HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Treatments
2.2. Western Blotting
2.3. Fluorescence Microscopy
2.4. Transmission Electron Microscopy
2.5. qRT-PCR
2.6. Statistical Analysis
3. Results
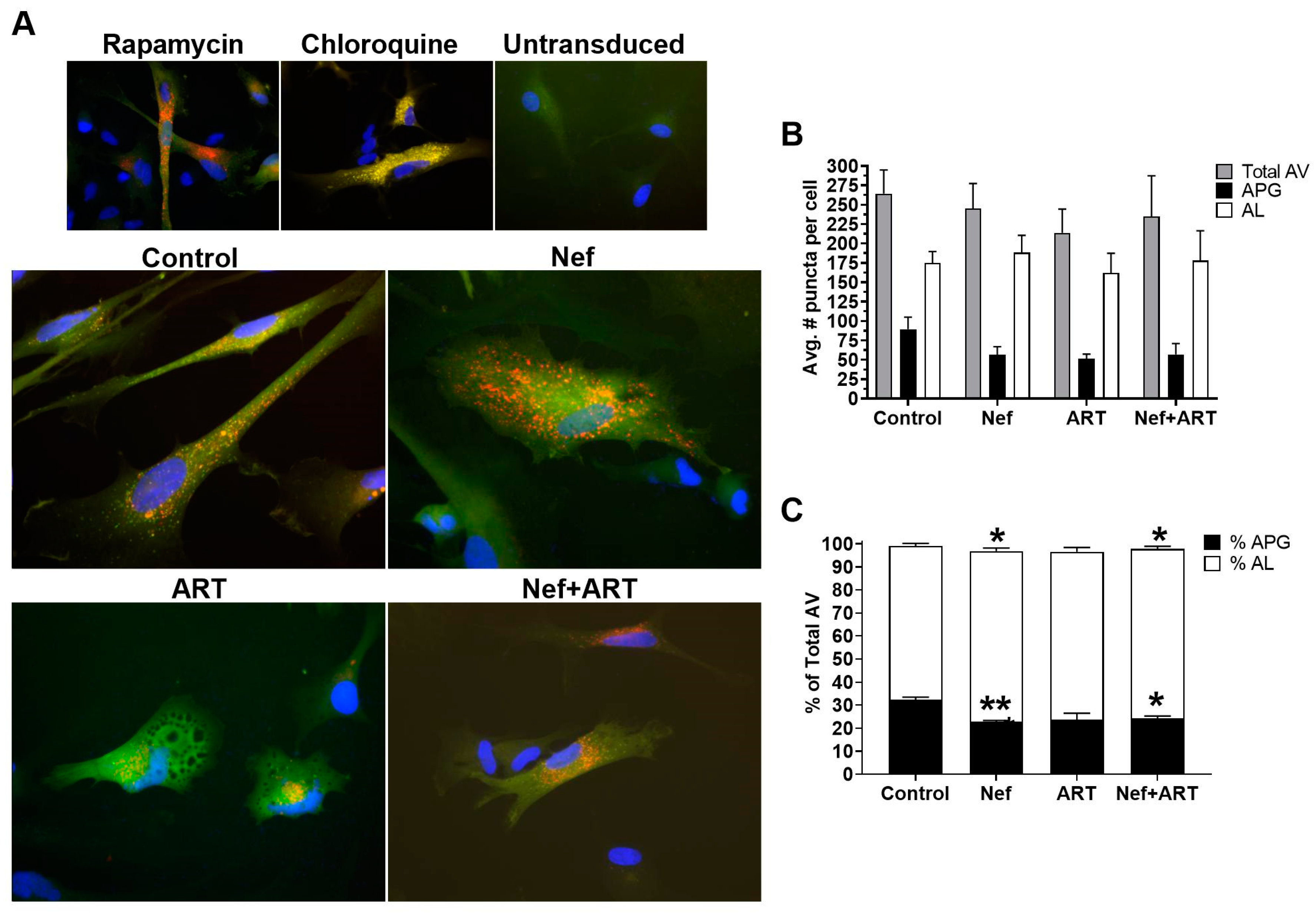
3.1. Nef and/or ART Imbalance Autophagy
3.2. Nef Alone or in Combination with ART Accelerates APG Maturation in Astrocytes
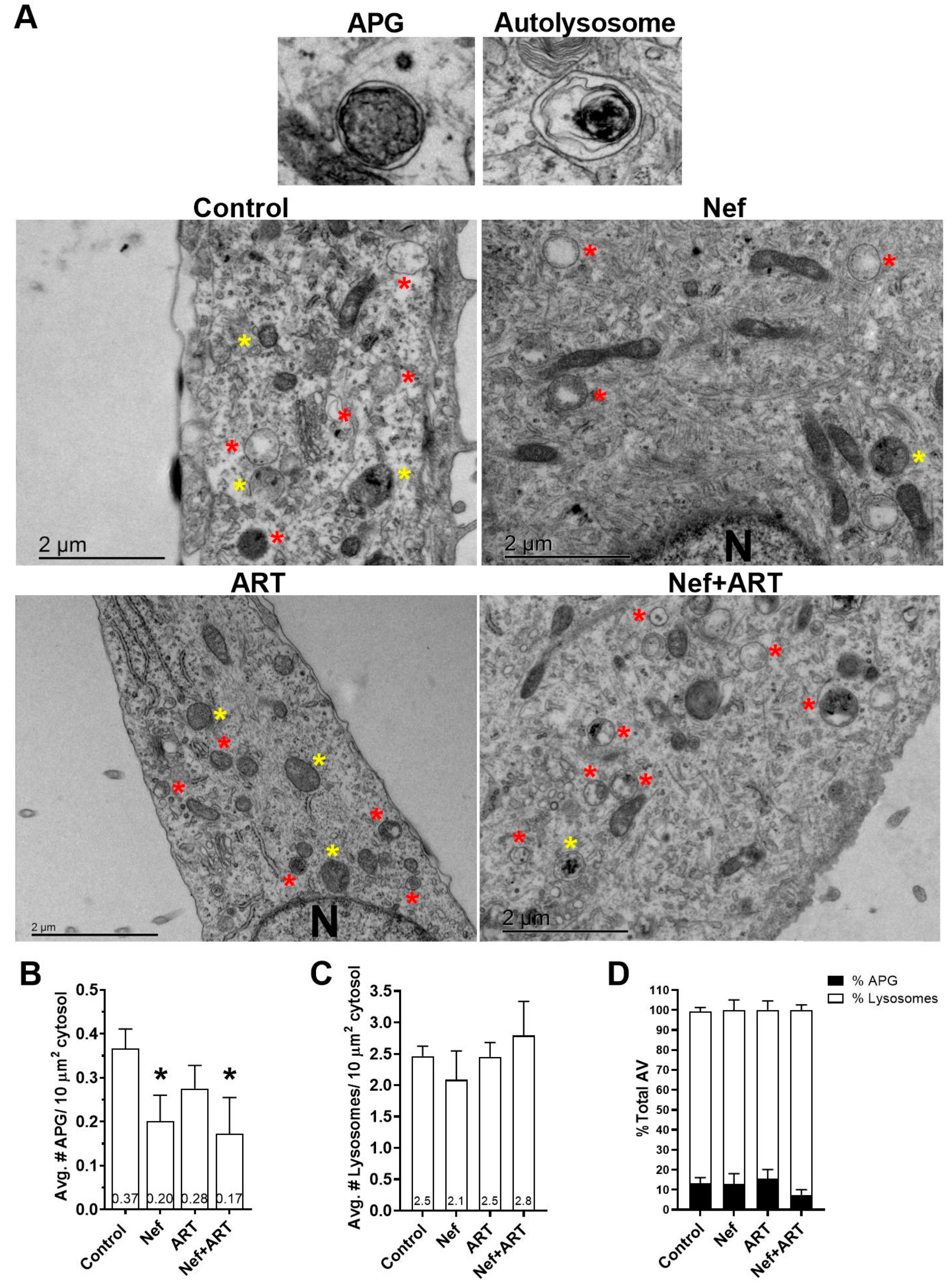
3.3. Nef and/or ART Decrease the Number of APG in Human Astrocytes
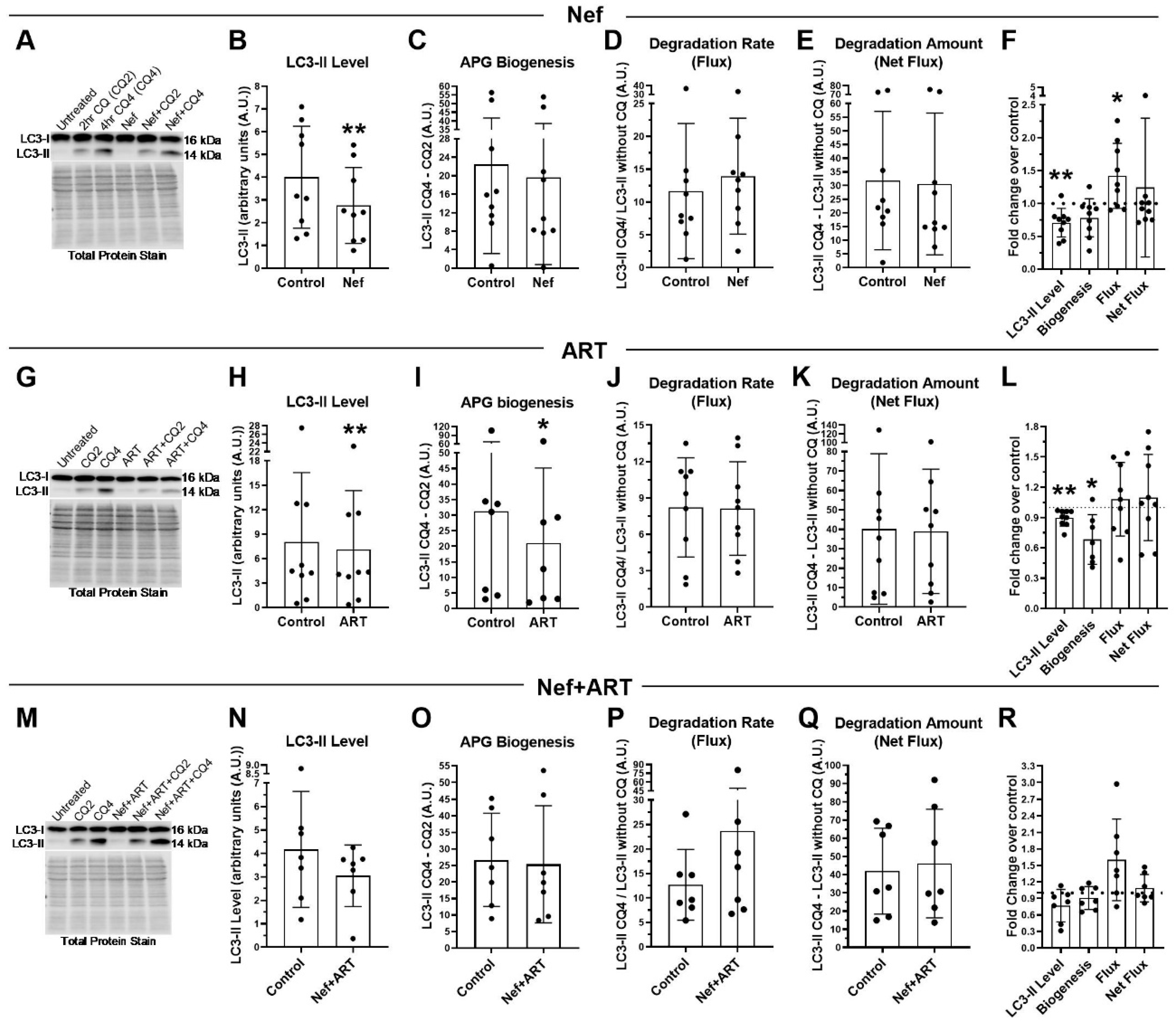
3.4. Nef and/or ART induce “Long-Term” Autophagy Imbalance in Astrocytes
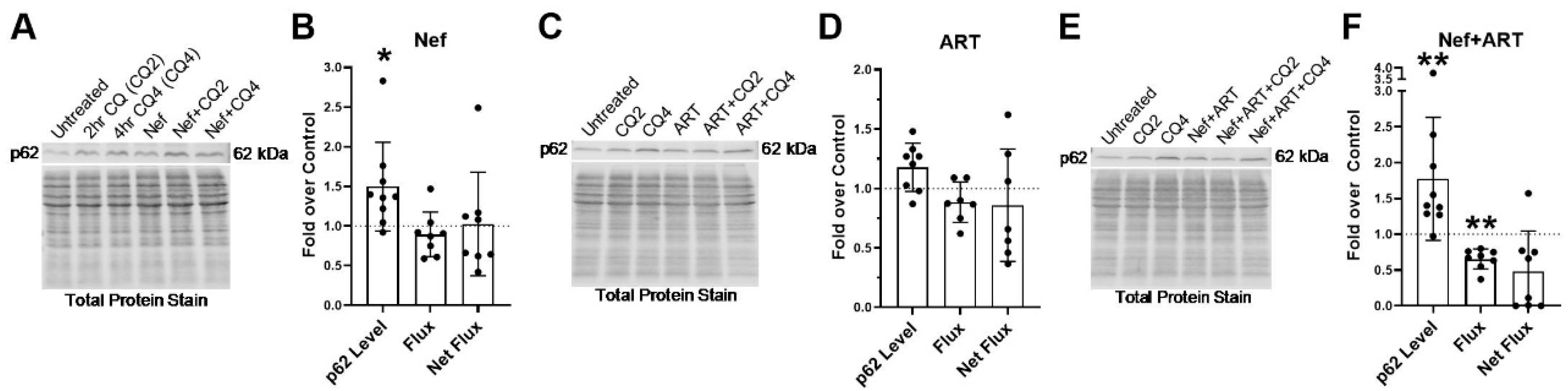
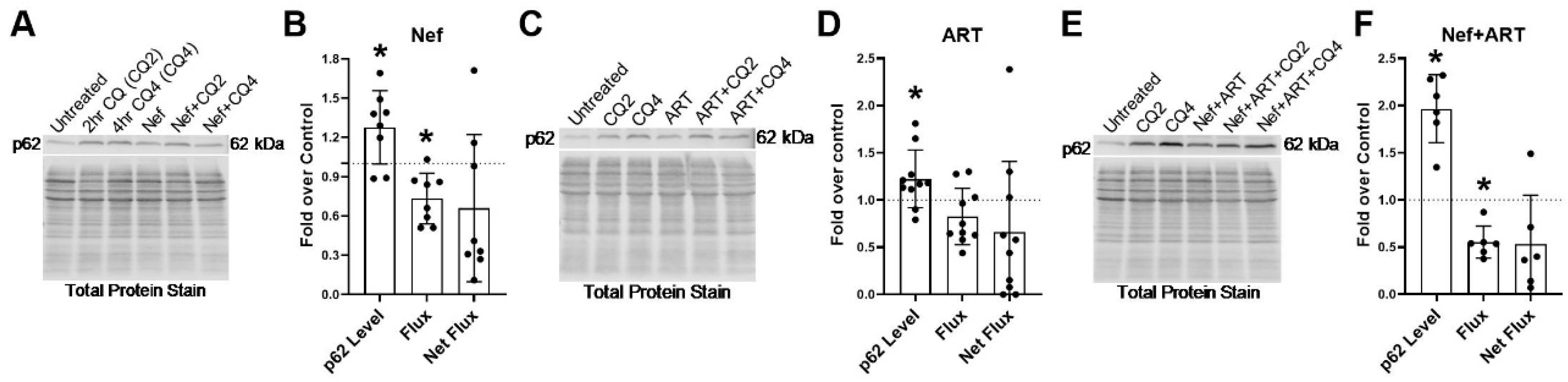
3.5. Nef and/or ART Compromise p62-Mediated Selective Autophagy Short and “Long Term”
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cysique, L.A.; Brew, B.J. Prevalence of non-confounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J. Neurovirol. 2011, 17, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R., Jr.; Deutsch, R.; Letendre, S.; Ellis, R.J.; Casaletto, K.; Marquine, M.J.; Woods, S.P.; Vaida, F.; Atkinson, J.H.; et al. Neurocognitive change in the era of HIV combination antiretroviral therapy: The longitudinal CHARTER study. Clin. Infect. Dis. 2015, 60, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Gott, C.; Gates, T.; Dermody, N.; Brew, B.J.; Cysique, L.A. Cognitive change trajectories in virally suppressed HIV-infected individuals indicate high prevalence of disease activity. PLoS ONE 2017, 12, e0171887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spudich, S.; Robertson, K.R.; Bosch, R.J.; Gandhi, R.T.; Cyktor, J.C.; Mar, H.; Macatangay, B.J.; Lalama, C.M.; Rinaldo, C.; Collier, A.C.; et al. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J. Clin. Investig. 2019, 129, 3339–3346. [Google Scholar] [CrossRef] [Green Version]
- Tozzi, V.; Balestra, P.; Lorenzini, P.; Bellagamba, R.; Galgani, S.; Corpolongo, A.; Vlassi, C.; Larussa, D.; Zaccarelli, M.; Noto, P.; et al. Prevalence and risk factors for human immunodeficiency virus-associated neurocognitive impairment, 1996 to 2002: Results from an urban observational cohort. J. Neurovirol. 2005, 11, 265–273. [Google Scholar] [CrossRef]
- Tozzi, V.; Balestra, P.; Murri, R.; Galgani, S.; Bellagamba, R.; Narciso, P.; Antinori, A.; Giulianelli, M.; Tosi, G.; Fantoni, M.; et al. Neurocognitive impairment influences quality of life in HIV-infected patients receiving HAART. Int. J. STD AIDS 2004, 15, 254–259. [Google Scholar] [CrossRef]
- Jones, J.D.; Kuhn, T.; Levine, A.; Sacktor, N.; Munro, C.A.; Teplin, L.A.; D’Souza, G.; Martin, E.M.; Becker, J.T.; Miller, E.N.; et al. Changes in cognition precede changes in HRQoL among HIV+ males: Longitudinal analysis of the multicenter AIDS cohort study. Neuropsychology 2019, 33, 370–378. [Google Scholar] [CrossRef]
- van Gorp, W.G.; Baerwald, J.P.; Ferrando, S.J.; McElhiney, M.C.; Rabkin, J.G. The relationship between employment and neuropsychological impairment in HIV infection. J. Int. Neuropsychol. Soc. 1999, 5, 534–539. [Google Scholar] [CrossRef]
- Heaton, R.K.; Marcotte, T.D.; Mindt, M.R.; Sadek, J.; Moore, D.J.; Bentley, H.; McCutchan, J.A.; Reicks, C.; Grant, I.; Group, H. The impact of HIV-associated neuropsychological impairment on everyday functioning. J. Int. Neuropsychol. Soc. 2004, 10, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Tozzi, V.; Balestra, P.; Serraino, D.; Bellagamba, R.; Corpolongo, A.; Piselli, P.; Lorenzini, P.; Visco-Comandini, U.; Vlassi, C.; Quartuccio, M.E.; et al. Neurocognitive impairment and survival in a cohort of HIV-infected patients treated with HAART. AIDS Res. Hum. Retrovir. 2005, 21, 706–713. [Google Scholar] [CrossRef]
- Vivithanaporn, P.; Heo, G.; Gamble, J.; Krentz, H.B.; Hoke, A.; Gill, M.J.; Power, C. Neurologic disease burden in treated HIV/AIDS predicts survival: A population-based study. Neurology 2010, 75, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Parikh, N.U.; Aalinkeel, R.; Reynolds, J.L.; Dmello, R.; Schwartz, S.A.; Mahajan, S.D. United States National Trends in Mortality, Length of Stay (LOS) and Associated Costs of Cognitive Impairment in HIV Population from 2005 to 2014. AIDS Behav. 2018, 22, 3198–3208. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef]
- Ranki, A.; Nyberg, M.; Ovod, V.; Haltia, M.; Elovaara, I.; Raininko, R.; Haapasalo, H.; Krohn, K. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS 1995, 9, 1001–1008. [Google Scholar] [CrossRef]
- Takahashi, K.; Wesselingh, S.L.; Griffin, D.E.; McArthur, J.C.; Johnson, R.T.; Glass, J.D. Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann. Neurol. 1996, 39, 705–711. [Google Scholar] [CrossRef]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef] [Green Version]
- Tornatore, C.; Chandra, R.; Berger, J.R.; Major, E.O. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology 1994, 44, 481–487. [Google Scholar] [CrossRef]
- Trillo-Pazos, G.; Diamanturos, A.; Rislove, L.; Menza, T.; Chao, W.; Belem, P.; Sadiq, S.; Morgello, S.; Sharer, L.; Volsky, D.J. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain. Pathol. 2003, 13, 144–154. [Google Scholar] [CrossRef]
- Wang, T.; Green, L.A.; Gupta, S.K.; Amet, T.; Byrd, D.J.; Yu, Q.; Twigg, H.L., 3rd; Clauss, M. Intracellular Nef detected in peripheral blood mononuclear cells from HIV patients. AIDS Res. Hum. Retrovir. 2015, 31, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef]
- Raymond, A.D.; Campbell-Sims, T.C.; Khan, M.; Lang, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV Type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res. Hum. Retrovir. 2011, 27, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Crumpacker, C.S. Human immunodeficiency virus type 1 RNA in peripheral blood mononuclear cells of patients receiving prolonged highly active antiretroviral therapy. J. Infect. Dis. 2001, 184, 1341–1344. [Google Scholar] [CrossRef]
- Olivetta, E.; Arenaccio, C.; Manfredi, F.; Anticoli, S.; Federico, M. The Contribution of Extracellular Nef to HIV-Induced Pathogenesis. Curr. Drug Targets 2016, 17, 46–53. [Google Scholar] [CrossRef]
- Puzar Dominkus, P.; Ferdin, J.; Plemenitas, A.; Peterlin, B.M.; Lenassi, M. Nef is secreted in exosomes from Nef.GFP-expressing and HIV-1-infected human astrocytes. J. Neurovirol. 2017, 23, 713–724. [Google Scholar] [CrossRef]
- Sami Saribas, A.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell Death Dis. 2017, 8, e2542. [Google Scholar] [CrossRef] [Green Version]
- Cheney, L.; Hou, J.C.; Morrison, S.; Pessin, J.; Steigbigel, R.T. Nef inhibits glucose uptake in adipocytes and contributes to insulin resistance in human immunodeficiency virus type I infection. J. Infect. Dis. 2011, 203, 1824–1831. [Google Scholar] [CrossRef]
- James, C.O.; Huang, M.B.; Khan, M.; Garcia-Barrio, M.; Powell, M.D.; Bond, V.C. Extracellular Nef protein targets CD4+ T cells for apoptosis by interacting with CXCR4 surface receptors. J. Virol. 2004, 78, 3099–3109. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; He, B.; Chiu, A.; Knowles, D.M.; Chadburn, A.; Cerutti, A. Human immunodeficiency virus 1 Nef suppresses CD40-dependent immunoglobulin class switching in bystander B cells. Nat. Immunol. 2006, 7, 302–310. [Google Scholar] [CrossRef]
- Quaranta, M.G.; Mattioli, B.; Spadaro, F.; Straface, E.; Giordani, L.; Ramoni, C.; Malorni, W.; Viora, M. HIV-1 Nef triggers Vav-mediated signaling pathway leading to functional and morphological differentiation of dendritic cells. FASEB J. 2003, 17, 2025–2036. [Google Scholar] [CrossRef]
- Chompre, G.; Cruz, E.; Maldonado, L.; Rivera-Amill, V.; Porter, J.T.; Noel, R.J., Jr. Astrocytic expression of HIV-1 Nef impairs spatial and recognition memory. Neurobiol. Dis. 2013, 49, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Trillo-Pazos, G.; McFarlane-Abdulla, E.; Campbell, I.C.; Pilkington, G.J.; Everall, I.P. Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain. Res. 2000, 864, 315–326. [Google Scholar] [CrossRef]
- Acheampong, E.A.; Parveen, Z.; Muthoga, L.W.; Kalayeh, M.; Mukhtar, M.; Pomerantz, R.J. Human Immunodeficiency virus type 1 Nef potently induces apoptosis in primary human brain microvascular endothelial cells via the activation of caspases. J. Virol. 2005, 79, 4257–4269. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.; D’Agostino, L.; Wilson, J.; Tuzer, F.; Torres, C. Astrocyte Senescence and Metabolic Changes in Response to HIV Antiretroviral Therapy Drugs. Front. Aging Neurosci. 2017, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Vivithanaporn, P.; Asahchop, E.L.; Acharjee, S.; Baker, G.B.; Power, C. HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS 2016, 30, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Pawitwar, S.; Karuppan, M.K.M.; Kashanchi, F.; El-Hage, N. Morphine counteracts the antiviral effect of antiretroviral drugs and causes upregulation of p62/SQSTM1 and histone-modifying enzymes in HIV-infected astrocytes. J. Neurovirol. 2019. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Ferguson, C.J.; Lenk, G.M.; Meisler, M.H. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum. Mol. Genet. 2009, 18, 4868–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrom, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergstrom, J.; Erlandsson, A. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.T.; Ureshino, R.P.; et al. Overexpression of alpha-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, L.; Chen, S.; Yang, D.; Wang, Y.; Zhang, X.; Wang, Z.; Le, W. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 2011, 7, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Kaminski, R.; Mullen, B.; Gordon, J.; Burdo, T.H.; Cheung, J.Y.; Feldman, A.M.; Madesh, M.; Khalili, K. HIV-1 Nef-induced cardiotoxicity through dysregulation of autophagy. Sci. Rep. 2017, 7, 8572. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, E.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [Green Version]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.J.; Lopiccolo, J.; Tsurutani, J.; Shoemaker, R.H.; Best, C.J.; Abu-Asab, M.S.; Borojerdi, J.; Warfel, N.A.; Gardner, E.R.; Danish, M.; et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin. Cancer Res. 2007, 13, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, K.; VanDeVen, N.A.; Sorenson, D.R.; Daudi, S.; Liu, J.R. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol. Oncol. 2009, 112, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy 2020, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.; Dumaop, W.; Rockenstein, E.; Mante, M.; Spencer, B.; Grant, I.; Ellis, R.; Letendre, S.; Patrick, C.; Adame, A.; et al. Age-dependent molecular alterations in the autophagy pathway in HIVE patients and in a gp120 tg mouse model: Reversal with beclin-1 gene transfer. J. Neurovirol. 2013, 19, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [Green Version]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS ONE 2008, 3, e2906. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kiosses, W.B.; Fox, H.S. Decreased neuronal autophagy in HIV dementia: A mechanism of indirect neurotoxicity. Autophagy 2008, 4, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Guo, M.L.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, L.; Toborek, M. Dysregulation of Endoplasmic Reticulum Stress and Autophagic Responses by the Antiretroviral Drug Efavirenz. Mol. Pharmacol. 2015, 88, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Estrada-Bueno, H.; Dever, S.M.; Gewirtz, D.A.; Kashanchi, F.; El-Hage, N. Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes. Viruses 2017, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Dever, S.M.; Rodriguez, M.; Lapierre, J.; Costin, B.N.; El-Hage, N. Differing roles of autophagy in HIV-associated neurocognitive impairment and encephalitis with implications for morphine co-exposure. Front. Microbiol. 2015, 6, 653. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.; Langford, D.; Masliah, E. HIV and antiretroviral therapy in the brain: Neuronal injury and repair. Nat. Rev. Neurosci. 2007, 8, 33–44. [Google Scholar] [CrossRef]
- Eugenin, E.A.; D’Aversa, T.G.; Lopez, L.; Calderon, T.M.; Berman, J.W. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem. 2003, 85, 1299–1311. [Google Scholar] [CrossRef]
- Yilmaz, A.; Price, R.W.; Gisslen, M. Antiretroviral drug treatment of CNS HIV-1 infection. J. Antimicrob. Chemother. 2012, 67, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [Green Version]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009, 452, 143–164. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. BioMed Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Zhang, Z.W.; Li, Z.L. Cell Death-Autophagy Loop and Glutamate-Glutamine Cycle in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 231. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, E.; Rodriguez-Navarro, J.A. Autophagy and amino acid metabolism in the brain: Implications for epilepsy. Amino Acids 2015, 47, 2113–2126. [Google Scholar] [CrossRef]
- Harris, J. Autophagy and cytokines. Cytokine 2011, 56, 140–144. [Google Scholar] [CrossRef]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherra, S.J., 3rd; Chu, C.T. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008, 3, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Qin, Z.H. Beclin 1, Bcl-2 and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Adefolaju, G.A.; Theron, K.E.; Hosie, M.J. BAX/BCL-2 mRNA and protein expression in human breast MCF-7 cells exposed to drug vehicles-methanol and dimethyl sulfoxide (DMSO) for 24 hrs. Niger. Med. J. 2015, 56, 169–174. [Google Scholar] [CrossRef]
- Adefolaju, G.A.; Theron, K.E.; Hosie, M.J. Effects of HIV protease, nucleoside/non-nucleoside reverse transcriptase inhibitors on Bax, Bcl-2 and apoptosis in two cervical cell lines. Biomed. Pharmacother. 2014, 68, 241–251. [Google Scholar] [CrossRef]
- Albuquerque, A.S.; Foxall, R.B.; Cortesao, C.S.; Soares, R.S.; Doroana, M.; Ribeiro, A.; Lucas, M.; Antunes, F.; Victorino, R.M.; Sousa, A.E. Low CD4 T-cell counts despite low levels of circulating HIV: Insights from the comparison of HIV-1 infected patients with a discordant response to antiretroviral therapy to patients with untreated advanced HIV-2 disease. Clin. Immunol. 2007, 125, 67–75. [Google Scholar] [CrossRef]
- Regamey, N.; Harr, T.; Battegay, M.; Erb, P. Downregulation of Bcl-2, but not of Bax or Bcl-x, is associated with T lymphocyte apoptosis in HIV infection and restored by antiretroviral therapy or by interleukin 2. AIDS Res. Hum. Retrovir. 1999, 15, 803–810. [Google Scholar] [CrossRef]
- Miro, O.; Villarroya, J.; Garrabou, G.; Lopez, S.; Rodriguez de la Concepcion, M.; Pedrol, E.; Martinez, E.; Giralt, M.; Gatell, J.M.; Cardellach, F.; et al. In vivo effects of highly active antiretroviral therapies containing the protease inhibitor nelfinavir on mitochondrially driven apoptosis. Antivir. Ther. 2005, 10, 945–951. [Google Scholar]
- Ehrhard, S.; Wernli, M.; Kaufmann, G.; Pantaleo, G.; Rizzardi, G.P.; Gudat, F.; Erb, P.; Battegay, M. Effect of antiretroviral therapy on apoptosis markers and morphology in peripheral lymph nodes of HIV-infected individuals. Infection 2008, 36, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Grelli, S.; Balestrieri, E.; Matteucci, C.; Minutolo, A.; D’Ettorre, G.; Lauria, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptotic cell signaling in lymphocytes from HIV+ patients during successful therapy. Ann. N. Y. Acad. Sci. 2006, 1090, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Gambardella, L.; Cassone, A.; Vella, S.; Cauda, R.; Malorni, W. Mitochondrial membrane hyperpolarization hijacks activated T lymphocytes toward the apoptotic-prone phenotype: Homeostatic mechanisms of HIV protease inhibitors. J. Immunol. 2003, 170, 6006–6015. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Gellera, C.; Tiloca, C.; Del Bo, R.; Corrado, L.; Pensato, V.; Agostini, J.; Cereda, C.; Ratti, A.; Castellotti, B.; Corti, S.; et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 183–187. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheney, L.; Guzik, H.; Macaluso, F.P.; Macian, F.; Cuervo, A.M.; Berman, J.W. HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders. Cells 2020, 9, 1426. https://doi.org/10.3390/cells9061426
Cheney L, Guzik H, Macaluso FP, Macian F, Cuervo AM, Berman JW. HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders. Cells. 2020; 9(6):1426. https://doi.org/10.3390/cells9061426
Chicago/Turabian StyleCheney, Laura, Hillary Guzik, Frank P. Macaluso, Fernando Macian, Ana Maria Cuervo, and Joan W. Berman. 2020. "HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders" Cells 9, no. 6: 1426. https://doi.org/10.3390/cells9061426
APA StyleCheney, L., Guzik, H., Macaluso, F. P., Macian, F., Cuervo, A. M., & Berman, J. W. (2020). HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders. Cells, 9(6), 1426. https://doi.org/10.3390/cells9061426