Cathelicidin-Related Antimicrobial Peptide Regulates CD73 Expression in Mouse Th17 Cells via p38

 , , , and
, , , and
Abstract
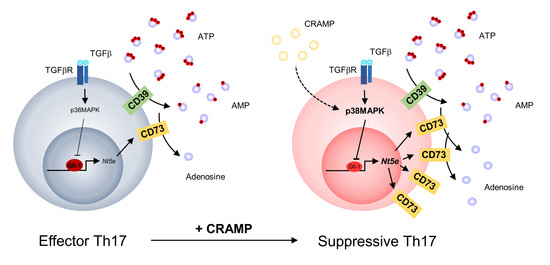
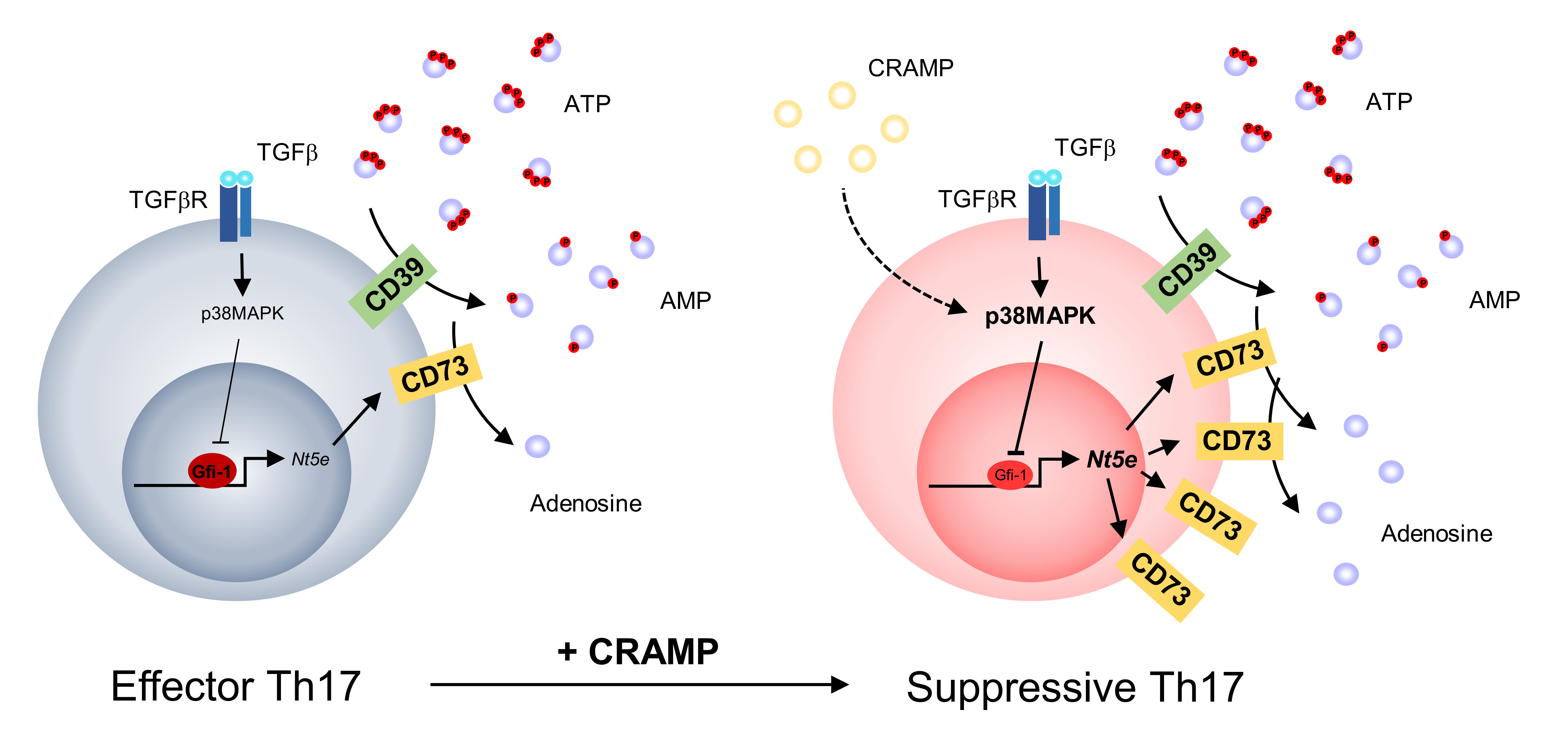
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Naïve CD73−CD4+ T Cell Isolation
2.3. In Vitro T Cell Culture System
2.4. Flow Cytometry Analysis
2.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Adenosine Quantification
2.7. Cell Death Assay
2.8. Plasmids and Retroviral Gene Transduction
2.9. Statistical Analysis
3. Results
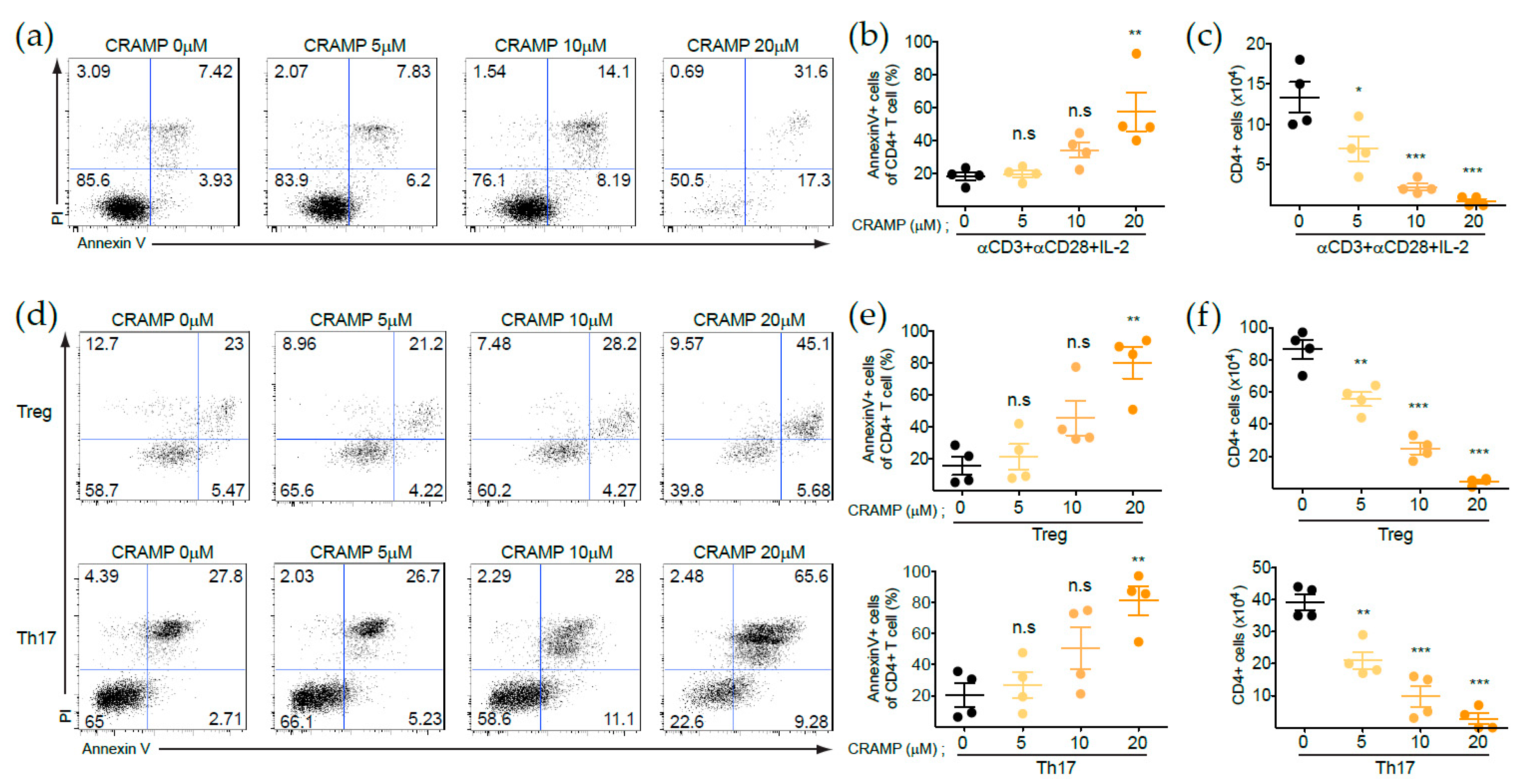
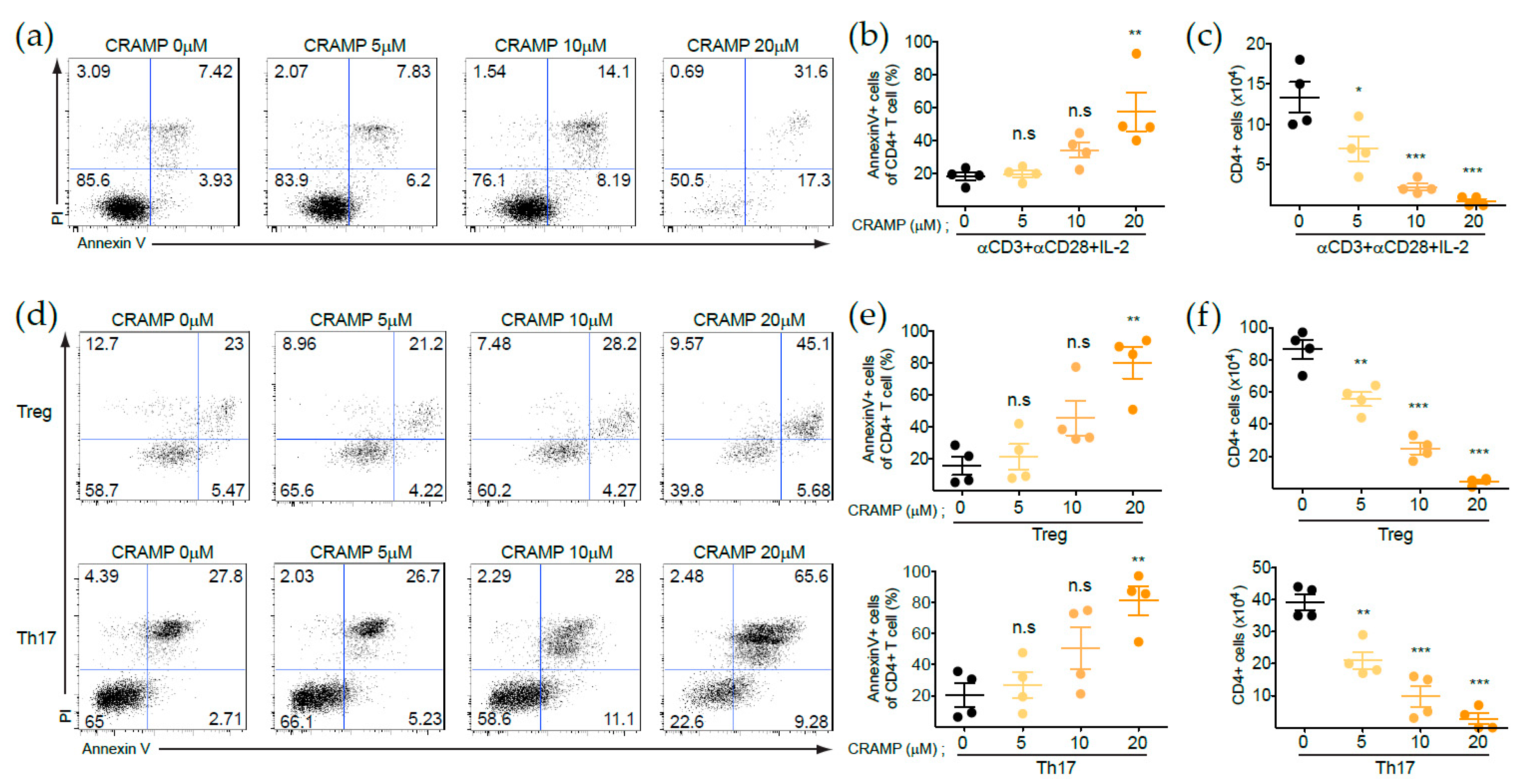
3.1. CRAMP Induces the Apoptosis of CD4+ T Cells
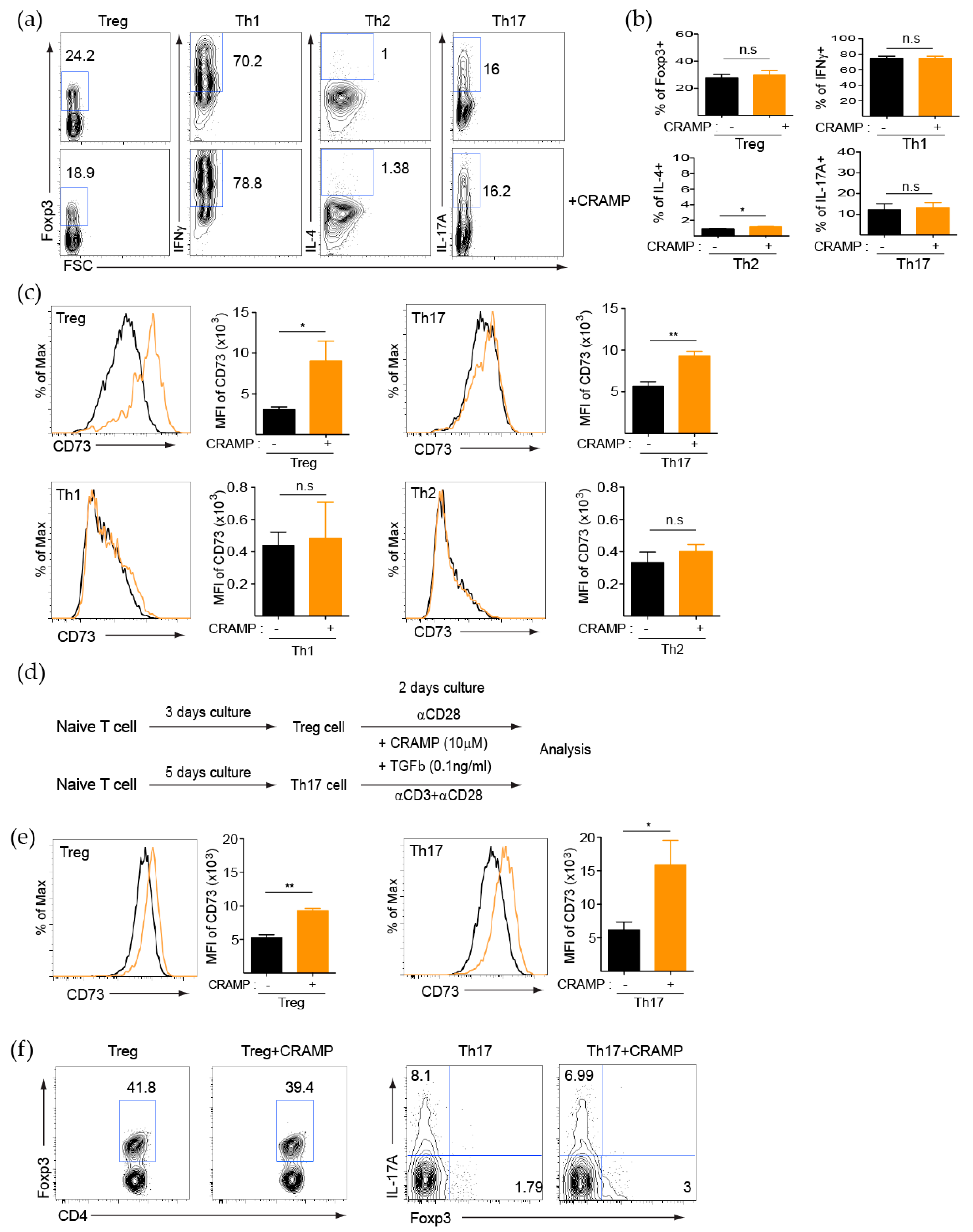
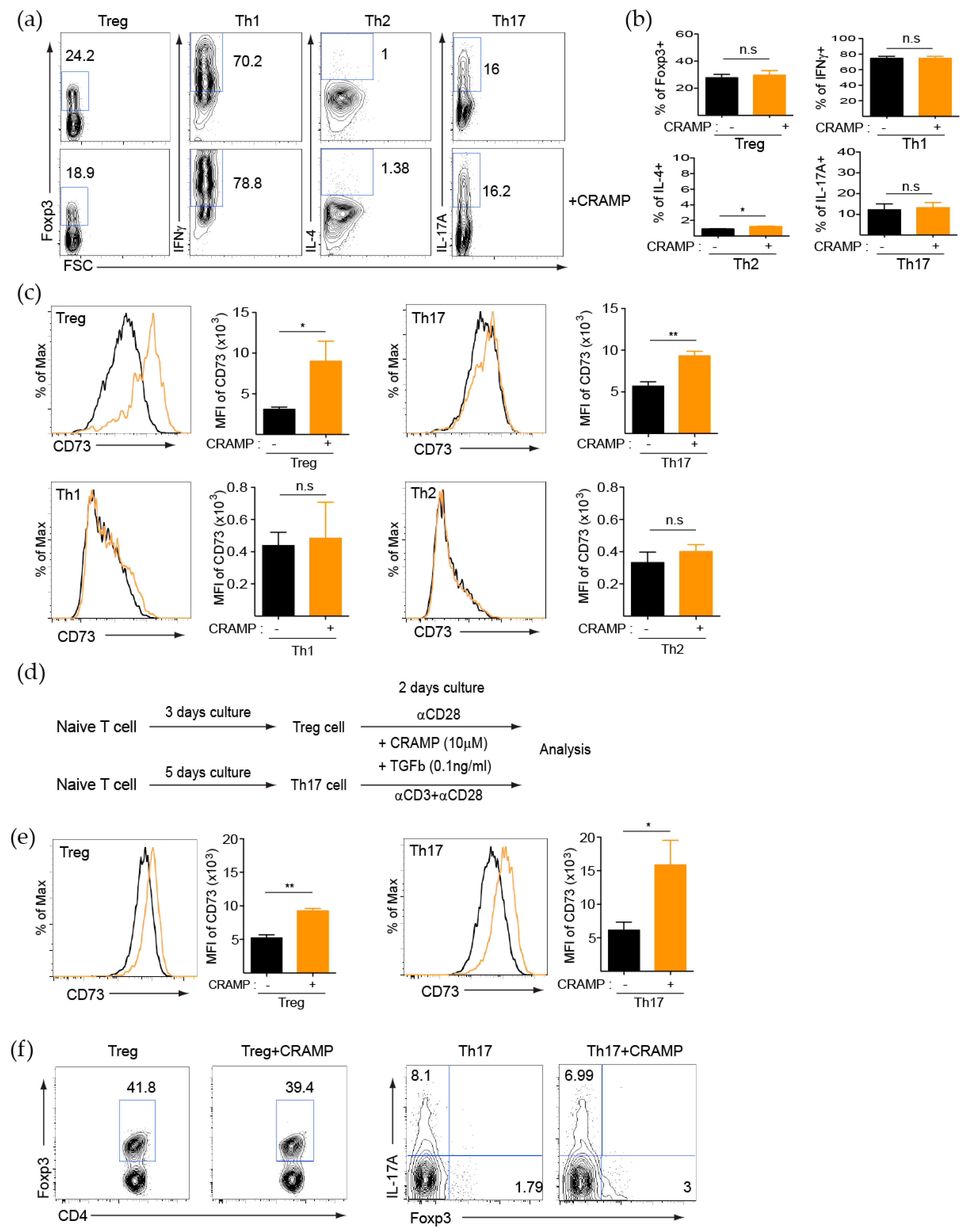
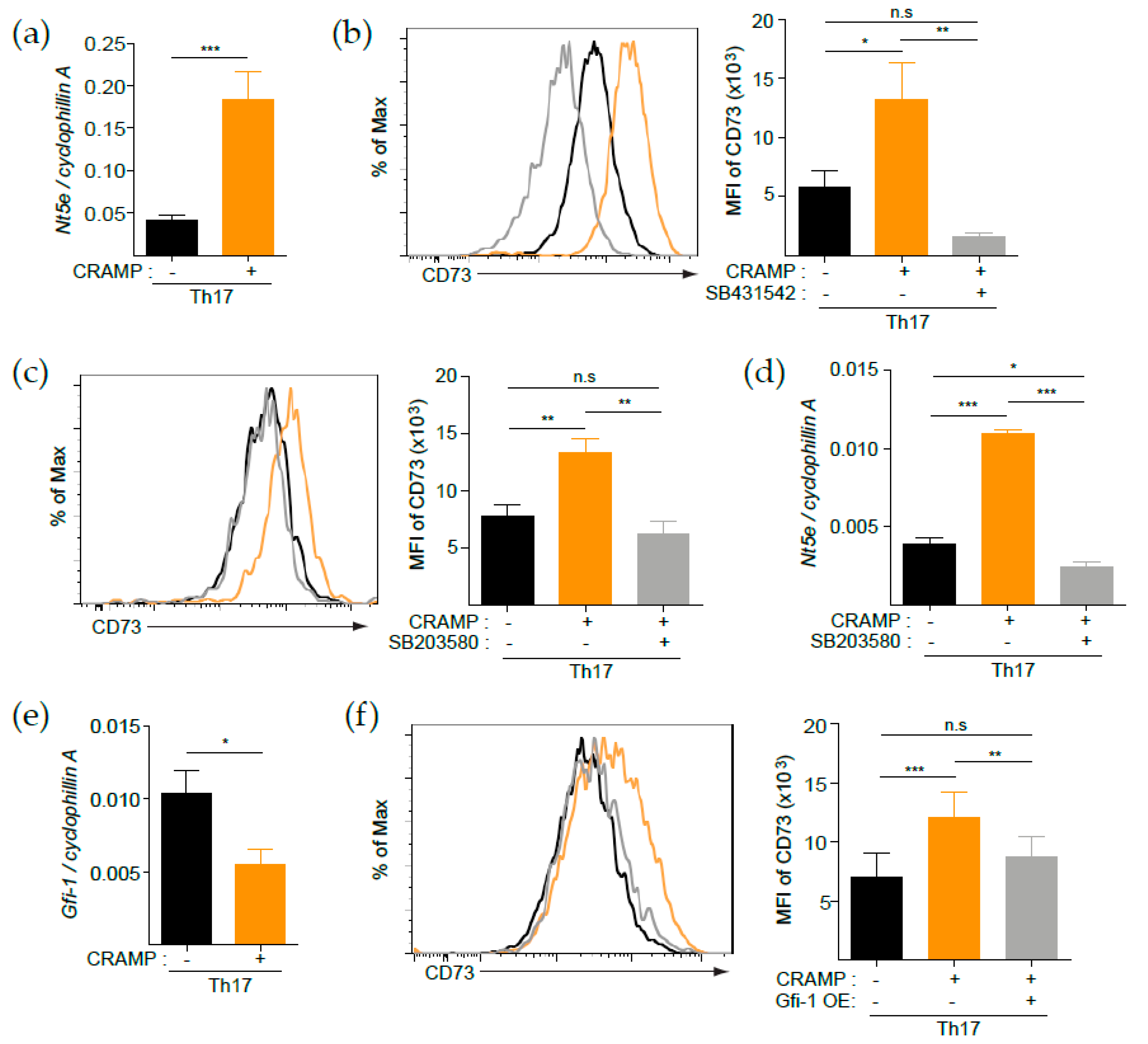
3.2. CRAMP Induces CD73 Expression on CD4+ T Cells
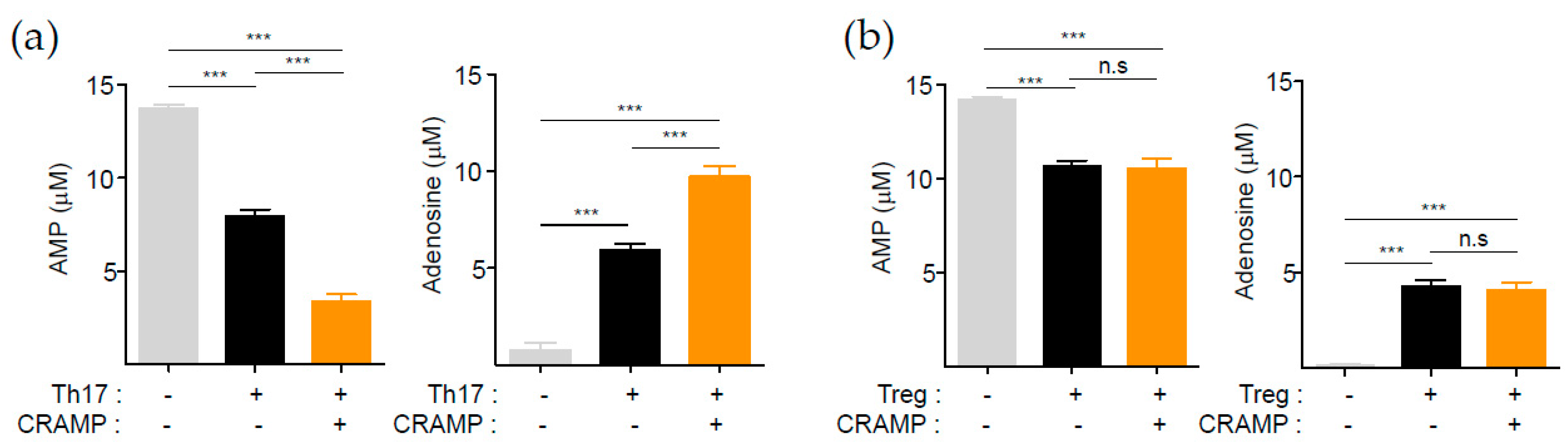
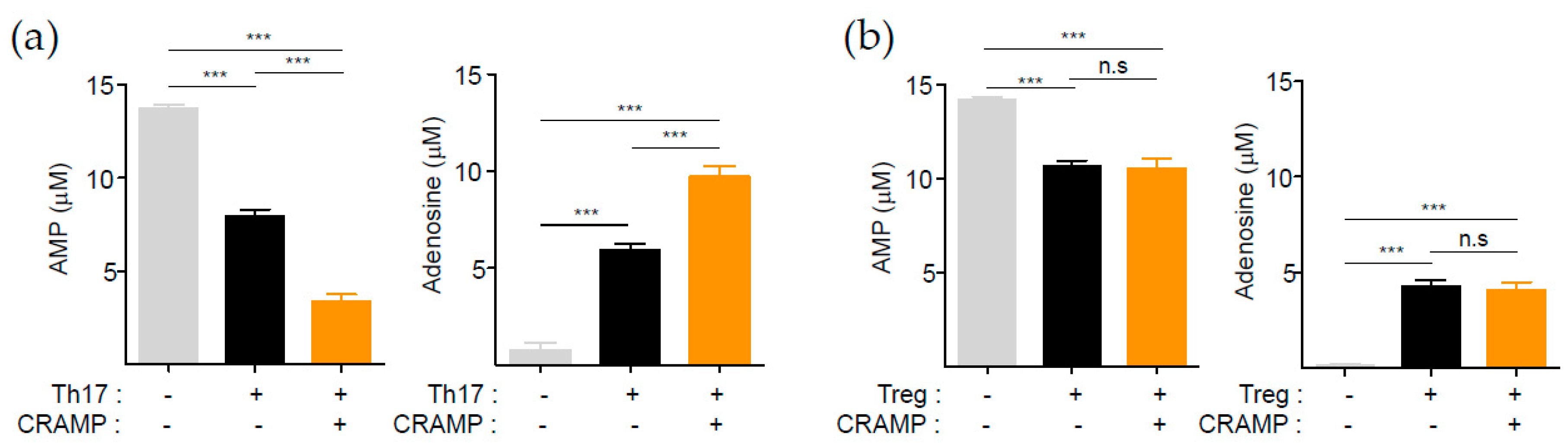
3.3. CRAMP-Induced CD73 Is Functional
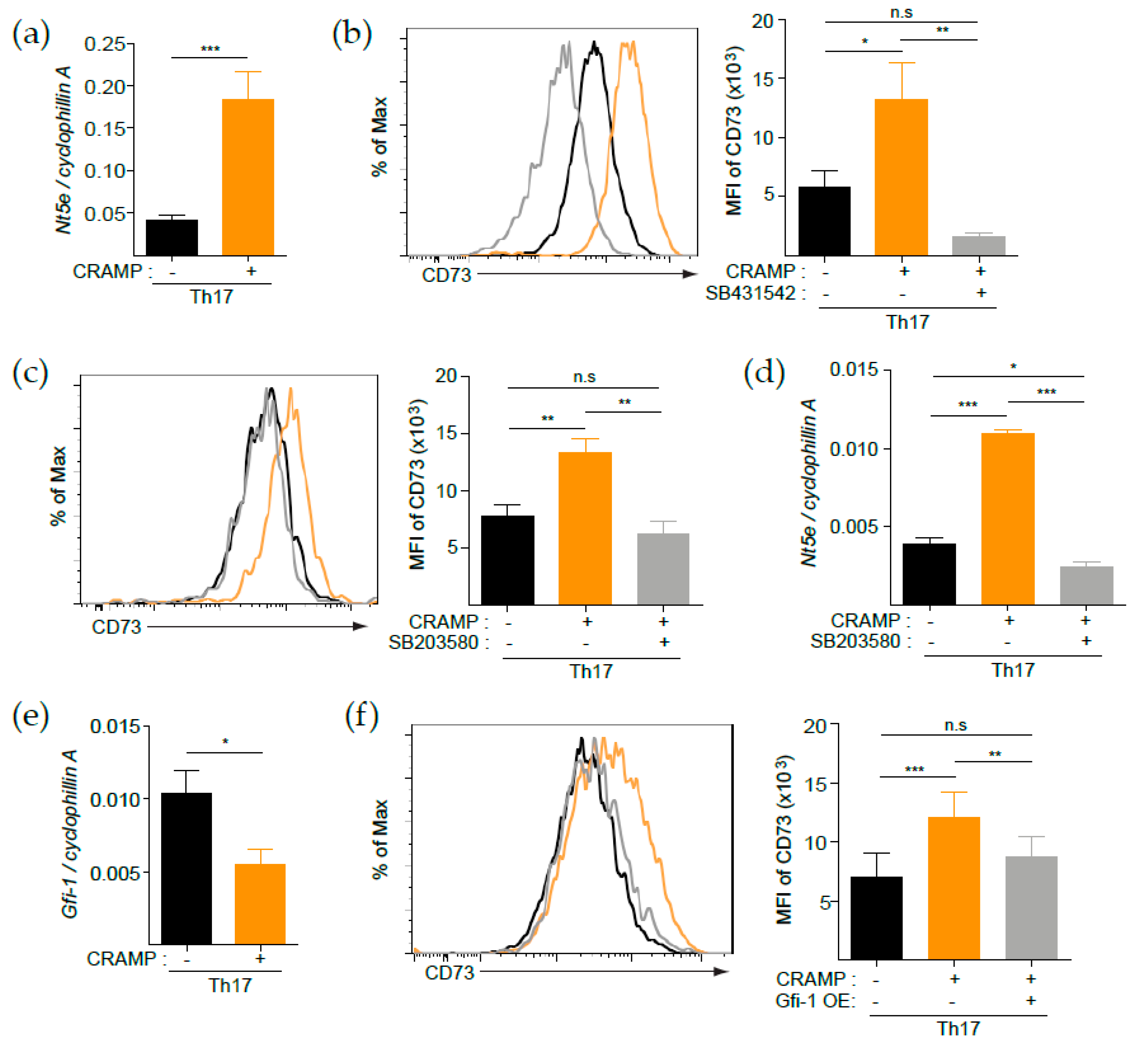
3.4. p38 Is Responsible for the CRAMP-Mediated Induction of CD73
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.; Celis, E. Multiple roles for CD4+ T cells in antitumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Pentcheva-Hoang, T.; Simpson, T.R.; Montalvo-Ortiz, W.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol. Res. 2014, 2, 970–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Babala, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Barkauskas, D.S.; Sult, E.; Hay, C.; Blake, S.J.; Huang, Q.; Liu, J.; Takeda, K.; Teng, M.W.L.; et al. Co-inhibition of CD73 and A2AR Adenosine Signaling Improves Antitumor Immune Responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Menetrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin Invest. 2011, 121, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regateiro, F.S.; Howie, D.; Nolan, K.F.; Agorogiannis, E.I.; Greaves, D.R.; Cobbold, S.P.; Waldmann, H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-beta. Eur. J. Immunol. 2011, 41, 2955–2965. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fan, J.; Zhang, M.; Qin, L.; Dominguez, D.; Long, A.; Wang, G.; Ma, R.; Li, H.; Zhang, Y.; et al. CD73 expression on effector T cells sustained by TGF-beta facilitates tumor resistance to anti-4-1BB/CD137 therapy. Nat. Commun. 2019, 10, 150. [Google Scholar] [CrossRef] [Green Version]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The human cathelicidin LL-37--A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta 2016, 1858, 546–566. [Google Scholar] [CrossRef]
- Kuroda, K.; Okumura, K.; Isogai, H.; Isogai, E. The Human Cathelicidin Antimicrobial Peptide LL-37 and Mimics are Potential Anticancer Drugs. Front. Oncol. 2015, 5, 144. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Kaplan, M.J. Little peptide, big effects: The role of LL-37 in inflammation and autoimmune disease. J. Immunol. 2013, 191, 4895–4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Martinez, N.; Avila, E.; Larrea, F.; Diaz, L. Calcitriol stimulates gene expression of cathelicidin antimicrobial peptide in breast cancer cells with different phenotype. J. Biomed. Sci. 2016, 23, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Marini, F.C.; Watson, K.; Zwezdaryk, K.J.; Dembinski, J.L.; LaMarca, H.L.; Tomchuck, S.L.; Honer zu Bentrup, K.; Danka, E.S.; Henkle, S.L.; et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl. Acad. Sci. USA 2009, 106, 3806–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girnita, A.; Zheng, H.; Gronberg, A.; Girnita, L.; Stahle, M. Identification of the cathelicidin peptide LL-37 as agonist for the type I insulin-like growth factor receptor. Oncogene 2012, 31, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Tjabringa, G.S.; Aarbiou, J.; Ninaber, D.K.; Drijfhout, J.W.; Sorensen, O.E.; Borregaard, N.; Rabe, K.F.; Hiemstra, P.S. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6690–6696. [Google Scholar] [CrossRef] [Green Version]
- Verjans, E.T.; Zels, S.; Luyten, W.; Landuyt, B.; Schoofs, L. Molecular mechanisms of LL-37-induced receptor activation: An overview. Peptides 2016, 85, 16–26. [Google Scholar] [CrossRef]
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic beta-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Petit, P.F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef]
- Lu, B.; Finn, O.J. T-cell death and cancer immune tolerance. Cell Death Differ. 2008, 15, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, O.; Cowland, J.B.; Askaa, J.; Borregaard, N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J. Immunol. Methods 1997, 206, 53–59. [Google Scholar] [CrossRef]
- Chen, C.I.; Schaller-Bals, S.; Paul, K.P.; Wahn, U.; Bals, R. Beta-defensins and LL-37 in bronchoalveolar lavage fluid of patients with cystic fibrosis. J. Cyst. Fibros 2004, 3, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zou, X.; Qi, G.; Tang, Y.; Guo, Y.; Si, J.; Liang, L. Roles and Mechanisms of Human Cathelicidin LL-37 in Cancer. Cell Physiol. Biochem. 2018, 47, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- van Harten, R.M.; van Woudenbergh, E.; van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory Antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowdish, D.M.; Davidson, D.J.; Speert, D.P.; Hancock, R.E. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J. Immunol. 2004, 172, 3758–3765. [Google Scholar] [CrossRef] [Green Version]
- Downs-Canner, S.; Berkey, S.; Delgoffe, G.M.; Edwards, R.P.; Curiel, T.; Odunsi, K.; Bartlett, D.L.; Obermajer, N. Suppressive IL-17A(+)Foxp3(+) and ex-Th17 IL-17A(neg)Foxp3(+) Treg cells are a source of tumour-associated Treg cells. Nat. Commun. 2017, 8, 14649. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Su, X.; Ye, J.; Hsueh, E.C.; Zhang, Y.; Hoft, D.F.; Peng, G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J. Immunol. 2010, 184, 1630–1641. [Google Scholar] [CrossRef]
- Chatterjee, S.; Thyagarajan, K.; Kesarwani, P.; Song, J.H.; Soloshchenko, M.; Fu, J.; Bailey, S.R.; Vasu, C.; Kraft, A.S.; Paulos, C.M.; et al. Reducing CD73 expression by IL1beta-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res. 2014, 74, 6048–6059. [Google Scholar] [CrossRef] [Green Version]
- Gourdin, N.; Bossennec, M.; Rodriguez, C.; Vigano, S.; Machon, C.; Jandus, C.; Bauche, D.; Faget, J.; Durand, I.; Chopin, N.; et al. Autocrine Adenosine Regulates Tumor Polyfunctional CD73(+)CD4(+) Effector T Cells Devoid of Immune Checkpoints. Cancer Res. 2018, 78, 3604–3618. [Google Scholar] [CrossRef] [Green Version]
- Fausther, M.; Sheung, N.; Saiman, Y.; Bansal, M.B.; Dranoff, J.A. Activated hepatic stellate cells upregulate transcription of ecto-5’-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am. J. Physiol. Gastrointest Liver Physiol. 2012, 303, G904–G914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.R.; Resta, R.; Webb, C.F.; Thompson, L.F. Isolation and characterization of the promoter of the human 5’-nucleotidase (CD73)-encoding gene. Gene 1995, 167, 307–312. [Google Scholar] [CrossRef]
- Pagnotta, S.M.; Laudanna, C.; Pancione, M.; Sabatino, L.; Votino, C.; Remo, A.; Cerulo, L.; Zoppoli, P.; Manfrin, E.; Colantuoni, V.; et al. Ensemble of gene signatures identifies novel biomarkers in colorectal cancer activated through PPARgamma and TNFalpha signaling. PLoS ONE 2013, 8, e72638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Shin, K.-O.; Kim, Y.; Cho, J.; Lim, H.W.; Yoon, S.-I.; Lee, G.-S.; Ko, H.-J.; Kim, P.-H.; Uchida, Y.; et al. Cathelicidin-Related Antimicrobial Peptide Regulates CD73 Expression in Mouse Th17 Cells via p38. Cells 2020, 9, 1561. https://doi.org/10.3390/cells9061561
Lee J, Shin K-O, Kim Y, Cho J, Lim HW, Yoon S-I, Lee G-S, Ko H-J, Kim P-H, Uchida Y, et al. Cathelicidin-Related Antimicrobial Peptide Regulates CD73 Expression in Mouse Th17 Cells via p38. Cells. 2020; 9(6):1561. https://doi.org/10.3390/cells9061561
Chicago/Turabian StyleLee, Jeonghyun, Kyong-Oh Shin, Yesol Kim, Jaewon Cho, Hyung W. Lim, Sung-Il Yoon, Geun-Shik Lee, Hyun-Jeong Ko, Pyeung-Hyeun Kim, Yoshikazu Uchida, and et al. 2020. "Cathelicidin-Related Antimicrobial Peptide Regulates CD73 Expression in Mouse Th17 Cells via p38" Cells 9, no. 6: 1561. https://doi.org/10.3390/cells9061561
APA StyleLee, J., Shin, K.-O., Kim, Y., Cho, J., Lim, H. W., Yoon, S.-I., Lee, G.-S., Ko, H.-J., Kim, P.-H., Uchida, Y., Park, K., & Kang, S. G. (2020). Cathelicidin-Related Antimicrobial Peptide Regulates CD73 Expression in Mouse Th17 Cells via p38. Cells, 9(6), 1561. https://doi.org/10.3390/cells9061561