Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death

 ,
, 
Abstract
:1. Introduction
2. Ferroptosis
2.1. The Hallmarks of Ferroptosis
2.2. The Role of Iron Metabolism in Ferroptosis
2.3. Ferroptosis and Cancer
2.4. Ferroptosis Is a New Promising Target for Cancer Treatment
3. Mitochondria at the Crossroad of Ferroptosis and Cancer Suppression
3.1. Mitochondrial Morphological Features in Ferroptosis
3.2. Mitochondrial Energetic Metabolism in Ferroptosis
3.3. Mitochondria and Iron Metabolism
3.4. Ferroptosis Mediated by Mitochondrial VDACs
3.5. Other Pathways
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and its potential role in human diseases. Front. Pharmacol. 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The hallmarks of ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooko, E.; Saeed, M.E.M.; Kadioglu, O.; Sarvi, S.; Colak, M.; Elmasaoudi, K.; Janah, R.; Greten, H.J.; Efferth, T. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 2015, 22, 1045–1054. [Google Scholar] [CrossRef]
- Green, D.R.; Victor, B. The pantheon of the fallen: Why are there so many forms of cell death? Trends Cell Biol. 2012, 22, 555–556. [Google Scholar] [CrossRef] [Green Version]
- Fearnhead, H.O.; Vandenabeele, P.; Berghe, T.V. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X. A physiological function for ferroptosis in tumor suppression by the immune system. Cell Metab. 2019, 30, 14–15. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Jiang, X. To eat or not to eat—The metabolic flavor of ferroptosis. Curr. Opin. Cell Biol. 2018, 51, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dan Dunn, J.; Alvarez, L.A.J.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauen, U.; Springer, A.; Weisheit, D.; Petrat, F.; Korth, H.G.; de Groot, H.; Sustmann, R. Assessment of chelatable mitochondrial iron by using mitochondrion- selective fluorescent iron indicators with different iron-binding affinities. ChemBioChem 2007, 8, 341–352. [Google Scholar] [CrossRef]
- Jhurry, N.D.; Chakrabarti, M.; McCormick, S.P.; Holmes-Hampton, G.P.; Lindahl, P.A. Biophysical investigation of the ironome of human Jurkat cells and mitochondria. Biochemistry 2012, 51, 5276–5284. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Stehling, O. The role of mitochondria in cellular iron—Sulfur processes, and diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, G.; Gentile, F.; Pizzimenti, S.; Canuto, R.A.; Daga, M.; Arcaro, A.; Cetrangolo, G.P.; Lepore, A.; Ferretti, C.; Dianzani, C.; et al. Mitochondrial dysfunction in cancer and neurodegenerative diseases: Spotlight on fatty acid oxidation and lipoperoxidation products. Antioxidants 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Urrutia, P.J.; Mena, N.P.; Núñez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell 2019, 73, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Yagoda, N.; Von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Agrawal, S.; Fox, J.H.J.; Thyagarajan, B.; Fox, J.H.J. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-dependent ER stress triggers glial dysfunction and nervous system degeneration in a drosophila model of friedreich’s ataxia. Front. Mol. Neurosci. 2018, 11, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.J.; Lyu, J.; et al. Mitoferrin-1 is involved in the progression of alzheimer’s disease through targeting mitochondrial iron metabolism in a caenorhabditis elegans model of alzheimer’s disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Song, J.; Wang, Y.; Wang, X.; Culmsee, C.; Zhu, C. The potential role of ferroptosis in neonatal brain injury. Front. Neurosci. 2019, 13, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef]
- DeHart, D.N.; Fang, D.; Heslop, K.; Li, L.; Lemasters, J.J.; Maldonado, E.N. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem. Pharmacol. 2018, 148, 155–162. [Google Scholar] [CrossRef]
- Agmon, E.; Solon, J.; Bassereau, P.; Stockwell, B.R. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci. Rep. 2018, 8, 5155. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Review Article reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef]
- Proneth, B.; Conrad, M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019, 26, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.; Liu, W.; Zhuo, Q.; Hu, Q.; Liu, M.; Sun, Q.; Zhang, Z.; Fan, G.; Xu, W.; Ji, S.; et al. Ferroptosis: Final destination for cancer? Cell Prolif. 2020, 53, e12761. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The Development of the Concept of Ferroptosis; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 133, pp. 130–143. [Google Scholar]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Warner, G.J.; Berry, M.J.; Moustafa, M.E.; Carlson, B.A.; Hatfield, D.L.; Faust, J.R. Inhibition of selenoprotein synthesis by selenocysteine tRNA([Ser]Sec) lacking isopentenyladenosine. J. Biol. Chem. 2000, 275, 28110–28119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2019, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am J Clin Nutr 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonte, F.; Battaglia, A.M.; Zolea, F.; Oliveira, D.M.; Aversa, I.; Santamaria, G.; Giovannone, E.D.; Rocco, G.; Viglietto, G.; Costanzo, F. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. 2018, 9, 1174. [Google Scholar] [CrossRef] [PubMed]
- Aversa, I.; Chirillo, R.; Chiarella, E.; Zolea, F.; Di Sanzo, M.; Biamonte, F.; Palmieri, C.; Costanzo, F. Chemoresistance in H-ferritin silenced cells: The role of NF-κB. Int. J. Mol. Sci. 2018, 19, 2969. [Google Scholar] [CrossRef] [Green Version]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxid. Med. Cell. Longev. 2019, 2019, 3461251. [Google Scholar] [CrossRef]
- di Sanzo, M.; Chirillo, R.; Aversa, I.; Biamonte, F.; Santamaria, G.; Giovannone, E.D.; Faniello, M.C.; Cuda, G.; Costanzo, F. shRNA targeting of ferritin heavy chain activates H19/miR-675 axis in K562 cells. Gene 2018, 657, 92–99. [Google Scholar] [CrossRef]
- Zolea, F.; Battaglia, A.M.; Chiarella, E.; Malanga, D.; De Marco, C.; Bond, H.M.; Morrone, G.; Costanzo, F.; Biamonte, F. Ferritin heavy subunit silencing blocks the erythroid commitment of K562 cells via miR-150 up-regulation and GATA-1 repression. Int. J. Mol. Sci. 2017, 18, 2167. [Google Scholar] [CrossRef] [Green Version]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J.; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Mumbauer, S.; Pascual, J.; Kolotuev, I.; Hamaratoglu, F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019, 15, e1008396. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, S.; Gong, X.; Tam, S.; Xiao, D.; Liu, S.; Tao, Y. The epigenetic regulators and metabolic changes in ferroptosis-Associated cancer progression. Mol. Cancer 2020, 19, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019, 176, 564–580. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Liao, H.; Liu, Y.; Lee, T.S.; Zhu, M.; Wang, X.; Stemerman, M.B.; Zhu, Y.; Shyy, J.Y.J. Sterol-responsive element-binding protein (SREBP) 2 down-regulates ATP-binding cassette transporter A1 in vascular endothelial cells: A novel role of SREBP in regulating cholesterol metabolism. J. Biol. Chem. 2004, 279, 48801–48807. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ma, M.Y.; Sun, M.; Jiang, L.Y.; Zhao, X.T.; Fang, X.X.; Lam, S.M.; Shui, G.H.; Luo, J.; Shi, X.J.; et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J. Lipid Res. 2019, 60, 1765–1775. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Affar, E.B.; Carbone, M. BAP1 regulates different mechanisms of cell death. Cell Death Dis. 2018, 9, 1151. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Yin, S.; Zhang, E.; Hu, H. Role of p62 in the regulation of cell death induction. Apoptosis 2018, 23, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Regulation of ferroptosis by MicroRNAs. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 125–145. [Google Scholar]
- Wang, M.; Mao, C.; Ouyang, L.; Liu, Y.; Lai, W.; Liu, N.; Shi, Y.; Chen, L.; Xiao, D.; Yu, F.; et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019, 26, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wang, X.; Liu, Y.; Wang, M.; Yan, B.; Jiang, Y.; Shi, Y.; Shen, Y.; Liu, X.; Lai, W.; et al. G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p. Cancer Res. 2018, 78, 3484–3496. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Zhao, W.; Yu, J.; Li, S.; Lin, L.; Chen, X. Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Poursaitidis, I.; Wang, X.; Crighton, T.; Labuschagne, C.; Mason, D.; Cramer, S.L.; Triplett, K.; Roy, R.; Pardo, O.E.; Seckl, M.J.; et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 2017, 18, 2547–2556. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Xie, Y.; Kang, R.; Hou, W.; Sun, X.; Epperly, M.W.; Greenberger, J.S.; Tang, D. FANCD2 protects against bone marrow injury from ferroptosis. Biochem. Biophys. Res. Commun. 2016, 480, 443–449. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, Z.; Song, R. Antisense lncRNA As-SLC7A11 suppresses epithelial ovarian cancer progression mainly by targeting SLC7A. Pharmazie 2017, 72, 403–407. [Google Scholar]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, C.F.; Ma, M.Z.; Chen, G.; Song, M.; Zeng, Z.L.; Lu, W.H.; Yang, J.; Wen, S.; Chiao, P.J.; et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget 2015, 6, 21148–21158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zheng, S.; Geng, Y.; Xue, J.; Wang, Z.; Xie, X.; Wang, J.; Zhang, S.; Hou, Y. MicroRNA profiling of atrial fibrillation in canines: MiR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD. PLoS ONE 2015, 10, e0122674. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Kyrychenko, S.; Kyrychenko, V.; Badr, M.A.; Ikeda, Y.; Sadoshima, J.; Shirokova, N. Pivotal role of MIR-448 in the development of ROS-induced cardiomyopathy. Cardiovasc. Res. 2015, 108, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.V.; Kupai, K.; Szucs, G.; Gáspár, R.; Pálóczi, J.; Faragó, N.; Zvara, Á.; Puskás, L.G.; Rázga, Z.; Tiszlavicz, L.; et al. MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J. Mol. Cell. Cardiol. 2013, 62, 111–121. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 404–414. [Google Scholar]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 drives ferroptosis through GPX4 inactivation and ros production in colorectal cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Yasui, H.; Higashikawa, K.; Miyamoto, N.; Kuge, Y. Erastin, a ferroptosis-inducing agent, sensitized cancer cells to X-ray irradiation via glutathione starvation in vitro and in vivo. PLoS ONE 2019, 14, e0225931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehm, T.; Fan, Z.; Ghoochani, A.; Rauh, M.; Engelhorn, T.; Minakaki, G.; Dörfler, A.; Klucken, J.; Buchfelder, M.; Eyüpoglu, I.Y.; et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget 2016, 7, 36021–36033. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of cancer therapy based on ferroptosis. Adv. Mater. 2018, 30, e1704007. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodríguez Martínez, M.; López, G.; Mattioli, M.; Realubit, R.; et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Zhu, J.; Liu, F.; Deng, J.; Meng, X.; Liu, G.; Wu, H.; Fan, A.; Wang, Z.; Zhao, Y. Boosting the ferroptotic antitumor efficacy via site-specific amplification of tailored lipid peroxidation. ACS Appl. Mater. Interfaces 2019, 11, 29655–29666. [Google Scholar] [CrossRef]
- Yao, X.; Yang, P.; Jin, Z.; Jiang, Q.; Guo, R.; Xie, R.; He, Q.; Yang, W. Multifunctional nanoplatform for photoacoustic imaging-guided combined therapy enhanced by CO induced ferroptosis. Biomaterials 2019, 197, 268–283. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, J.; Zhang, Z.; Dong, D.; Shen, Y.; Fang, Y.; Hu, L.; Liu, M.; Dai, C.; Peng, S.; et al. Iron and magnetic: New research direction of the ferroptosis-based cancer therapy. Am. J. Cancer Res. 2018, 8, 1933–1946. [Google Scholar] [PubMed]
- Kim, S.E.; Zhang, L.; Ma, K.; Riegman, M.; Chen, F.; Ingold, I.; Conrad, M.; Turker, M.Z.; Gao, M.; Jiang, X.; et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 2016, 11, 977–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, D.; Wang, X.; Lu, M.; Gao, B.; Qiao, X. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. Mol. Med. Rep. 2014, 9, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 217–532. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef] [Green Version]
- Weïwer, M.; Bittker, J.A.; Lewis, T.A.; Shimada, K.; Yang, W.S.; MacPherson, L.; Dandapani, S.; Palmer, M.; Stockwell, B.R.; Schreiber, S.L.; et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 2012, 22, 1822–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; van Oppen, L.M.P.E.; Schöckel, L.; Bossenbroek, H.M.; van de Vries, S.E.E.; Hermeling, J.C.W.; Grefte, S.; Kopitz, C.; Heroult, M.; Willems, P.H.G.M.; et al. Mitochondrial complex i inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Szwed, M.; Sønstevold, T.; Øverbye, A.; Engedal, N.; Grallert, B.; Mørch, Ý.; Sulheim, E.; Iversen, T.G.; Skotland, T.; Sandvig, K.; et al. Small variations in nanoparticle structure dictate differential cellular stress responses and mode of cell death. Nanotoxicology 2019, 13, 761–782. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Lewerenz, J.; Ates, G.; Methner, A.; Conrad, M.; Maher, P. Oxytosis/ferroptosis-(Re-) emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 2018, 12, 214. [Google Scholar] [CrossRef]
- Strzyz, P. Iron expulsion by exosomes drives ferroptosis resistance. Nat. Rev. Mol. Cell Biol. 2020, 21, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J.R.; White, A.R. Nexus between mitochondrial function, iron, copper and glutathione in Parkinson’s disease. Neurochem. Int. 2018, 117, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Valette, J.; Pépin, J.; Flament, J.; Brouillet, E. Energy defects in Huntington’s disease: Why “in vivo” evidence matters. Biochem. Biophys. Res. Commun. 2017, 483, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kosaras, B.; del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, M.M.; Kauffman, M.M.; Traore, K.; Zhu, H.; Trush, M.; Jia, Z.; Li, Y. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zielonka, M.; Dranka, B.; Kumar, S.N.; Myers, C.R.; Bennett, B.; Garces, A.M.; Machado, L.G.D.D.; Thiebaut, D.; Ouari, O.; et al. Detection of mitochondria-generated reactive oxygen species in cells using multiple probes and methods: Potentials, pitfalls, and the future. J. Biol. Chem. 2018, 293, 10363–10380. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.M.; Kim, A.; Yang, W.S. Detection of ferroptosis by BODIPYTM 581/591 C. In Methods in Molecular Biology; Vancurova, I., Zhu, Y., Eds.; Humana: New York, NY, USA, 2020; Volume 2108, pp. 125–130. [Google Scholar]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harb. Protoc. 2016, 2016, 1092–1096. [Google Scholar] [CrossRef]
- Grohm, J.; Plesnila, N.; Culmsee, C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain. Behav. Immun. 2010, 24, 831–838. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; von Karstedt, S. Ferroptosis in cancer cell biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botman, D.; Tigchelaar, W.; van Noorden, C.J.F. Determination of phosphate-activated glutaminase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem. Cytochem. 2014, 62, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.; Lee, J.; You, J.H.; Kim, D.; Roh, J.L. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020, 30, 101418. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ždralević, M.; Vučetić, M.; Daher, B.; Marchiq, I.; Parks, S.K.; Pouysségur, J. Disrupting the ‘warburg effect’ re-routes cancer cells to OXPHOS offering a vulnerability point via ‘ferroptosis’-induced cell death. Adv. Biol. Regul. 2018, 68, 55–63. [Google Scholar] [CrossRef]
- Atkinson, A.; Winge, D.R. Metal acquisition and availability in the mitochondria. Chem. Rev. 2009, 109, 4708–4721. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A.; Tong, W.H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Zhang, A.S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar]
- Wu, J.R.; Tuo, Q.Z.; Lei, P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J. Mol. Neurosci. 2018, 66, 197–206. [Google Scholar] [CrossRef]
- Colombini, M. VDAC: The channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 2004, 256, 107–115. [Google Scholar] [CrossRef]
- Lange, H.; Kispal, G.; Lill, R. Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. J. Biol. Chem. 1999, 274, 18989–18996. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, E.N.; Sheldon, K.L.; Dehart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: Regulation by free tubulin and erastin. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [Green Version]
- Lill, R.; Mühlenhoff, U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef] [Green Version]
- Cardenas-Rodriguez, M.; Chatzi, A.; Tokatlidis, K. Iron-sulfur clusters: From metals through mitochondria biogenesis to disease. J. Biol. Inorg. Chem. 2018, 23, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Missirlis, F.; Holmberg, S.; Georgieva, T.; Dunkov, B.C.; Rouault, T.A.; Law, J.H. Characterization of mitochondrial ferritin in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 5893–5898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Zhang, N.; Wang, Y.Q.; Wu, Q.; Yu, P.; Shi, Z.H.; Duan, X.L.; Zhao, B.L.; Wu, W.S.; Chang, Y.Z. Mitochondrial ferritin protects hydrogen peroxide-induced neuronal cell damage. Aging Dis. 2017, 8, 458–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A new link between iron metabolism, reactive oxygen species, and cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) knockout mice show signs of striatal mitochondrial dysfunction and a parkinson’s disease phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Maldonado, E.N. VDAC regulation: A mitochondrial target to stop cell proliferation. Adv. Cancer Res. 2018, 138, 41–69. [Google Scholar]
- Sampson, V.B.; Vetter, N.S.; Zhang, W.; Patil, P.U.; Mason, R.W.; George, E.; Gorlick, R.; Kolb, E.A. Integrating mechanisms of response and resistance against the tubulin binding agent Eribulin in preclinical models of osteosarcoma. Oncotarget 2016, 7, 86594–86607. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. J. Biochem. Mol. Biol. 2008, 43, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Tu, H.C.; Ren, D.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.D.; Cheng, E.H.Y. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 2009, 36, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugarli, E.I.; Langer, T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012, 31, 1336–1349. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, C.; Zhao, Y.; Zhang, W.; Xu, K.; Li, D.; Zhou, Y.; Li, H.; Xiao, G.; Lu, B.; et al. Inhibition of LONP1 protects against erastin-induced ferroptosis in Pancreatic ductal adenocarcinoma PANC1 cells. Biochem. Biophys. Res. Commun. 2020, 522, 1063–1068. [Google Scholar] [CrossRef]
- Edenharter, O.; Clement, J.; Schneuwly, S.; Navarro, J.A. Overexpression of Drosophila frataxin triggers cell death in an iron-dependent manner. J. Neurogenet. 2017, 31, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Pondarré, C.; Antiochos, B.B.; Campagna, D.R.; Clarke, S.L.; Greer, E.L.; Deck, K.M.; McDonald, A.; Han, A.P.; Medlock, A.; Kutok, J.L.; et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 2006, 15, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Naik, T.J.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Besse, E.K.; Ha, D.; Kovtunovych, G.; Rouault, T.A. Iron-dependent regulation of frataxin expression: Implications for treatment of Friedreich ataxia. Hum. Mol. Genet. 2008, 17, 2265–2273. [Google Scholar] [CrossRef] [Green Version]
- Nie, G.; Sheftel, A.D.; Kim, S.F.; Ponka, P. Overexpression of mitochondrial ferritin causes cytosolic iron depletion and changes cellular iron homeostasis. Blood 2005, 105, 2161–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läsche, M.; Emons, G.; Gründker, C. Shedding new light on cancer metabolism: A metabolic tightrope between life and death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Cerella, C.; Radogna, F.; Dicato, M.; Diederich, M. Natural compounds as regulators of the cancer cell metabolism. Int. J. Cell Biol. 2013, 2013, 639401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Wei, B.; Lu, C.; Li, P.; Chen, L. Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol. Lett. 2017, 14, 3117–3123. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
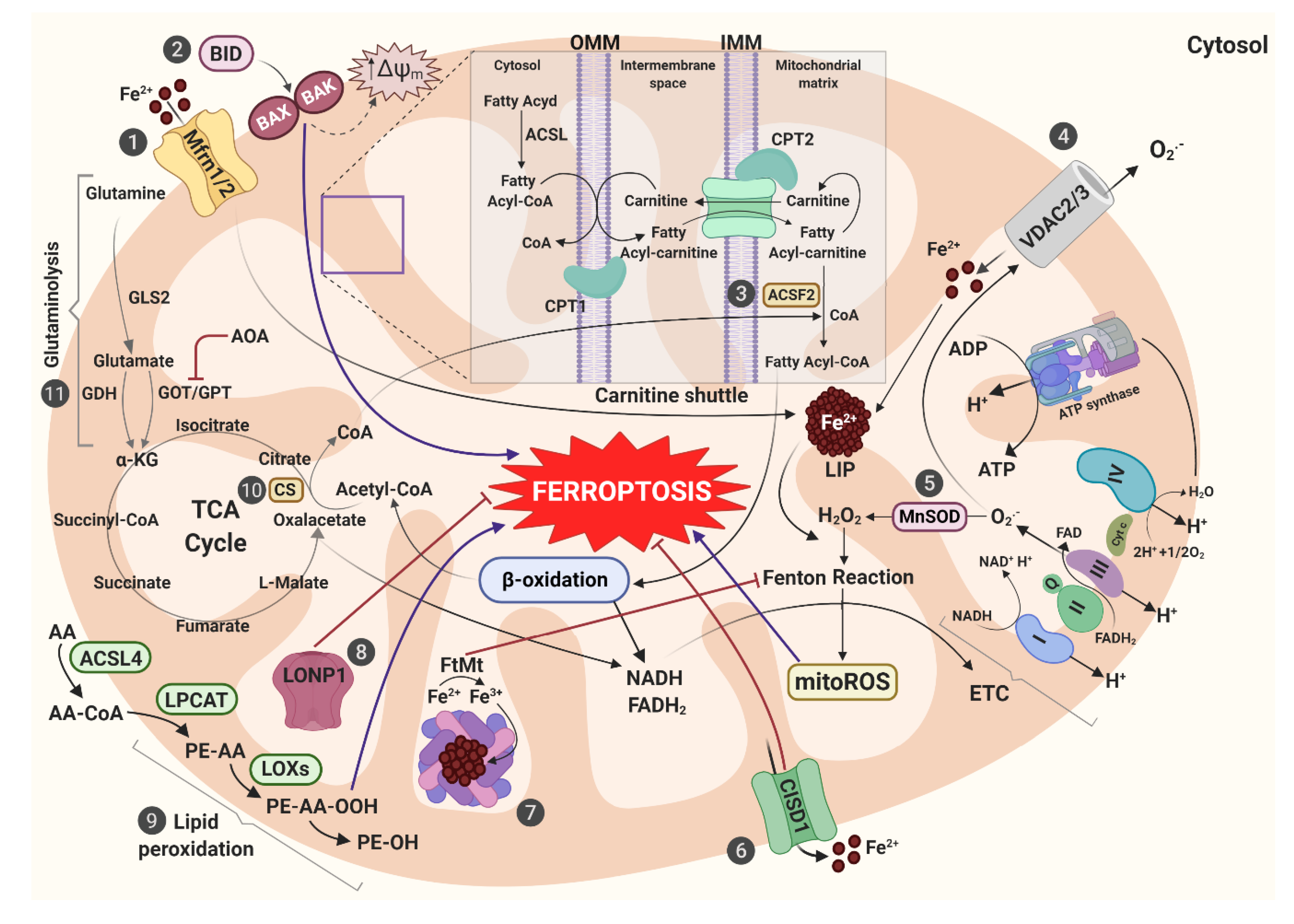{kind=link}
| Molecule | Targets | Mechanisms | Effect | References |
|---|---|---|---|---|
| p53 | SLC7A11 | Inhibition of System xc− | Pro-ferroptosis | [2,83] |
| BAP1 | SLC7A11 | Inhibition of System xc− | [91] | |
| IFNγ | SLC7A11 SLC3A2 | Induction of lipid peroxidation | [80] | |
| EGFR | MAPK | Cystine deprivation | [100] | |
| HO-1 | Heme | Heme degradation: cellular iron availability | [101] | |
| P53RRA lncRNA | G3BP1 | p53 nucleus retention | [96] | |
| FANCD2 | GPX4 Iron metabolism genes | GPX4 inhibition and cellular iron availability | [102] | |
| as-lncRNA SLC7A11 | SLC7A11 | Inhibition of System xc− | [103] | |
| SAT1 | lipoxygenases | Lipid peroxidation | [104] | |
| DPP4 | NOXs | Lipid peroxidation | [86] | |
| ACSL4 | fatty acids | Conversion of free fatty acids into fatty CoA ester | [105] | |
| miR-155 | ROS | Increase of ROS levels through inhibiting FOXO3a expression | [106] | |
| miR-206 | ROS | Increase of ROS production by targeting SOD1 | [107] | |
| HSPB1 | actin dynamics | Cellular iron availability | [108] | |
| NRF2 | Iron metabolism genes SLC7A11, HO-1, GPX4, G6PD | Inhibition of System xc−, Cellular iron availability | Anti-ferroptosis | [109] |
| miR-137 | SLC1A5 | Accumulation of MDA | [97] | |
| miR-448-3p | ROS | Reduces of NOX2-dependent ROS production | [110] | |
| miR-25 | ROS | Restrains ROS level by targeting NOX4 | [111] | |
| p53 | P21 | Boosts antioxidant defense | [85] |
| Drugs | Targets | Mechanisms | References |
|---|---|---|---|
| erastin | VDAC2/3 and System xc− | inhibition of the cystine–glutamate antiporter | [28,43] |
| sorafenib | VEGFR, PDGFR, RAF, GSH, and System xc− | inhibition of the cystine–glutamate antiporter | [126,127,128] |
| sulfasalazine (SAS) | System xc− | inhibition of the cystine–glutamate antiporter | [129] |
| cisplatin | GSH | depletion of intracellular GSH | [119] |
| l-buthionine sulfoximine (BSO) | GCLC | inhibition of GSH synthesis through γ- glutamylcysteine synthetase | [47,130] |
| artesunate (ART) | lysosomal iron | iron-mediated ROS generation | [6,131,132] |
| lanperisone | GSH and System xc− | inhibition of cysteine-glutamate antiporter | [132] |
| RSL3 | GPX4 | inhibition of GPX4 | [79] |
| altretamine (hexamethylmelamine) | GPX4 | inhibition of GPX4 | [120] |
| ML162, DPI compounds | GPX4 | GPX4 inactivation and GSH deletion | [133] |
| FIN56 | CoQ10 and GPX4 | CoQ10 deletion and GPX4 inactivation | [51] |
| FINO2 | GPX4 | GPX4 inactivation and lipid peroxides accumulation | [134] |
| Statins | HMG | CoQ10 deletion | [135] |
| trigonelline, brusatol | NRF2 | NRF2 inhibition | [86] |
| siramesine, lapatinib | Ferroportin, TF | increased cellular iron | [136] |
| BAY 87-2243 | ETC | inhibition of mitochondrial respiratory chain | [137] |
| iron ionophores | sequestration of iron into lysosomes and stimulation of ferritin degradation | [138] | |
| poly(butylcyanoacrylate) and zero-valent iron nanoparticles and arginine-rich manganese silicate nanobubbles | induction of oxidative stress and lipid peroxidation | [139] | |
| Nanocarriers (doxorubicin into mesoporous carbon nanoparticles/withaferin A/poly(ethylene glycol)-coated (PEGylated) silica nanoparticles (C’ dots ) | induction of oxidative stress | [122,123,125] |
| Biological Context | Reagents | Functions | References |
|---|---|---|---|
| Morphological changes | TEM | detects ultrastructural mitochondrial morphology changes in the occurrence of ferroptosis | [142] |
| Mitochondrial oxidative stress | MitoSOX | detects mitochondrial superoxide formation in live cells | [149] |
| MitoTEMPO | mitochondrially targeted antioxidant, a specific scavenger of mitochondrial superoxide; it can be used in combination with MitoSOX reagent as positive control | [150] | |
| Mitotracker | fluorescent dye that stains mitochondria in live cells and its accumulation is dependent upon membrane potential; in can be also used coupled with MitoSOX, in order to stain mitochondrial superoxide and mitochondria together | [26] | |
| Lipid peroxidation | BODIPY | detects reactive oxygen species generated by lipid peroxidation in mitochondrial and plasma membranes using flow cytometry | [151] |
| ΔΨm | TMRE | quantifies changes in mitochondrial transmembrane potential (ΔΨm) in live cells by flow cytometry, microplate spectrophotometry and fluorescent microscopy | [152] |
| Classification | Molecule | Mechanisms | References |
|---|---|---|---|
| Energetic metabolism markers | FH | its loss of function mutation confers resistance to cysteine-deprivation induced ferroptosis | [26] |
| DLD | blocks the increase of L-ROS amount and ΔΨm caused by cystine deprivation- or sulfasalazine treatment-induced ferroptosis in head and neck cancer | [159] | |
| GLS1/2 | catalyze the conversion of glutamine into glutamate | [26,34] | |
| TRANSAMINASES | convert glutamate into -KG through the transamination process | [34,158] | |
| AOA | inhibits cystine deprivation-induced ferroptosis in MEFs | [34,158] | |
| GOT1 | its knockdown inhibits CDI ferroptosis in MEFs | [34,158] | |
| ACSF2 | forms an activating thioester bond between the fatty acid and CoA | [34] | |
| CS | catalyzes the first reaction of the TCA, condensing acetyl-CoA and oxaloacetate to form citrates | [26,34] | |
| Iron metabolism markers | IRON | alterations in (Fe–S) clusters and LIP amount contribute to accumulation of ROS | [14,29] |
| Mfrn1/2 | iron accumulation and oxidative damage | [30,32,167,188] | |
| FtMt | protects against the increase of mitochondrial ROS though its storage and ferroxidase activity | [76] | |
| FLVCR1b | iron export mechanism out of mitochondria | [189] | |
| ABCB7/8 | mitochondrial Fe–S cluster export | [190,191] | |
| CISD1 | regulates mitochondrial iron uptake and generation of mitochondrial lipid peroxides | [29] | |
| Others | VDAC2/3 | control the trafficking of ions and metabolites between cytosol and mitochondria, leading an enhanced absorption of mitochondrial iron | [169,170] |
| FSP1 | mitochondrial effector of apoptotic cell death, able to convert CoQ10 in ubiquinol, that traps lipid peroxyl radicals | [57] | |
| BID | acts as a connection bridge between surface death receptors and the core apoptotic pathway in mitochondria | [27] | |
| LONP1 | mediates the selective degradation of misfolded or oxidatively damaged polypeptides in the mitochondrial matrix and maintain the integrity of the mitochondrial genome | [187] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. https://doi.org/10.3390/cells9061505
Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells. 2020; 9(6):1505. https://doi.org/10.3390/cells9061505
Chicago/Turabian StyleBattaglia, Anna Martina, Roberta Chirillo, Ilenia Aversa, Alessandro Sacco, Francesco Costanzo, and Flavia Biamonte. 2020. "Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death" Cells 9, no. 6: 1505. https://doi.org/10.3390/cells9061505
APA StyleBattaglia, A. M., Chirillo, R., Aversa, I., Sacco, A., Costanzo, F., & Biamonte, F. (2020). Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells, 9(6), 1505. https://doi.org/10.3390/cells9061505