Moonlighting in Mitosis: Analysis of the Mitotic Functions of Transcription and Splicing Factors

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Transcription Factors (TFs) with Mitotic Functions
2.1. TFs That Control Mitosis by Regulating the Expression of Mitotic Genes
2.2. TFs That Localize to Centrosomes
2.3. TFs with Moonlighting Functions at the Midbody
2.4. TFs That Localize to the Spindle
2.5. TFs with Roles in the Spindle Assembly Checkpoint (SAC)
2.6. Mitotic and Meiotic Roles of the TFIIH Complex Components
2.7. Mitotic Functions of the KANSL and NSL Complexes
3. Mitotic Functions of Splicing Factors (SFs)
3.1. SFs with Indirect Mitotic Functions
3.2. SFs That Control Cohesin Behavior
3.3. The Indirect and Direct Mitotic Functions of the PRP19 Complex
3.4. SFs with Roles in the SAC and in Kinetochore-MT Interactions
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11. [Google Scholar] [CrossRef]
- Chen, C.; Zabad, S.; Liu, H.; Wang, W.; Jeffery, C.J. MoonProt 2.0: An expansion and update of the moonlighting proteins database. Nucleic Acids Res. 2018, 46, D640–D644. [Google Scholar] [CrossRef]
- Ribeiro, D.; Briere, G.; Bely, B.; Spinelli, L.; Brun, C. MoonDB 2.0: An updated database of extreme multifunctional and moonlighting proteins. Nucleic Acids Res. 2018, 47, D398–D402. [Google Scholar] [CrossRef] [PubMed]
- Copley, S.D. An evolutionary perspective on protein moonlighting. Biochem. Soc. Trans. 2014, 42, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Fares, M.; Martin, A.C.R. Protein Moonlighting in Biology and Medicine; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Yokoyama, H.; Gruss, O.J. New mitotic regulators released from chromatin. Front. Oncol. 2013, 3, 308. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H. Chromatin-binding proteins moonlight as mitotic microtubule regulators. Trends Cell Biol. 2016, 26, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Rybina, S.; Santarella-Mellwig, R.; Mattaj, I.; Karsenti, E. ISWI is a RanGTP-dependent MAP required for chromosome segregation. J. Cell Biol. 2009, 187, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Nakos, K.; Santarella-Mellwig, R.; Rybina, S.; Krijgsveld, J.; Koffa, M.; Mattaj, I. CHD4 is a RanGTP-dependent MAP that stabilizes microtubules and regulates bipolar spindle formation. Curr. Biol. 2013, 23, 2443–2451. [Google Scholar] [CrossRef]
- Hur, S.-K.; Park, E.-J.; Han, J.-E.; Kim, Y.-A.; Kim, J.-D.; Kang, D.; Kwon, J. Roles of human INO80 chromatin remodeling enzyme in DNA replication and chromosome segregation suppress genome instability. Cell. Mol. Life Sci. 2010, 67, 2283–2296. [Google Scholar] [CrossRef]
- Park, E.-J.; Hur, S.-K.; Lee, H.-S.; Lee, S.-A.; Kwon, J. The human Ino80 binds to microtubule via the E-hook of tubulin: Implications for the role in spindle assembly. Biochem. Biophys. Res. Commun. 2011, 416, 416–420. [Google Scholar] [CrossRef]
- Gottesfeld, J.M. Mitotic repression of the transcriptional machinery. Trends Biochem. Sci. 1997, 22, 197–202. [Google Scholar] [CrossRef]
- Hofmann, J.C.; Husedzinovic, A.; Gruss, O.J. The function of spliceosome components in open mitosis. Nucleus 2010, 1, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Palozola, K.C.; Donahue, G.; Liu, H.; Grant, G.R.; Becker, J.S.; Coté, A.; Yu, H.; Raj, A.; Zaret, K.S. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 2017, 358, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Eggert, U.S.; Kiger, A.A.; Richter, C.; Perlman, Z.E.; Perrimon, N.; Mitchison, T.J.; Field, C.M. Parallel chemical genetic and genome-wide RNAi screens identify cytokinesis inhibitors and targets. PLoS Biol. 2004, 2, e379. [Google Scholar] [CrossRef] [PubMed]
- Kittler, R.; Putz, G.; Pelletier, L.; Poser, I.; Heninger, A.-K.; Drechsel, D.; Fischer, S.; Konstantinova, I.; Habermann, B.; Grabner, H.; et al. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature 2004, 432, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Goshima, G.; Wollman, R.; Goodwin, S.; Zhang, N.; Scholey, J.M.; Vale, R.D.; Stuurman, N. Genes required for mitotic spindle assembly in drosophila S2 cells. Science 2007, 316, 417–421. [Google Scholar] [CrossRef]
- Dobbelaere, J.; Josué, F.; Suijkerbuijk, S.; Baum, B.; Tapon, N.; Raff, J. A Genome-wide RNAi screen to dissect centriole duplication and centrosome maturation in drosophila. PLoS Biol. 2008, 6, e224. [Google Scholar] [CrossRef]
- Somma, M.P.; Ceprani, F.; Bucciarelli, E.; Naim, V.; De Arcangelis, V.; Piergentili, R.; Palena, A.; Ciapponi, L.; Giansanti, M.G.; Pellacani, C.; et al. Identification of drosophila mitotic genes by combining co-expression analysis and RNA interference. PLoS Genet. 2008, 4, e1000126. [Google Scholar] [CrossRef]
- Neumann, B.; Walter, T.; Hériché, J.-K.; Bulkescher, J.; Erfle, H.; Conrad, C.; Rogers, P.; Poser, I.; Held, M.; Liebel, U.; et al. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464, 721–727. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Martínez-A, C.; Burgering, B.M.T.; Carrera, A. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature 2001, 413, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Futcher, B.; Leatherwood, J. A new transcription factor for mitosis: In Schizosaccharomyces pombe, the RFX transcription factor Sak1 works with forkhead factors to regulate mitotic expression. Nucleic Acids Res. 2015, 43, 6874–6888. [Google Scholar] [CrossRef]
- Schiklenk, C.; Petrova, B.; Kschonsak, M.; Hassler, M.; Klein, C.; Gibson, T.J.; Haering, C.H. Control of mitotic chromosome condensation by the fission yeast transcription factor Zas1. J. Cell Biol. 2018, 217, 2383–2401. [Google Scholar] [CrossRef] [PubMed]
- Rambout, X.; Detiffe, C.; Bruyr, J.; Mariavelle, E.; Cherkaoui, M.; Brohée, S.; Demoitié, P.; Lebrun, M.; Soin, R.; Lesage, B.; et al. The transcription factor ERG recruits CCR4–NOT to control mRNA decay and mitotic progression. Nat. Struct. Mol. Biol. 2016, 23, 663–672. [Google Scholar] [CrossRef]
- Dias, M.B.; Hildebrandt, F.; Pellman, D.; Woods, G.; Godinho, S.A. Centrosomes and cilia in human disease. Trends Genet. 2011, 27, 307–315. [Google Scholar] [CrossRef]
- Tillery, M.; Blake-Hedges, C.; Zheng, Y.; Buchwalter, R.A.; Megraw, T.L. Centrosomal and non-centrosomal microtubule-organizing centers (MTOCs) in drosophila melanogaster. Cells 2018, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Anderson, S.F.; Chao, D.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2017, 23, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.-C.; White, R.L. BRCA1 is associated with the centrosome during mitosis. Proc. Natl. Acad. Sci. USA 1998, 95, 12983–12988. [Google Scholar] [CrossRef]
- Lotti, L.V.; Ottini, L.; D’Amico, C.; Gradini, R.; Cama, A.; Belleudi, F.; Frati, L.; Torrisi, M.R.; Mariani-Costantini, R. Subcellular localization of theBRCA1 gene product in mitotic cells. Genes Chromosom. Cancer 2002, 35, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Kais, Z.; Parvin, J.D. Regulation of centrosomes by the BRCA1-dependent ubiquitin ligase. Cancer Biol. Ther. 2008, 7, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Lipkowitz, S.; Weissman, A.M. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 2011, 11, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, A.; Kanno, S.-I.; Nakayama, M.; Mochiduki, H.; Wei, L.; Shimaoka, T.; Furukawa, Y.; Kato, K.; Shibata, S.; Yasui, A.; et al. The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Mol. Cell 2014, 53, 101–114. [Google Scholar] [CrossRef]
- Starita, L.M.; Machida, Y.; Sankaran, S.; Elias, J.E.; Griffin, K.; Schlegel, B.P.; Gygi, S.P.; Parvin, J.D. BRCA1-dependent ubiquitination of γ-tubulin regulates centrosome number. Mol. Cell. Biol. 2004, 24, 8457–8466. [Google Scholar] [CrossRef]
- Sankaran, S.; Starita, L.M.; Simons, A.M.; Parvin, J.D. Identification of domains of BRCA1 critical for the ubiquitin-dependent inhibition of centrosome function. Cancer Res. 2006, 66, 4100–4107. [Google Scholar] [CrossRef]
- Xu, X.; Weaver, Z.; Linke, S.; Li, C.; Gotay, J.; Wang, X.; Harris, C.C.; Ried, T.; Deng, C.-X. Centrosome amplification and a defective G2–M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform–deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Cross, M.K.; Powers, M.A. Learning about cancer from frogs: Analysis of mitotic spindles in Xenopus egg extracts. Dis. Model. Mech. 2009, 2, 541–547. [Google Scholar] [CrossRef]
- Joukov, V.; Groen, A.C.; Prokhorova, T.; Gerson, R.; White, E.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006, 127, 539–552. [Google Scholar] [CrossRef]
- Murray, A.W.; Kirschner, M.W. Cyclin synthesis drives the early embryonic cell cycle. Nature 1989, 339, 275–280. [Google Scholar] [CrossRef]
- Hofmann, J.C.; Tegha-Dunghu, J.; Dräger, S.; Will, C.L.; Lührmann, R.; Gruss, O.J. The Prp19 complex directly functions in mitotic spindle assembly. PLoS ONE 2013, 8, e74851. [Google Scholar] [CrossRef] [PubMed]
- Sears, T.K.; Angelastro, J.M. The transcription factor ATF5: Role in cellular differentiation, stress responses, and cancer. Oncotarget 2017, 8, 84595–84609. [Google Scholar] [CrossRef] [PubMed]
- Madarampalli, B.; Yuan, Y.; Liu, D.; Lengel, K.; Xu, Y.; Li, G.; Yang, J.; Liu, X.; Lu, Z.; Liu, D.X. ATF5 connects the pericentriolar materials to the proximal end of the mother centriole. Cell 2015, 162, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gaither, K.; Kim, E.; Liu, E.; Hu, M.; Lengel, K.; Qian, N.; Xu, Y.; Wang, B.; Knipprath, H.; et al. SUMO2/3 modification of activating transcription factor 5 (ATF5) controls its dynamic translocation at the centrosome. J. Biol. Chem. 2018, 293, 2939–2948. [Google Scholar] [CrossRef]
- Hu, C.-K.; Coughlin, M.; Mitchison, T.J. Midbody assembly and its regulation during cytokinesis. Mol. Biol. Cell 2012, 23, 1024–1034. [Google Scholar] [CrossRef]
- D’Avino, P.P.; Giansanti, M.G.; Petronczki, M. Cytokinesis in animal cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a015834. [Google Scholar] [CrossRef]
- Elia, N.; Ott, C.; Lippincott-Schwartz, J. Incisive imaging and computation for cellular mysteries: Lessons from abscission. Cell 2013, 155, 1220–1231. [Google Scholar] [CrossRef]
- Ortega, G.C.; Falk, S.; Johansson, P.A.; Peyre, E.; Broix, L.; Sahu, S.K.; Hirst, W.; Schlichthaerle, T.; Romero, C.D.J.; Draganova, K.; et al. The centrosome protein AKNA regulates neurogenesis via microtubule organization. Nature 2019, 567, 113–117. [Google Scholar] [CrossRef]
- Davies, A.H.; Barrett, I.; Pambid, M.R.; Hu, K.; Stratford, A.L.; Freeman, S.; Berquin, I.M.; Pelech, S.; Hieter, P.; Maxwell, C.A.; et al. YB-1 evokes susceptibility to cancer through cytokinesis failure, mitotic dysfunction and HER2 amplification. Oncogene 2011, 30, 3649–3660. [Google Scholar] [CrossRef][Green Version]
- Kawaguchi, A.; Asaka, M.N.; Matsumoto, K.; Nagata, K. Centrosome maturation requires YB-1 to regulate dynamic instability of microtubules for nucleus reassembly. Sci. Rep. 2015, 5, 8768. [Google Scholar] [CrossRef]
- Kang, J.; Goodman, B.; Zheng, Y.; Tantin, D. Dynamic regulation of Oct1 during mitosis by phosphorylation and ubiquitination. PLoS ONE 2011, 6, e23872. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Jin, S.; Tong, T.; Zhao, H.; Fan, F.; Antinore, M.J.; Rajasekaran, B.; Wu, M.; Zhan, Q. BRCA1 regulates GADD45 through its interactions with the OCT-1 and CAAT motifs. J. Biol. Chem. 2002, 277, 8061–8067. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Yu, H.; Deng, C.-X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 17108–17113. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Sakamoto, S.; Song, D.; Qu, Z.; Ota, K.; Taniguchi, T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998, 424, 27–32. [Google Scholar] [CrossRef]
- Tang, X.; Zhao, Y.; Buchon, N.; Engstrom, Y. The POU/Oct transcription factor nubbin controls the balance of intestinal stem cell maintenance and differentiation by isoform-specific regulation. Stem Cell Rep. 2018, 10, 1565–1578. [Google Scholar] [CrossRef]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef]
- Zajdel, M.E.B.; Blair, G.E. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 1988, 2, 579–584. [Google Scholar]
- Morris, V.B.; Brammall, J.; Noble, J.; Reddel, R.R. p53 localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines, and a human primary culture: An immunofluorescence study. Exp. Cell Res. 2000, 256, 122–130. [Google Scholar] [CrossRef]
- Ciciarello, M. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 2001, 276, 19205–19213. [Google Scholar] [CrossRef]
- Tritarelli, A.; Oricchio, E.; Ciciarello, M.; Mangiacasale, R.; Palena, A.; LaVia, P.; Soddu, S.; Cundari, E. p53 localization at centrosomes during mitosis and postmitotic checkpoint are ATM-dependent and require serine 15 phosphorylation. Mol. Biol. Cell 2004, 15, 3751–3757. [Google Scholar] [CrossRef]
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Woude, G.F.V. Abnormal centrosome amplification in the absence of p53. Science 1996, 271, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K.; Wiener, F.; Woude, G.F.V.; Mai, S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene 1997, 15, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2006, 26, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 2007, 7, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.A.; Mesquita, M.; Cunha, A.I.; Cardoso, J.; Carapeta, S.; Laranjeira, C.; Pinto, A.E.; Pereira-Leal, J.B.; Dias-Pereira, A.; Dias, M.B.; et al. Centrosome amplification arises before neoplasia and increases upon p53 loss in tumorigenesis. J. Cell Biol. 2018, 217, 2353–2363. [Google Scholar] [CrossRef]
- Contadini, C.; Monteonofrio, L.; Virdia, I.; Prodosmo, A.; Valente, D.; Chessa, L.; Musio, A.; Fava, L.L.; Rinaldo, C.; Di Rocco, G.; et al. p53 mitotic centrosome localization preserves centrosome integrity and works as sensor for the mitotic surveillance pathway. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Astrinidis, A.; Kim, J.; Kelly, C.M.; Olofsson, B.A.; Torabi, B.; Sorokina, E.M.; Azizkhan-Clifford, J. The transcription factor SP1 regulates centriole function and chromosomal stability through a functional interaction with the mammalian target of rapamycin/raptor complex. Genes Chromosom. Cancer 2009, 49, 282–297. [Google Scholar] [CrossRef]
- Lai, P.-Y.; Wang, C.-Y.; Chen, W.-Y.; Kao, Y.-H.; Tsai, H.-M.; Tachibana, T.; Chang, W.-C.; Chung, B.-C. Steroidogenic factor 1 (NR5A1) resides in centrosomes and maintains genomic stability by controlling centrosome homeostasis. Cell Death Differ. 2011, 18, 1836–1844. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Kao, Y.-H.; Lai, P.-Y.; Chen, W.-Y.; Chung, B.-C. Steroidogenic factor 1 (NR5A1) maintains centrosome homeostasis in steroidogenic cells by restricting centrosomal DNA-dependent protein kinase activation. Mol. Cell. Biol. 2012, 33, 476–484. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Kamalyukova, I.M.; Razin, S.V. Association of the mammalian transcriptional regulator Kaiso with centrosomes and the midbody. Cell Cycle 2009, 8, 2303–2304. [Google Scholar] [CrossRef]
- Soubry, A.; Staes, K.; Parthoens, E.; Noppen, S.; Stove, C.; Bogaert, P.; Van Hengel, J.; Van Roy, F. The transcriptional repressor kaiso localizes at the mitotic spindle and is a constituent of the pericentriolar material. PLoS ONE 2010, 5, e9203. [Google Scholar] [CrossRef] [PubMed]
- Defossez, P.-A.; Kelly, K.F.; Filion, G.J.; Torrado, R.P.; Magdinier, F.; Menoni, H.; Nordgaard, C.L.; Daniel, J.M.; Gilson, E. The human enhancer blocker CTC-binding factor interacts with the transcription factor kaiso. J. Biol. Chem. 2005, 280, 43017–43023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Burke, L.J.; Rasko, J.E.; Lobanenkov, V.V.; Renkawitz, R. Dynamic association of the mammalian insulator protein CTCF with centrosomes and the midbody. Exp. Cell Res. 2004, 294, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.; Hernandez, N. Mitotic functions for SNAP45, a subunit of the small nuclear RNA-activating protein complex SNAPc. J. Biol. Chem. 2008, 283, 14845–14856. [Google Scholar] [CrossRef]
- Demir, O.; Kurnaz, I.A. Phospho-Ser383-Elk-1 is localized to the mitotic spindles during cell cycle and interacts with mitotic kinase Aurora-A. Cell Biochem. Funct. 2013, 31, 591–598. [Google Scholar] [CrossRef]
- Ito, Y.; Bae, S.-C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef]
- Ben-Ami, O.; Friedman, D.; Leshkowitz, D.; Goldenberg, D.; Orlovsky, K.; Pencovich, N.; Lotem, J.; Tanay, A.; Groner, Y. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep. 2013, 4, 1131–1143. [Google Scholar] [CrossRef]
- Chuang, L.S.H.; Khor, J.M.; Lai, S.K.; Garg, S.; Krishnan, V.; Koh, C.-G.; Lee, S.H.; Ito, Y. Aurora kinase-induced phosphorylation excludes transcription factor RUNX from the chromatin to facilitate proper mitotic progression. Proc. Natl. Acad. Sci. USA 2016, 113, 6490–6495. [Google Scholar] [CrossRef]
- Chuang, L.S.H.; Lai, S.K.; Murata-Hori, M.; Yamada, A.; Li, H.Y.; Gunaratne, J.; Ito, Y. RUNX3 interactome reveals novel centrosomal targeting of RUNX family of transcription factors. Cell Cycle 2012, 11, 1938–1947. [Google Scholar] [CrossRef]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef]
- Doxsey, S.; Mccollum, D.; Theurkauf, W. Centrosomes in cellular regulation. Annu. Rev. Cell Dev. Biol. 2005, 21, 411–434. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Cubizolles, F.; Schmidt, A.; Nigg, E.A. Quantitative analysis of human centrosome architecture by targeted proteomics and fluorescence imaging. EMBO J. 2016, 35, 2152–2166. [Google Scholar] [CrossRef] [PubMed]
- Rale, M.; Kadzik, R.S.; Petry, S. Phase transitioning the centrosome into a microtubule nucleator. Biochemistry 2017, 57, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Mullee, L.I.; Morrison, C.G. Centrosomes in the DNA damage response—The hub outside the centre. Chromosom. Res. 2015, 24, 35–51. [Google Scholar] [CrossRef]
- Sabat-Pośpiech, D.; Fabian-Kolpanowicz, K.; Prior, I.; Coulson, J.M.; Fielding, A. Targeting centrosome amplification, an Achilles’ heel of cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef]
- Skop, A.R.; Liu, H.; Yates, J.; Meyer, B.J.; Heald, R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 2004, 305, 61–66. [Google Scholar] [CrossRef]
- López-Camacho, C.; Van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Stein, G.S. Core binding factor β (CBFβ) is retained in the midbody during cytokinesis. J. Cell. Physiol. 2014, 229, 1466–1474. [Google Scholar] [CrossRef]
- She, Z.-Y.; Wei, Y.; Lin, Y.; Li, Y.; Lu, M. Mechanisms of the Ase1/PRC1/MAP65 family in central spindle assembly. Biol. Rev. 2019, 94, 2033–2048. [Google Scholar] [CrossRef]
- Bui, D.A.; Lee, W.; White, A.E.; Harper, J.W.; Schackmann, R.C.J.; Overholtzer, M.; Selfors, L.M.; Brugge, J.S. Cytokinesis involves a nontranscriptional function of the Hippo pathway effector YAP. Sci. Signal. 2016, 9, ra23. [Google Scholar] [CrossRef]
- Maizels, Y.; Gerlitz, G. Shaping of interphase chromosomes by the microtubule network. FEBS J. 2015, 282, 3500–3524. [Google Scholar] [CrossRef]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Benoist, C. Aire. Annu. Rev. Immunol. 2009, 27, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.; Lambert, J.-P.; Cockburn, K.; Gingras, A.-C.; Rossant, J. AIRE is a critical spindle-associated protein in embryonic stem cells. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, P.; Wong, J. HDAC3: Taking the SMRT-N-CoRrect road to repression. Oncogene 2007, 26, 5439–5449. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kurasawa, Y.; Wong, J.; Yu-Lee, L.-Y. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore–microtubule attachment. Proc. Natl. Acad. Sci. USA 2008, 105, 4179–4184. [Google Scholar] [CrossRef]
- Shin, H.; Kwon, S.; Song, H.; Lim, H.J. The transcription factor Egr3 is a putative component of the microtubule organizing center in mouse oocytes. PLoS ONE 2014, 9, e94708. [Google Scholar] [CrossRef]
- Liu, H.; Yin, F.-X.; Bai, C.-L.; Shen, Q.-Y.; Wei, Z.-Y.; Li, X.-X.; Liang, H.; Bou, S.; Li, G.-P. TFIIB co-localizes and interacts with α-tubulin during oocyte meiosis in the mouse and depletion of TFIIB causes arrest of subsequent embryo development. PLoS ONE 2013, 8, e80039. [Google Scholar] [CrossRef]
- Bae, I.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Hassan, B.A.; Kirilyuk, A.; Fan, S.; Avantaggiati, M.L.; Rosen, E.M. BRCA1 regulates gene expression for orderly mitotic progression. Cell Cycle 2005, 4, 1641–1666. [Google Scholar] [CrossRef]
- Kops, G.J.; Weaver, B.A.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar] [CrossRef]
- Shandilya, J.; Toska, E.; Richard, D.J.; Medler, K.F.; Roberts, S. WT1 interacts with MAD2 and regulates mitotic checkpoint function. Nat. Commun. 2014, 5, 4903. [Google Scholar] [CrossRef]
- Shandilya, J.; Roberts, S. A role of WT1 in cell division and genomic stability. Cell Cycle 2015, 14, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, G.G.; Townsend, K.; Martin, A.; Shimwell, N.J.; Grand, R.J.A.; Stewart, G.S.; Nilsson, J.; Turnell, A.S. Transcriptional intermediary factor 1γ binds to the anaphase-promoting complex/cyclosome and promotes mitosis. Oncogene 2012, 32, 4622–4633. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, C.; Ding, Z.; Liang, H.; Zhang, B.; Chen, X. The roles of TIF1γ in cancer. Front. Oncol. 2019, 9, 979. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Egly, J.-M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Rimel, J.K.; Taatjes, D.J. The essential and multifunctional TFIIH complex. Protein Sci. 2018, 27, 1018–1037. [Google Scholar] [CrossRef] [PubMed]
- Zurita, M.; Murillo-Maldonado, J.M. Drosophila as a model organism to understand the effects during development of TFIIH-related human diseases. Int. J. Mol. Sci. 2020, 21, 630. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Jones, R.S.; Liang, B.-C.; Gelbart, W.; Fuller, M.T. A Drosophila model for xeroderma pigmentosum and Cockayne’s syndrome: Haywire encodes the fly homolog of ERCC3, a human excision repair gene. Cell 1992, 71, 925–937. [Google Scholar] [CrossRef]
- Regan, C.L.; Fuller, M.T. Interacting genes that affect microtubule function: The nc2 allele of the haywire locus fails to complement mutations in the testis-specific beta-tubulin gene of Drosophila. Genes Dev. 1988, 2, 82–92. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Fuller, M.T. Molecular characterization of mutant alleles of the DNA repair/basal transcription factor haywire/ERCC3 in Drosophila. Genetics 1999, 152, 291–297. [Google Scholar]
- Weber, A.; Chung, H.-J.; Springer, E.; Heitzmann, D.; Warth, R. The TFIIH subunit p89 (XPB) localizes to the centrosome during mitosis. Cell. Oncol. 2010, 32, 121–130. [Google Scholar]
- Ito, S.; Tan, L.J.; Andoh, D.; Narita, T.; Seki, M.; Hirano, Y.; Narita, K.; Kuraoka, I.; Hiraoka, Y.; Tanaka, K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol. Cell 2010, 39, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Becerra, G.; Valerio-Cabrera, S.; Juarez, M.; Bucio-Mendez, A.; Zurita, M. TFIIH localization is highly dynamic during zygotic genome activation in Drosophila, and its depletion causes catastrophic mitosis. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Urwyler, O.; Suter, B. Drosophila Xpd regulates Cdk7 localization, mitotic kinase activity, spindle dynamics, and chromosome segregation. PLoS Genet. 2010, 6, e1000876. [Google Scholar] [CrossRef] [PubMed]
- Chelmicki, T.; Dündar, F.; Turley, M.J.; Khanam, T.; Aktas, T.; Ramírez, F.; Gendrel, A.-V.; Wright, P.R.; Videm, P.; Backofen, R.; et al. MOF-associated complexes ensure stem cell identity and Xist repression. eLife 2014, 3, e02024. [Google Scholar] [CrossRef]
- Ravens, S.; Fournier, M.; Ye, T.; Stierlé, M.; Dembélé, D.; Chavant, V.; Tora, L. Mof-associated complexes have overlapping and unique roles in regulating pluripotency in embryonic stem cells and during differentiation. eLife 2014, 3. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Guhathakurta, S.; Akhtar, A. The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep. 2019, 20, e47630. [Google Scholar] [CrossRef]
- Feller, C.; Prestel, M.; Hartmann, H.; Straub, T.; Soeding, J.; Becker, P.B. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res. 2011, 40, 1509–1522. [Google Scholar] [CrossRef][Green Version]
- Lam, K.C.; Mühlpfordt, F.; Vaquerizas, J.M.; Raja, S.J.; Holz, H.; Luscombe, N.M.; Manke, T.; Akhtar, A. The NSL complex regulates housekeeping genes in Drosophila. PLoS Genet. 2012, 8, e1002736. [Google Scholar] [CrossRef]
- Meunier, S.; Vernos, I. K-fibre minus ends are stabilized by a RanGTP-dependent mechanism essential for functional spindle assembly. Nature 2011, 13, 1406–1414. [Google Scholar] [CrossRef]
- Meunier, S.; Shvedunova, M.; Nguyen, N.V.; Avila, L.; Vernos, I.; Akhtar, A. An epigenetic regulator emerges as microtubule minus-end binding and stabilizing factor in mitosis. Nat. Commun. 2015, 6, 7889. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kitabayashi, I.; Ayton, P.M.; Cleary, M.L.; Ohki, M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 2002, 100, 3710–3718. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 2008, 283, 32162–32175. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.K.; Fields, A.T.; Cheng, K.; Lee, A.; Wagenaar, E.; Lagrois, R.; Schmidt, B.; Xia, B.; Ma, D. WD repeat-containing protein 5 (WDR5) localizes to the midbody and regulates abscission. J. Biol. Chem. 2015, 290, 8987–9001. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Veeranki, S.N.; Chinchole, A.; Tyagi, S. MLL/WDR5 complex regulates Kif2A localization to ensure chromosome congression and proper spindle assembly during mitosis. Dev. Cell 2017, 41, 605–622.e7. [Google Scholar] [CrossRef]
- Karole, A.M.; Chodisetty, S.; Ali, A.; Kumari, N.; Tyagi, S. Novel sub-cellular localizations and intra-molecular interactions may define new functions of Mixed Lineage Leukemia protein. Cell Cycle 2018, 17, 2684–2696. [Google Scholar] [CrossRef]
- Pavlova, G.A.; Popova, J.V.; Andreyeva, E.N.; Yarinich, L.A.; Lebedev, M.O.; Razuvaeva, A.V.; Dubatolova, T.D.; Oshchepkova, A.L.; Pellacani, C.; Somma, M.P.; et al. RNAi-mediated depletion of the NSL complex subunits leads to abnormal chromosome segregation and defective centrosome duplication in Drosophila mitosis. PLoS Genet. 2019, 15, e1008371. [Google Scholar] [CrossRef]
- Blower, M.D.; Karpen, G.H. The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. Nat. Cell Biol. 2001, 3, 730–739. [Google Scholar] [CrossRef]
- Pesenti, M.; Weir, J.; Musacchio, A. Progress in the structural and functional characterization of kinetochores. Curr. Opin. Struct. Biol. 2016, 37, 152–163. [Google Scholar] [CrossRef]
- Burns, C.G.; Ohi, R.; Mehta, S.; O’Toole, E.T.; Winey, M.; Clark, T.A.; Sugnet, C.W.; Ares, M.; Gould, K.L. Removal of a single α-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 801–815. [Google Scholar] [CrossRef]
- Pacheco, T.; Coelho, M.B.; Desterro, J.; Mollet, I.; Carmo-Fonseca, M. In vivo requirement of the small subunit of U2AF for recognition of a weak 3’ splice site. Mol. Cell. Biol. 2006, 26, 8183–8190. [Google Scholar] [CrossRef]
- Ahn, E.-Y.; DeKelver, R.C.; Lo, M.-C.; Nguyen, T.A.; Matsuura, S.; Boyapati, A.; Pandit, S.; Fu, X.-D.; Zhang, D.-E. SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol. Cell 2011, 42, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Park, E.M.; Scott, P.M.; Clutario, K.; Cassidy, K.; Zhan, K.; Gerber, S.A.; Holland, A.J. WBP11 is required for splicing the TUBGCP6 pre-mRNA to promote centriole duplication. J. Cell Biol. 2019, 219. [Google Scholar] [CrossRef] [PubMed]
- Jiao, A.L.; Perales, R.; Umbreit, N.T.; Haswell, J.R.; Piper, M.E.; Adams, B.D.; Pellman, D.; Kennedy, S.; Slack, F.J. Human nuclear RNAi-defective 2 (NRDE2) is an essential RNA splicing factor. RNA 2018, 25, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Varmark, H.; Vitting-Seerup, K.; Beli, P.; Waage, J.; Hakobyan, A.; Mistrik, M.; Choudhary, C.; Rohde, M.; Bekker-Jensen, S.; et al. UBL5 is essential for pre- mRNA splicing and sister chromatid cohesion in human cells. EMBO Rep. 2014, 15, 956–964. [Google Scholar] [CrossRef]
- Sundaramoorthy, S.; Vázquez-Novelle, M.D.; Lekomtsev, S.; Howell, M.; Petronczki, M. Functional genomics identifies a requirement of pre-m RNA splicing factors for sister chromatid cohesion. EMBO J. 2014, 33, 2623–2642. [Google Scholar] [CrossRef]
- Van der Lelij, P.; Stocsits, R.R.; Ladurner, R.; Petzold, G.; Kreidl, E.; Koch, B.; Schmitz, J.; Neumann, B.; Ellenberg, J.; Peters, J.-M. SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J. 2014, 33, 2643–2658. [Google Scholar] [CrossRef]
- Watrin, E.; Demidova, M.; Watrin, T.; Hu, Z.; Prigent, C. Sororin pre- mRNA splicing is required for proper sister chromatid cohesion in human cells. EMBO Rep. 2014, 15, 948–955. [Google Scholar] [CrossRef]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef]
- Kim, J.-S.; He, X.; Liu, J.; Duan, Z.; Kim, T.; Gerard, J.; Kim, B.; Pillai, M.M.; Lane, W.S.; Noble, W.S.; et al. Systematic proteomics of endogenous human cohesin reveals an interaction with diverse splicing factors and RNA-binding proteins required for mitotic progression. J. Biol. Chem. 2019, 294, 8760–8772. [Google Scholar] [CrossRef]
- Chanarat, S.; Sträßer, K. Splicing and beyond: The many faces of the Prp19 complex. Biochim. Biophys. Acta 2013, 1833, 2126–2134. [Google Scholar] [CrossRef]
- De Moura, T.R.; Mozaffari-Jovin, S.; Szabó, C.Z.K.; Schmitzová, J.; Dybkov, O.; Cretu, C.; Kachala, M.; Svergun, D.I.; Urlaub, H.; Lührmann, R.; et al. Prp19/Pso4 is an autoinhibited ubiquitin ligase activated by stepwise assembly of three splicing factors. Mol. Cell 2018, 69, 979–992.e6. [Google Scholar] [CrossRef] [PubMed]
- Mu, R.; Wang, Y.-B.; Wu, M.; Yang, Y.; Song, W.; Li, T.; Zhang, W.-N.; Tan, B.; Li, A.-L.; Wang, N.; et al. Depletion of pre-mRNA splicing factor Cdc5L inhibits mitotic progression and triggers mitotic catastrophe. Cell Death Dis. 2014, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Lutzelberger, M.; Wiegmann, H.; Klingenhoff, A.; Shenoy, S.; Kaufer, N.F. functional analysis of the fission yeast Prp4 protein kinase involved in pre-mrna splicing and isolation of a putative mammalian homologue. Nucleic Acids Res. 1997, 25, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Forman-Kay, J.D.; Luo, Y.; Luo, M.; Chow, Y.-H.; Plumb, J.; Friesen, J.D.; Tsui, L.-C.; Heng, H.H.Q.; Woolford, J.L.; et al. Identification and characterization of human genes encoding Hprp3p and Hprp4p, interacting components of the spliceosome. Hum. Mol. Genet. 1997, 6, 2117–2126. [Google Scholar] [CrossRef][Green Version]
- Bertram, K.; Agafonov, D.E.; Dybkov, O.; Haselbach, D.; Leelaram, M.N.; Will, C.L.; Urlaub, H.; Kastner, B.; Lührmann, R.; Stark, H. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 2017, 170, 701–713.e11. [Google Scholar] [CrossRef]
- Montembault, E.; Dutertre, S.; Prigent, C.; Giet, R. PRP4 is a spindle assembly checkpoint protein required for MPS1, MAD1, and MAD2 localization to the kinetochores. J. Cell Biol. 2007, 179, 601–609. [Google Scholar] [CrossRef]
- Liu, S.; Rauhut, R.; Vornlocher, H.-P.; Lührmann, R. The network of protein–protein interactions within the human U4/U6.U5 tri-snRNP. RNA 2006, 12, 1418–1430. [Google Scholar] [CrossRef]
- Herold, N.; Will, C.L.; Wolf, E.; Kastner, B.; Urlaub, H.; Lührmann, R. Conservation of the protein composition and electron microscopy structure of drosophila melanogaster and human spliceosomal complexes. Mol. Cell. Biol. 2008, 29, 281–301. [Google Scholar] [CrossRef]
- Nguyen, T.H.D.; Galej, W.; Bai, X.-C.; Savva, C.G.; Newman, A.J.; Scheres, S.H.; Nagai, K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 2015, 523, 47–52. [Google Scholar] [CrossRef]
- Pellacani, C.; Bucciarelli, E.; Renda, F.; Hayward, D.; Palena, A.; Chen, J.; Bonaccorsi, S.; Wakefield, J.G.; Gatti, M.; Somma, M.P. Splicing factors Sf3A2 and Prp31 have direct roles in mitotic chromosome segregation. eLife 2018, 7. [Google Scholar] [CrossRef]
- Takenaka, K.; Nakagawa, H.; Miyamoto, S.; Miki, H. The pre-mRNA-splicing factor SF3a66 functions as a microtubule-binding and -bundling protein. Biochem. J. 2004, 382, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.A.; O’Farrell, P.H. From egg to gastrula: How the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu. Rev. Genet. 2014, 48, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Tadros, W.; Lipshitz, H.D. The maternal-to-zygotic transition: A play in two acts. Dev. Camb. Engl. 2009, 136, 3033–3042. [Google Scholar] [CrossRef]
- Guilgur, L.G.; Prudêncio, P.; Sobral, D.; Liszeková, D.; Rosa, A.; Martinho, R.G. Requirement for highly efficient pre-mRNA splicing during Drosophila early embryonic development. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]


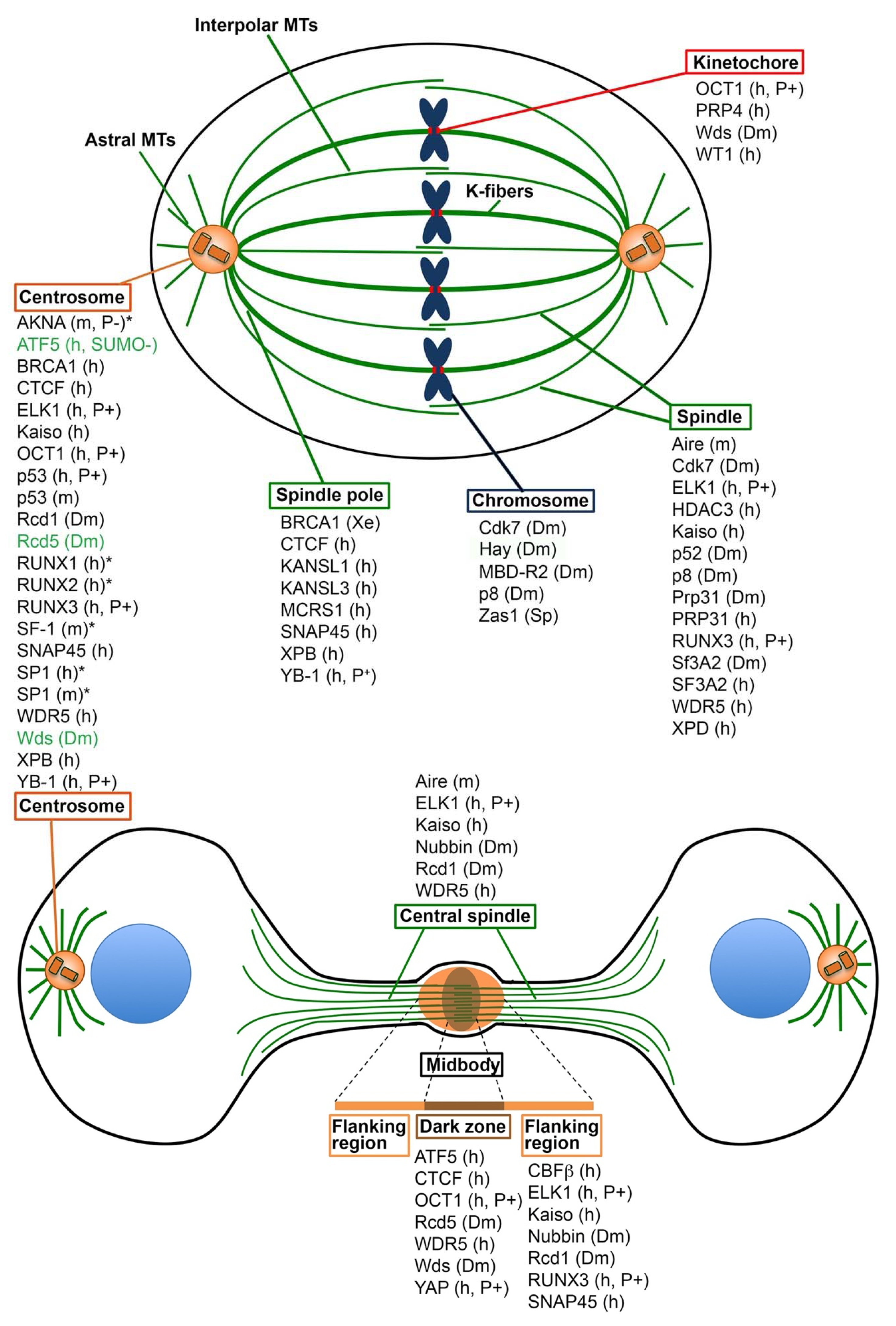{kind=link}
| Name (Organism) | Mitotic Localization | Interacting Mitotic Proteins | Loss-of-Function Mitotic Phenotype | Mitotic Function (Role in Cancer) |
|---|---|---|---|---|
| Sak1, Fkh2 (Sp) | ND | ND | Regulate mitotic gene transcription. Defective septation [24]. | Indirect |
| FKHRL1/FOXO3 (h) | ND | ND | Regulates mitotic gene transcription. Defective mitotic exit and cytokinesis [23]. | Indirect (TS) |
| Zas1 (Sp) | Chromosomes | ND | Regulates mitotic gene transcription. Defective chromosome condensation and segregation [25]. | Indirect |
| ERG (h) | RNA processing bodies (PBs) | ND | Degrades AURKA and AURKB mRNAs. Multipolar spindles and aberrant spindle structure [26]. | Indirect (TP) |
| BRCA1 (h) | Not relevant | Not relevant | Regulates transcription of multiple mitotic genes, including BUB1, BUBR1/BUB1B, AURKA, ESPL1, PTTG1, ASPM, PRC1, and PLK1 [99]. | Indirect (TS) |
| BRCA1 (m) | Not relevant | Not relevant | Regulates SAC gene transcription, including MAD2, BUB1, BUBR1, and ZW10 [54,100]. | Indirect (TS) |
| BRCA1 (h) | Centrosomes | γ-tubulin | Centrosome amplification and fragmentation [31,32,33,34,35,36,37]. | DIRECT (TS) |
| BRCA1 (Xe) | Spindle poles | TPX2, NuMa, XRHAMM | Morphologically abnormal spindles in BRCA1-immunodepleted Xenopus egg extracts [40]. | DIRECT |
| ATF5 (h) | Centrosomes (mother centriole) Midbody DZ | γ-tubulin, PCNT | Defective accumulation of PCM; centriolar fragmentation, multipolar spindles; unknown function at the midbody [44,45]. | DIRECT |
| AKNA (m) | Centrosomes (mother centriole) | γTuRC, EB1, DCTN1 | Defective centrosome-driven MT regrowth after MT depolymerization [49]. | DIRECT |
| YB-1/YBX1 (h) | Centrosomes Spindle poles | γ-tubulin, PCNT | Structurally abnormal centrosomes with reduced MT nucleation ability [50,51]. | DIRECT (TP) |
| OCT1/POU2F1 (h) Interacts with BRCA1 | Centrosomes Kinetochores Midbody DZ | PARP-1, APC1 | Abnormal mitosis; unknown function at the midbody [52,53,54,55]. | DIRECT (TP) |
| Oct1 (Xe) | ND | ND | Morphologically abnormal spindles in Oct1-immunodepleted Xenopus egg extracts [52]. | DIRECT |
| Nubbin (Dm) (OCT1 ortholog) | Central spindle Midbody FR | ND | Unknown function at the midbody [56]. | P-direct ND |
| p53/TP53 (h) | Centrosomes | ND | Centrosome amplification and fragmentation [60,61,65,66,67]. | P-direct (TS) |
| p53 (m) | Centrosomes | ND | Centrosome amplification [58,62,63,64]. | P-direct |
| SP1 (h) | Centrosomes | ND | Centrosome amplification and decreased centrosome-driven MT nucleation [68]. | P-direct (TP) |
| SP1 (m) | Centrosomes | ND | Centrosome amplification and decreased centrosome-driven MT nucleation [68]. | P-direct |
| SF-1/NR5A1 (m) | Centrosomes | ND | Centrosome amplification [69,70]. | P-direct |
| Kaiso/ZBTB33 (h) | Centrosomes Spindle Central spindle Midbody FR | γ-tubulin, PCNT | No obvious mitotic defects [71,72]. | P-direct MR (TS) |
| CTCF (h) Interacts with Kaiso | Centrosomes Spindle poles Midbody DZ | ND | Mitotic phenotype not investigated [73,74]. | P-direct ND (TS) |
| SNAP45/SNAPC2 (h) | Centrosomes Spindle poles Midbody FR | ND | Multiple mitotic defects; defective chromosome condensation [75]. | P-direct |
| ELK1 (h) | Centrosomes Spindle Central spindle Midbody FR | AURKA | No obvious mitotic defects [20,76]. | P-direct MR (TP, TS) |
| RUNX1 (h) | Not relevant | Not relevant | Regulates transcription of BUBR1, BUB1, and NEK6 [78]. | Indirect (TP) |
| RUNX1, RUNX2, RUNX3 (h) | Centrosomes Spindle Midbody FR | γ-tubulin, rootletin | Reduced cyclin B1 accumulation and delayed mitotic entry. No specific mitotic defects [79,80]. | P-direct MR (TP) |
| CBFβ/CBFB (h) Binds RUNX proteins | Midbody FR | MRLC3 | Defective midbody structure and cytokinesis (abscission); polyploid cells [88]. | DIRECT (TS, TP) |
| YAP/YAP1 (h) Binds RUNX proteins | Midbody DZ | PATJ | Defective cytokinesis (abscission) [90]. | DIRECT (TS, TP) |
| Aire (m) (AIRE in humans) | Spindle Central spindle | AURKB, CEP55, CNTROB, HAUS5, HAUS8, CLASP1, CLASP2 | Abnormal spindle poles and centrosome amplification [94]. | DIRECT |
| HDAC3 (h) (in complex with NCOR1, TBL1X and TBL1XR1) | Spindle | ND | Morphologically abnormal spindles and chromosome misalignment [96]. | P-direct (TP) |
| Egr3 (m) | Meiotic spindles of mouse females | ND | Meiotic phenotype not investigated [97]. | P-direct ND |
| TFIIB (m) (GTF2B in humans) | Meiotic spindles of mouse females | ND | Morphologically abnormal spindles and chromosomes misalignment after antibody injection [98]. | P-direct |
| WT1 (h) | Kinetochores (colocalizes with MAD2) | MAD2 | Accelerated metaphase-to-anaphase transition; defective chromosome segregation [101,102]. | DIRECT (TS, TP) |
| TIF1γ/TRIM33 (h) | ND |
CDC20, APC/C | Chromosome misalignment; delay in metaphase-to-anaphase transition [103,104]. | P-direct (TS, TP) |
| XPB/ERCC3 (h) (TFIIH subunit) | Centrosomes Spindle poles | γ-tubulin | Mitotic phenotype not investigated [111]. | Direct ND (TS) |
| Hay (Dm) (TFIIH subunit, orthologous to XPB) | Chromosome/ spindle area in embryos | Testis-specific β2-tubulin | Defective meiotic spindles in males; abnormal mitotic spindles and defective chromosome segregation in embryos from hay mutant mothers [108,109,110,113]. | P-direct |
| p8, p52, Cdk7 (Dm) (TFIIH subunits) | Chromosome/ spindle area in embryos | ND | Abnormal mitotic spindles and defective chromosome segregation in embryos from mutant or RNAi mothers [113]. | P-direct |
| XPD/ERCC2 (h) (TFIIH subunit forms another complex with MIP18 and MMS19) | Spindle (with MIP18 and MMS19) | ND | Multipolar spindles and multinucleated cells [112]. | P-direct |
| Xpd (Dm) (TFIIH subunit) | ND | Regulates Cdk7 localization | Abnormal mitotic spindles and defective chromosome segregation in embryos from Xpd mutant mothers [114]. | P-direct |
| KANSL1, KANSL3, MCRS1 (h) (KANSL complex subunits) | Spindle poles; KANSL1 and KANSL3 bind MT minus ends | TPX2, MCAK | Chromosome misalignment and defective segregation [120,121]. | DIRECT (TP) |
| WDR5 (h) (KANSL subunit that forms another complex with MLL1/KMT2A) | Centrosomes Spindle Central spindle Midbody DZ | KIF2A, PRC1, MKLP1, CYK4, CEP55 | Morphologically abnormal and elongated spindles, aberrant chromosome segregation, and failures in cytokinesis [124,125,126]. | DIRECT (TP) |
| Dgt1 (Dm) (NSL subunit orthologous to KANSL2) | Diffuse (weak on spindle) | ND | Diminished γ-tubulin at centrosomes; long spindles [17]. | P-direct |
| Rcd1 (Dm) (NSL subunit orthologous to KANSL3) | Centrosomes Central spindle Midbody FR | ND | Defective centriole replication; frequent failures in chromosome alignment and segregation [18,19,127]. | Indirect (P-direct MR) |
| Rcd5 (Dm) (NSL subunit orthologous to MCRS1) | Centrosomes Midbody DZ | ND | Defective centriole replication; frequent failures in chromosome alignment and segregation [17,18,127]. | Indirect (P-direct MR) |
| MBD-R2 (Dm) (NSL subunit orthologous to PHF20) | Chromosomes | ND | Defective centriole replication; frequent failures in chromosome alignment and segregation [19,127]. | Indirect (P-direct MR) |
| Wds (Dm) (NSL subunit orthologous to WDR5) | Centrosomes Kinetochores Midbody DZ | ND | Defective centriole replication; frequent failures in chromosome alignment and segregation [127]. | Indirect (P-direct MR) |
| Name (Organism) | Mitotic Localization | Interacting Mitotic Proteins | Loss-of-Function Mitotic Phenotype | Mitotic Function (Role in Cancer) |
|---|---|---|---|---|
| CEF1 (Sc) | ND | ND | Inefficient splicing of α-tubulin pre-mRNA. Defective spindle assembly. Mutant phenotype rescued by an intronless α-tubulin gene [130]. | Indirect |
| U2F35/U2AF1 (h) | ND | ND | Inefficient splicing of the CDC27 pre-mRNA. Defective spindle morphology and chromosome segregation [131]. | Indirect (TS, TP) |
| SON (h) | ND | ND | Inefficient splicing of TUBG1, TUBGCP2, TUBGCP4, and AKT1 pre-mRNAs. Impaired centrosome separation at prophase; defective chromosome segregation and cytokinesis [132]. | Indirect (TS) |
| WBP11 (h) | ND | ND | Inefficient splicing of TUBGCP6 pre-mRNA. Impaired centriole duplication; mutant phenotype rescued by an intronless TUBGCP6 transgene [133]. | Indirect |
| NRDE2 (h) | Diffuse in the cytoplasm | ND | Inefficient splicing of CEP131 pre-mRNA. Impaired centrosomal recruitment of γ-tubulin, AURKA, and CEP192; defective chromosome segregation [134]. | Indirect (TS, TP) |
| 27 different human SFs, including PRPF8, NHP2L1/SNU13, SART1, MFAP1, CDC5L, SNW1, PRP19/PRPF19, UBL5, SF3B1, SNRNP200, PRPF6 | Some colocalize with the Cohesin complex | Some copurify with the Cohesin subunits | Intron retention in the sororin mRNA. Parallel sister chromatids at metaphase and defective chromosome segregation; mutant phenotype partially rescued by an intronless sororin transgene [135,136,137,138,140]. | Indirect, (possibly also DIRECT) (TP: CDC5L, SNW1, U2AF2) |
| PRP19/PRPF19 (h), CDC5L (h), SPF27/BCAS2 (h), PLRG1 (h) (PRP19 complex) | Not relevant | Not relevant | Inefficient splicing of DYNC1H1, DYNLRB2, and DCTN4 pre-mRNAs. Impaired kinetochore-MT attachment, defective chromosome segregation [143]. | Indirect (TP: CDC5L, BCAS2) |
| PRP19/PRPF19 (h), CDC5L (h), SPF27/BCAS2 (h) (PRP19 complex) | Diffuse | ND | Defective kinetochore-MT interaction; abnormal spindles and defective chromosome alignment in human cells [42,138]. | P-direct (TP: CDC5L, BCAS2) |
| Prp19 (Xe), Bcas2 (Xe) (PRP19 complex) | ND | ND | Defective chromatin-MT interaction; morphologically abnormal spindles in Xenopus egg extracts [42]. | DIRECT |
| PRP4/PRPF4 (h) | Kinetochores | ND | Defective recruitment at kinetochores of the MPS1, MAD1, and MAD2 SAC proteins. Precocious anaphase onset; lagging anaphase chromosomes and aneuploidy [147]. | P-direct (TP) |
| SF3A2 (h), PRP31/PRPF31 (h) | Spindle (Mouse Sf3A2 binds MTs in vitro) | Ndc80/HEC1 | Morphologically irregular spindles; defective chromosome congression at metaphase; reduced frequency of ana-telophases [151,152]. | DIRECT |
| Sf3A2 (Dm), Prp31 (Dm) | Spindle (Binds MTs in vitro) | Ndc80, Mitch, Nuf2 (Ndc80 complex) | Reduced accumulation of Ndc80 at kinetochores and severe defects in chromosome alignment and segregation. Anti-Sf3A2 and anti-Prp31 injections disrupt mitosis in embryos [151]. | DIRECT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somma, M.P.; Andreyeva, E.N.; Pavlova, G.A.; Pellacani, C.; Bucciarelli, E.; Popova, J.V.; Bonaccorsi, S.; Pindyurin, A.V.; Gatti, M. Moonlighting in Mitosis: Analysis of the Mitotic Functions of Transcription and Splicing Factors. Cells 2020, 9, 1554. https://doi.org/10.3390/cells9061554
Somma MP, Andreyeva EN, Pavlova GA, Pellacani C, Bucciarelli E, Popova JV, Bonaccorsi S, Pindyurin AV, Gatti M. Moonlighting in Mitosis: Analysis of the Mitotic Functions of Transcription and Splicing Factors. Cells. 2020; 9(6):1554. https://doi.org/10.3390/cells9061554
Chicago/Turabian StyleSomma, Maria Patrizia, Evgeniya N. Andreyeva, Gera A. Pavlova, Claudia Pellacani, Elisabetta Bucciarelli, Julia V. Popova, Silvia Bonaccorsi, Alexey V. Pindyurin, and Maurizio Gatti. 2020. "Moonlighting in Mitosis: Analysis of the Mitotic Functions of Transcription and Splicing Factors" Cells 9, no. 6: 1554. https://doi.org/10.3390/cells9061554
APA StyleSomma, M. P., Andreyeva, E. N., Pavlova, G. A., Pellacani, C., Bucciarelli, E., Popova, J. V., Bonaccorsi, S., Pindyurin, A. V., & Gatti, M. (2020). Moonlighting in Mitosis: Analysis of the Mitotic Functions of Transcription and Splicing Factors. Cells, 9(6), 1554. https://doi.org/10.3390/cells9061554