFRSF1A | P19438 | nd |
| TNF receptor Superfamily Member-1β | TNFRSF1B | P20333 | nd |
| Chemokines | |||
| C-C motif chemokine-1 | CCL1 | P22362 | nd |
| C-C motif chemokine-5/RANTES | CCL5 | P13501 | + |
| C-X-C motif chemokine-9 | CXCL9 | Q07325 | nd |
| C-X-C motif chemokine-10 | CXCL10 | P02778 | nd |
| Eotaxin-1 | CCL11 | P51671 | nd |
| Eotaxin-2 | CCL24 | O00175 | nd |
| Interleukin-8 | CXCL8 | P10145 | + |
| Monocyte chemoattractant protein-1 | CCL2/MCP1 | P13500 | + |
| Monocyte chemoattractant protein-2 | CCL8/MCP2 | P80075 | + |
| Macrophage inflammatory protein-1α | CCL3 | P10147 | nd |
| Macrophage inflammatory protein-1β | CCL4 | P13236 | nd |
| Macrophage inflammatory protein-5 | CCL15 | Q16663 | nd |
| Other agents | |||
| Soluble intercellular adhesion molecule-1 (s-ICAM-1] | ICAM1 | P05362 | + |
| Metalloproteinase inhibitor-2 | TIMP2 | P16035 | + |
| Platelet-derived growth factor subunit-B | PDGFB | P01127 | + |
| Treatments | Cumulative Secretion § | % Changes vs. CTR | % Changes vs. fAβ25–35 | |
|---|---|---|---|---|
| IL-6 | CTR | 502.1 ± 39.6 | ||
| fAβ25–35 | 905.6 ± 48.4 | +80.4 * | ||
| fAβ25–35 +NPS 2143 | 576.9 ± 41.0 | +14.9 | −81.5 * | |
| s-ICAM-1 | CTR | 30.6 ± 2.3 | ||
| fAβ25–35 | 59.7 ± 2.7 | +95.1 * | ||
| fAβ25–35 + NPS 2143 | 41 ± 2.6 | +34.0 * | −64.3 * | |
| RANTES | CTR | 42.9 ± 13.3 | ||
| fAβ25–35 | 226 ± 17.2 | +426.8 * | ||
| fAβ25–35 + NPS 2143 | 149.3 ± 7.4 | +248.0 * | −41.9 * | |
| MCP-2 | CTR | 185.9 ± 29.3 | ||
| fAβ25–35 | 6897.9 ± 248 | +3611.0 * | ||
| fAβ25–35 + NPS 2143 | 2717.6 ± 320 | +1361.9 * | −62.3 * |
| Agent | Proinflammatory Roles |
| IL-6 | Induces extensive gliosis and microglial phagocytosis of Aβs deposits in vivo [77] Increases Tau protein hyperphosphorylation in neurons [78] Increases the levels of hippocampal and cerebellar GFAP, glutamine synthase, STAT-3, phosphorylated STAT-3, and phosphorylated pp42/44 MAPK [79] Decreases the levels of Syn 1, GAD65/67, GluA1, and GluN1 [79] Upregulates CASR gene transcription [75] Upregulates the transcription of several other genes including ICAM-1, RANTES, MCP-2, VEGF-A [73,74] |
| s-ICAM-1 | Promotes lymphocytes and leukocyte trafficking through the BBB [87] Partakes in BBB dysfunction and CNS infiltration by immune cells [87] Localizes to vessels, early and late stage amyloid senile plaques, and peri-plaque astrocytes in AD brains [96,97] |
| RANTES | Attracts and activates eosinophil and basophil leukocytes [104] Is relevant to the neuroinflammatory cascade that contributes to neurodegeneration in AD brains [62,104] Stimulates astrocytes’ production and release of proinflammatory mediators [93,106] The RANTES/CCR3 signaling lowers the endogenous levels of cAMP [106] May contribute to upregulating the s-ICAM-1 levels [93] |
| MCP-2 | Enhances chemotaxis of proinflammatory cells (monocytes or other leukocyte populations) to inflamed CNS areas [109] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells 2020, 9, 1386. https://doi.org/10.3390/cells9061386
Chiarini A, Armato U, Hu P, Dal Prà I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells. 2020; 9(6):1386. https://doi.org/10.3390/cells9061386
Chicago/Turabian StyleChiarini, Anna, Ubaldo Armato, Peng Hu, and Ilaria Dal Prà. 2020. "CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes" Cells 9, no. 6: 1386. https://doi.org/10.3390/cells9061386
APA StyleChiarini, A., Armato, U., Hu, P., & Dal Prà, I. (2020). CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells, 9(6), 1386. https://doi.org/10.3390/cells9061386