Oxidative Inactivation of the Proteasome Augments Alveolar Macrophage Secretion of Vesicular SOCS3


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of CSE
2.2. Isolation and Culture of Primary and Immortalized AMs
2.3. EV Isolation
2.4. Western Blot
2.5. ROS Assay
2.6. Nanoparticle Tracking Analysis (NTA)
2.7. Carboxyfluorescein Succinimidyl Ester (CFSE)-Based Vesicular SOCS3 Packaging Assay
2.8. 20S Proteasome Activity Assay
2.9. Data Collection and Analysis
3. Results
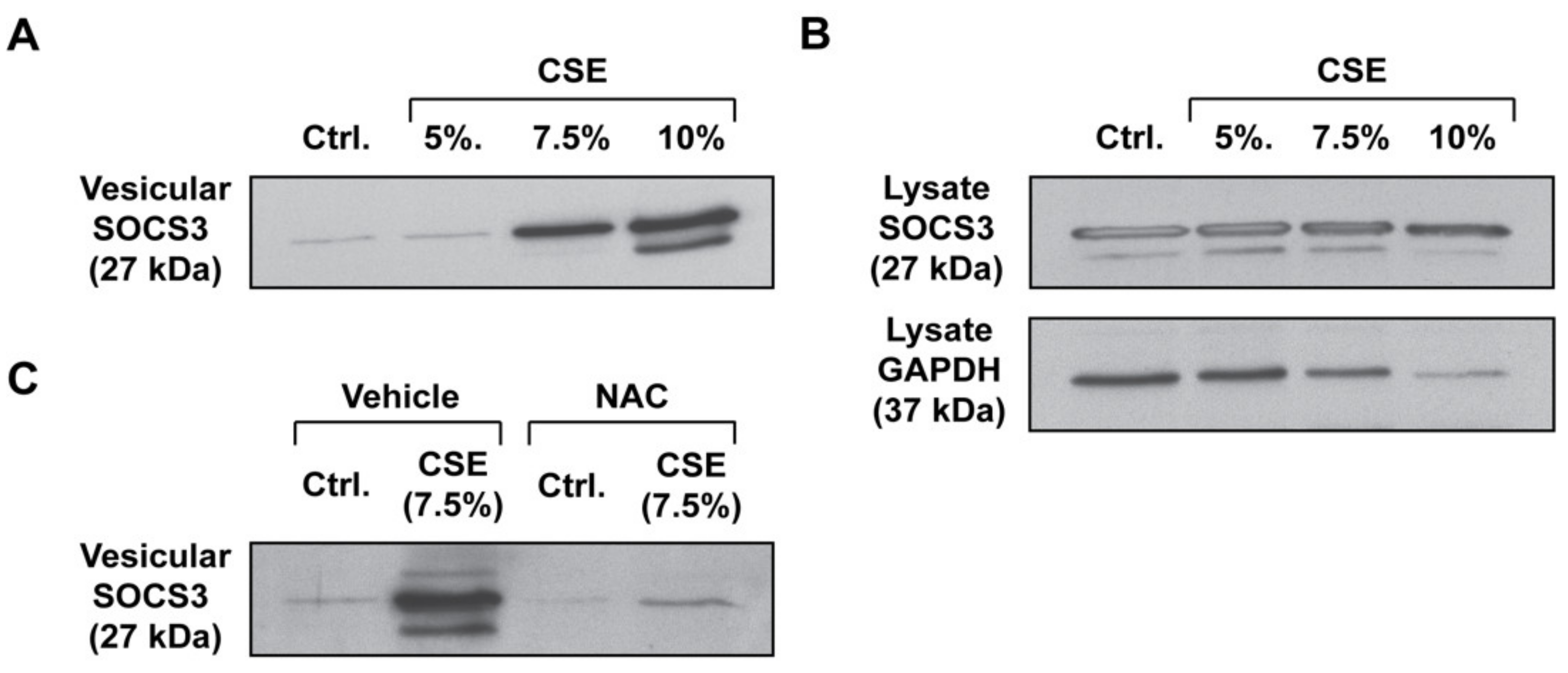
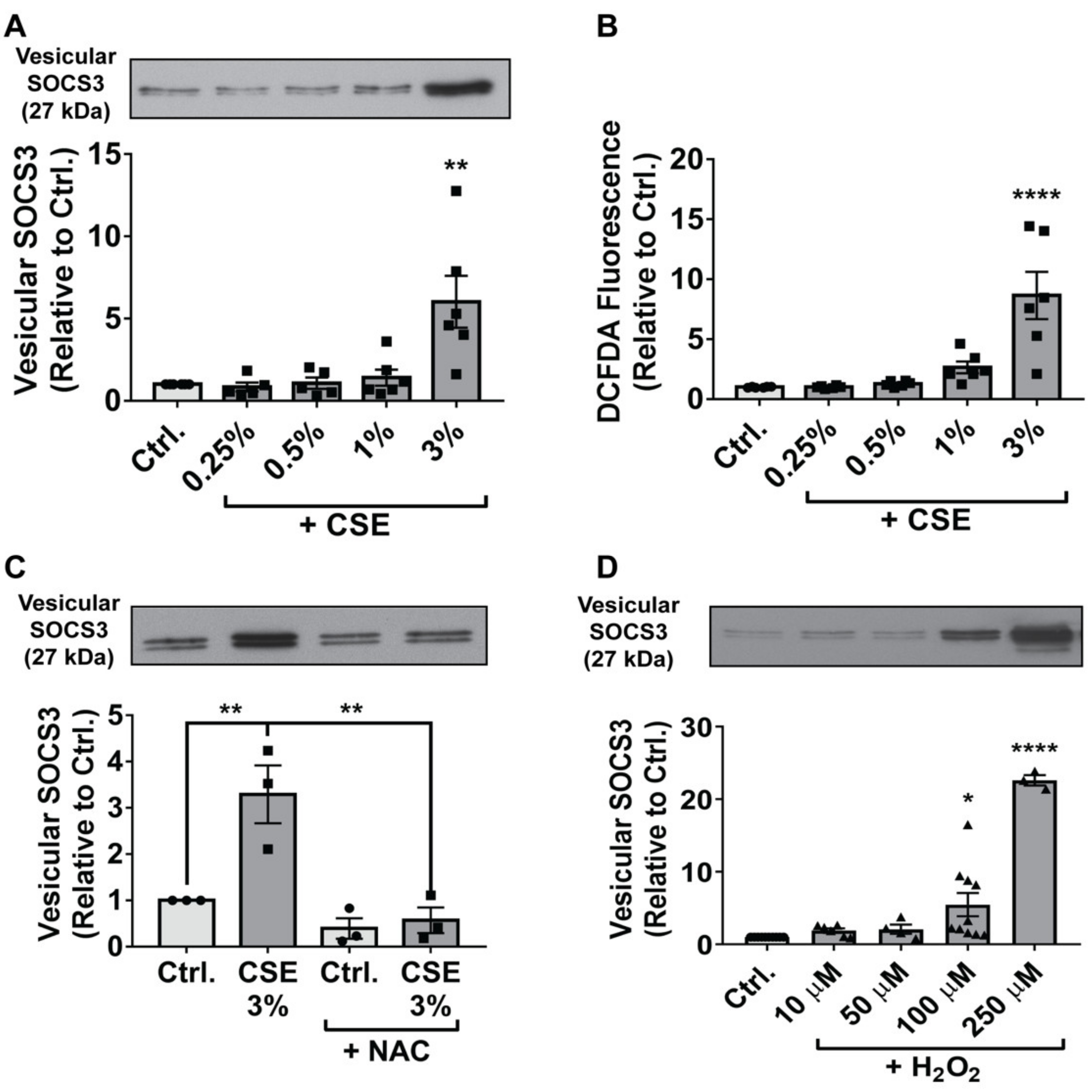
3.1. CSE Enhances SOCS3 Secretion by Primary AMs in an ROS-Dependent Manner
3.2. Endogenous and Exogenous ROS Stimulate SOCS3 Release by Immortalized AMs
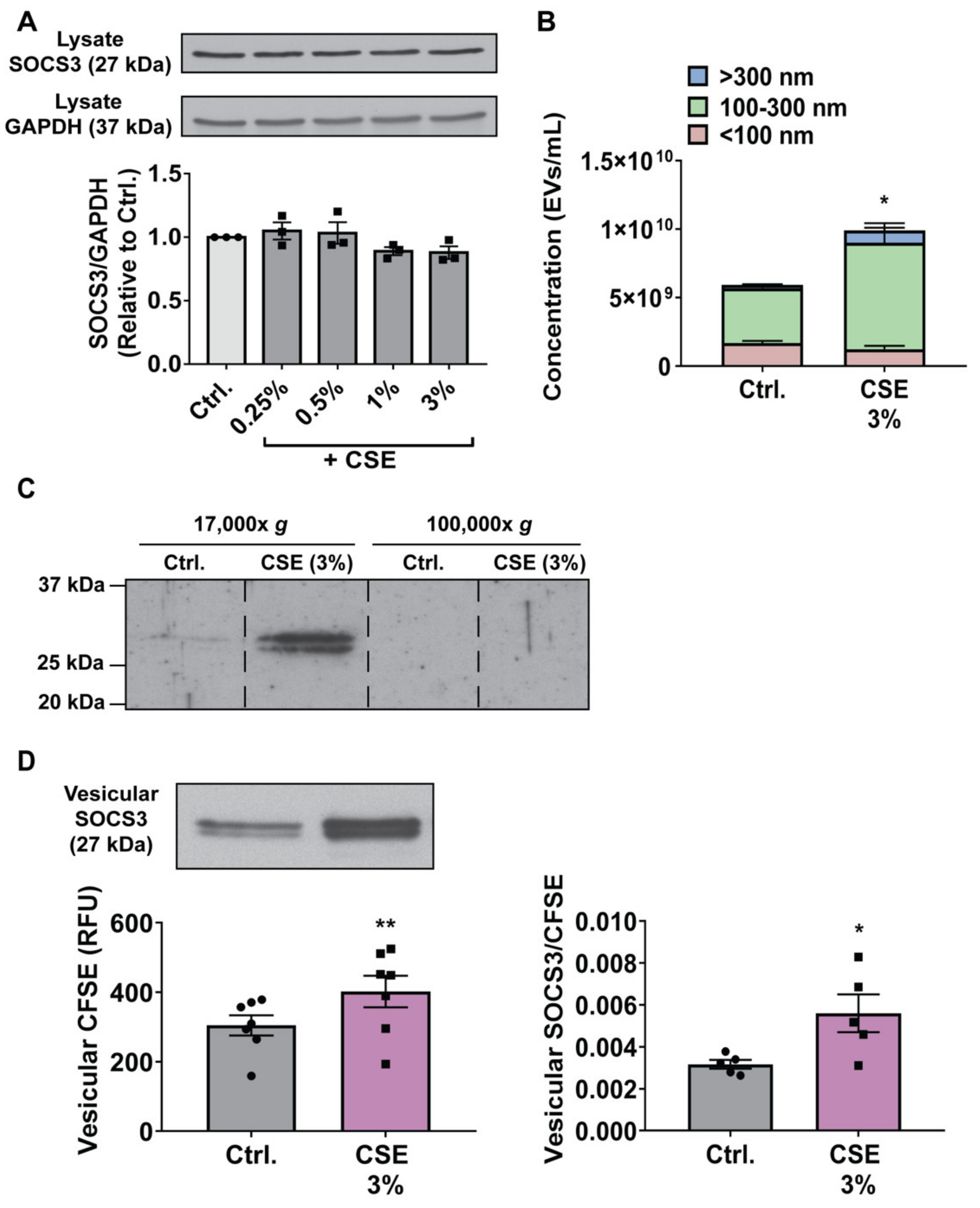
3.3. CSE Augments MH-S Cell Secretion of SOCS3 by Stimulating Production of, and Packaging of SOCS3 into, EVs
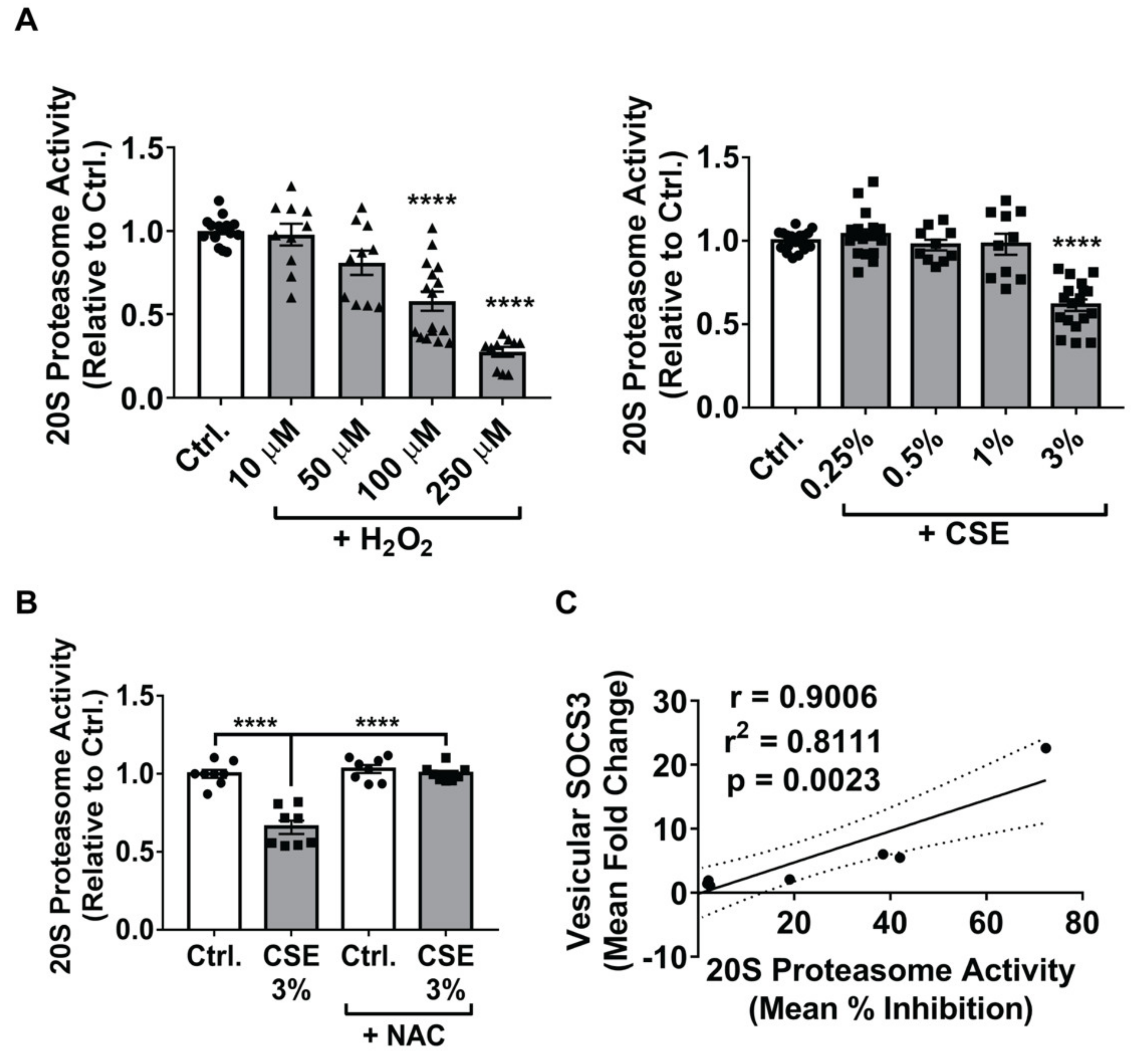
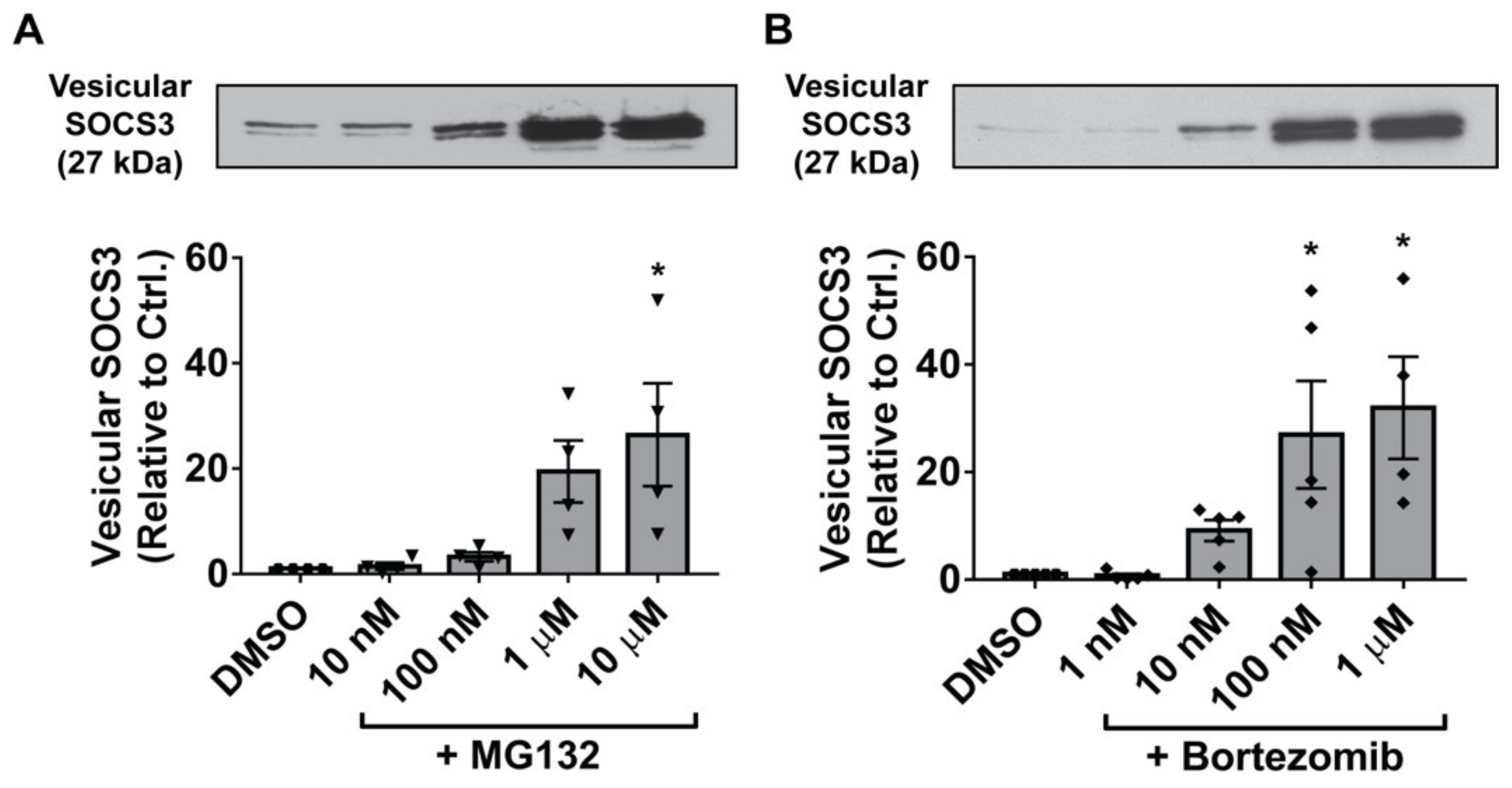
3.4. Inhibition of the 20S Proteasome Mimics the Stimulatory Effects of ROS on Vesicular SOCS3 Secretion by MH-S Cells
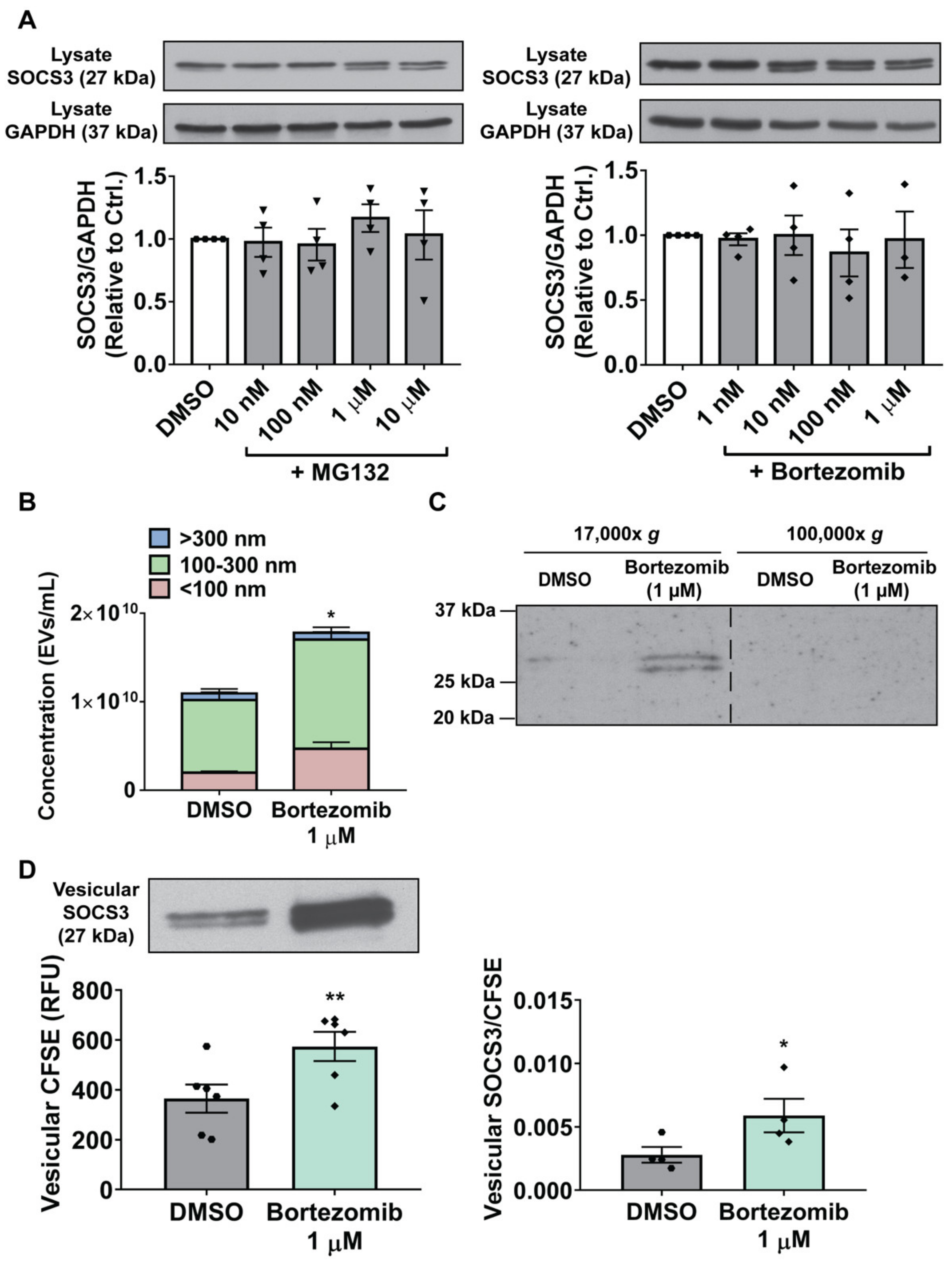
3.5. Proteasome Inactivation in MH-S Cells Augments SOCS3 Secretion by Stimulating Production of, and Packaging of SOCS3 into, EVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of exosome composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmidou, I.; Vassilakopoulos, T.; Xagorari, A.; Zakynthinos, S.; Papapetropoulos, A.; Roussos, C. Production of interleukin-6 by skeletal myotubes: Role of reactive oxygen species. Am. J. Respir. Cell Mol. Biol. 2002, 26, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Kwon, M.H.; Park, S.; Shin, H.J.; Gwak, H.S.; Chwae, Y.J. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010, 6, 1125–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Zhang, D.; Zhu, Z.; Dela Cruz, C.S.; Jin, Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci. Rep. 2016, 6, 35250. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.G.; Cao, Y.; Yang, J.; Lee, J.H.; Choi, H.S.; Jin, Y. Lung epithelial cell-derived extracellular vesicles activate macrophage-mediated inflammatory responses via ROCK1 pathway. Cell Death Dis. 2015, 6, e2016. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; Wilson, M.R.; O’Dea, K.P.; Yoshida, M.; Katbeh, U.; Woods, S.J.; Takata, M. Alveolar macrophage-derived microvesicles mediate acute lung injury. Thorax 2016, 71, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Bourdonnay, E.; Zaslona, Z.; Penke, L.R.; Speth, J.M.; Schneider, D.J.; Przybranowski, S.; Swanson, J.A.; Mancuso, P.; Freeman, C.M.; Curtis, J.L.; et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J. Exp. Med. 2015, 212, 729–742. [Google Scholar] [CrossRef]
- Schneider, D.J.; Speth, J.M.; Penke, L.R.; Wettlaufer, S.H.; Swanson, J.A.; Peters-Golden, M. Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. J. Biol. Chem. 2017, 292, 20897–20910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speth, J.M.; Bourdonnay, E.; Penke, L.R.; Mancuso, P.; Moore, B.B.; Weinberg, J.B.; Peters-Golden, M. Alveolar epithelial cell-derived prostaglandin E2 serves as a request signal for macrophage secretion of suppressor of cytokine signaling 3 during innate inflammation. J. Immunol. 2016, 196, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Speth, J.M.; Penke, L.R.; Bazzill, J.D.; Park, K.S.; de Rubio, R.G.; Schneider, D.J.; Ouchi, H.; Moon, J.J.; Keshamouni, V.G.; Zemans, R.L.; et al. Alveolar macrophage secretion of vesicular SOCS3 represents a platform for lung cancer therapeutics. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.L.; Lundberg, K.C.; Humphries, K.M.; Sadek, H.A.; Szweda, P.A.; Friguet, B.; Szweda, L.I. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J. Biol. Chem. 2001, 276, 30057–30063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Sakurai, T.; Usami, H.; Uchida, K. Oxidative modification of proteasome: Identification of an oxidation-sensitive subunit in 26 S proteasome. Biochemistry 2005, 44, 13893–13901. [Google Scholar] [CrossRef] [PubMed]
- Paramore, A.; Frantz, S. Bortezomib. Nat. Rev. Drug Discov. 2003, 2, 611–612. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Zarfati, M.; Avivi, I.; Brenner, B.; Katz, T.; Aharon, A. Extracellular vesicles of multiple myeloma cells utilize the proteasome inhibitor mechanism to moderate endothelial angiogenesis. Angiogenesis 2019, 22, 185–196. [Google Scholar] [CrossRef]
- Phipps, J.C.; Aronoff, D.M.; Curtis, J.L.; Goel, D.; O’Brien, E.; Mancuso, P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect. Immun. 2010, 78, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- Peters-Golden, M.; Thebert, P. Inhibition by methylprednisolone of zymosan-induced leukotriene synthesis in alveolar macrophages. Am. Rev. Respir. Dis. 1987, 135, 1020–1026. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.; Lugg, S.T.; Aldridge, K.; Lewis, K.E.; Bowden, A.; Mahida, R.Y.; Grudzinska, F.S.; Dosanjh, D.; Parekh, D.; Foronjy, R.; et al. Pro-inflammatory effects of e-cigarette vapour condensate on human alveolar macrophages. Thorax 2018, 73, 1161–1169. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Joseph, J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Pryor, W.A.; Prier, D.G.; Church, D.F. Electron-spin resonance study of mainstream and sidestream cigarette smoke: Nature of the free radicals in gas-phase smoke and in cigarette tar. Environ. Health Perspect. 1983, 47, 345–355. [Google Scholar] [CrossRef]
- Pryor, W.A.; Terauchi, K.; Davis, W.H., Jr. Electron spin resonance (ESR) study of cigarette smoke by use of spin trapping techniques. Environ. Health Perspect. 1976, 16, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Haralambous, E. A comparative study by electron paramagnetic resonance of free radical species in the mainstream and sidestream smoke of cigarettes with conventional acetate filters and ‘bio-filters’. Redox Rep. 2001, 6, 161–171. [Google Scholar] [CrossRef]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koeter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinska, D.; Szymanski, J.; Patalas-Krawczyk, P.; Michalska, B.; Wojtala, A.; Prill, M.; Partyka, M.; Drabik, K.; Walczak, J.; Sewer, A.; et al. Assessment of mitochondrial function following short- and long-term exposure of human bronchial epithelial cells to total particulate matter from a candidate modified-risk tobacco product and reference cigarettes. Food Chem. Toxicol. 2018, 115, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Cheng, S.E.; Hsiao, L.D.; Kou, Y.R.; Yang, C.M. Cigarette smoke extract upregulates heme oxygenase-1 via PKC/NADPH oxidase/ROS/PDGFR/PI3K/Akt pathway in mouse brain endothelial cells. J. Neuroinflamm. 2011, 8, 104. [Google Scholar] [CrossRef] [Green Version]
- Starke, R.M.; Thompson, J.W.; Ali, M.S.; Pascale, C.L.; Martinez Lege, A.; Ding, D.; Chalouhi, N.; Hasan, D.M.; Jabbour, P.; Owens, G.K.; et al. Cigarette smoke initiates oxidative stress-induced cellular phenotypic modulation leading to cerebral aneurysm pathogenesis. Arter. Thromb. Vasc. Biol. 2018, 38, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, H.; Horinouchi, T.; Mai, Y.; Sawada, O.; Fujii, S.; Nishiya, T.; Minami, M.; Katayama, T.; Iwanaga, T.; Terada, K.; et al. Nicotine- and tar-free cigarette smoke induces cell damage through reactive oxygen species newly generated by PKC-dependent activation of NADPH oxidase. J. Pharmacol. Sci. 2012, 118, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, E.A.; DeMaster, E.G.; Tian, R.X.; Raij, L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arter. Thromb. Vasc. Biol. 2004, 24, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordazzo, C.; Petrini, S.; Neri, T.; Lombardi, S.; Carmazzi, Y.; Pedrinelli, R.; Paggiaro, P.; Celi, A. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on Ca2+ mobilization. Inflamm. Res. 2014, 63, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Liu, Y.; Chen, Y.; Yu, D.; Williams, K.J.; Liu, M.L. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am. J. Pathol. 2013, 182, 1552–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Yu, D.; Williams, K.J.; Liu, M.L. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arter. Thromb. Vasc. Biol. 2010, 30, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.E.; Scruggs, B.S.; Schaffer, J.E.; Hanson, P.I. Effects of inhibiting VPS4 support a general role for ESCRTs in extracellular vesicle biogenesis. Biophys. J. 2017, 113, 1342–1352. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, M.; Nagaeva, O.; Kargl, D.; Baranov, V.; Mincheva-Nilsson, L. Thermal- and oxidative stress causes enhanced release of NKG2D ligand-bearing immunosuppressive exosomes in leukemia/lymphoma T and B cells. PLoS ONE 2011, 6, e16899. [Google Scholar] [CrossRef]
- Harmati, M.; Gyukity-Sebestyen, E.; Dobra, G.; Janovak, L.; Dekany, I.; Saydam, O.; Hunyadi-Gulyas, E.; Nagy, I.; Farkas, A.; Pankotai, T.; et al. Small extracellular vesicles convey the stress-induced adaptive responses of melanoma cells. Sci. Rep. 2019, 9, 15329. [Google Scholar] [CrossRef] [Green Version]
- Saeed-Zidane, M.; Linden, L.; Salilew-Wondim, D.; Held, E.; Neuhoff, C.; Tholen, E.; Hoelker, M.; Schellander, K.; Tesfaye, D. Cellular and exosome mediated molecular defense mechanism in bovine granulosa cells exposed to oxidative stress. PLoS ONE 2017, 12, e0187569. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.; Foot, N.J.; Anand, S.; Dalton, H.E.; Chaudhary, N.; Collins, B.M.; Mathivanan, S.; Kumar, S. Regulation of the divalent metal ion transporter via membrane budding. Cell Discov. 2016, 2, 16011. [Google Scholar] [CrossRef] [Green Version]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Lu, Q. Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat. Commun. 2017, 8, 709. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat. Commun. 2018, 9, 960. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 2010, 3, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhou, J.; Fernandes, A.F.; Sparrow, J.R.; Pereira, P.; Taylor, A.; Shang, F. The proteasome: A target of oxidative damage in cultured human retina pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3622–3630. [Google Scholar] [CrossRef]
- Yu, C.; Wang, X.; Huszagh, A.S.; Viner, R.; Novitsky, E.; Rychnovsky, S.D.; Huang, L. Probing H2O2-mediated structural dynamics of the human 26S proteasome using quantitative cross-linking mass spectrometry (QXL-MS). Mol. Cell. Proteom. 2019, 18, 954–967. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Callard, A.; Goldberg, A.L. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J. Biol. Chem. 2006, 281, 8582–8590. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.; O’Dea, K.P.; Tan, Y.Y.; Cho, K.; Abe, E.; Romano, R.; Cui, J.; Ma, D.; Sarathchandra, P.; Wilson, M.R.; et al. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. 2019, 33, 6442–6455. [Google Scholar] [CrossRef] [Green Version]
- Eldh, M.; Ekstrom, K.; Valadi, H.; Sjostrand, M.; Olsson, B.; Jernas, M.; Lotvall, J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE 2010, 5, e15353. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Pan, Y.; Li, X.H.; Yang, X.Y.; Feng, Y.L.; Tan, H.H.; Jiang, L.; Feng, J.; Yu, X.Y. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016, 7, e2277. [Google Scholar] [CrossRef] [Green Version]
- Grindheim, A.K.; Hollas, H.; Raddum, A.M.; Saraste, J.; Vedeler, A. Reactive oxygen species exert opposite effects on Tyr23 phosphorylation of the nuclear and cortical pools of annexin A2. J. Cell Sci. 2016, 129, 314–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy-Feitosa, E.; Pinto, R.F.; Pires, K.M.; Monteiro, A.P.; Machado, M.N.; Santos, J.C.; Ribeiro, M.L.; Zin, W.A.; Canetti, C.A.; Romana-Souza, B.; et al. The influence of 5-lipoxygenase on cigarette smoke-induced emphysema in mice. Biochim. Biophys. Acta 2014, 1840, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kayyali, U.S.; Budhiraja, R.; Pennella, C.M.; Cooray, S.; Lanzillo, J.J.; Chalkley, R.; Hassoun, P.M. Upregulation of xanthine oxidase by tobacco smoke condensate in pulmonary endothelial cells. Toxicol. Appl. Pharmacol. 2003, 188, 59–68. [Google Scholar] [CrossRef]
- Kim, B.S.; Serebreni, L.; Hamdan, O.; Wang, L.; Parniani, A.; Sussan, T.; Scott Stephens, R.; Boyer, L.; Damarla, M.; Hassoun, P.M.; et al. Xanthine oxidoreductase is a critical mediator of cigarette smoke-induced endothelial cell DNA damage and apoptosis. Free Radic. Biol. Med. 2013, 60, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, E.; Stinson, A.; Shan, L.; Yang, J.; Gietl, D.; Albino, A.P. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 2008, 8, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, Y.; Hiramatsu, N.; Kasai, A.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Radic. Biol. Med. 2008, 45, 50–59. [Google Scholar] [CrossRef]
- Tucher, C.; Bode, K.; Schiller, P.; Classen, L.; Birr, C.; Souto-Carneiro, M.M.; Blank, N.; Lorenz, H.M.; Schiller, M. Extracellular vesicle subtypes released from activated or apoptotic T-lymphocytes carry a specific and stimulus-dependent protein cargo. Front. Immunol. 2018, 9, 534. [Google Scholar] [CrossRef] [Green Version]
- Dieude, M.; Bell, C.; Turgeon, J.; Beillevaire, D.; Pomerleau, L.; Yang, B.; Hamelin, K.; Qi, S.; Pallet, N.; Beland, C.; et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci. Transl. Med. 2015, 7, 318ra200. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.C.; Tan, S.S.; Teh, B.J.; Sze, S.K.; Arslan, F.; de Kleijn, D.P.; Choo, A.; Lim, S.K. Proteolytic potential of the MSC exosome proteome: Implications for an exosome-mediated delivery of therapeutic proteasome. Int. J. Proteom. 2012, 2012, 971907. [Google Scholar] [CrossRef] [Green Version]
- Bochmann, I.; Ebstein, F.; Lehmann, A.; Wohlschlaeger, J.; Sixt, S.U.; Kloetzel, P.M.; Dahlmann, B. T lymphocytes export proteasomes by way of microparticles: A possible mechanism for generation of extracellular proteasomes. J. Cell. Mol. Med. 2014, 18, 59–68. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Mutlu, G.M.; Budinger, G.R.; Wu, M.; Lam, A.P.; Zirk, A.; Rivera, S.; Urich, D.; Chiarella, S.E.; Go, L.H.; Ghosh, A.K.; et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-beta(1) signalling. Thorax 2012, 67, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeniya, M.; Mori, T.; Yui, N.; Nomura, N.; Mandai, S.; Isobe, K.; Chiga, M.; Sohara, E.; Rai, T.; Uchida, S. The proteasome inhibitor bortezomib attenuates renal fibrosis in mice via the suppression of TGF-beta1. Sci. Rep. 2017, 7, 13086. [Google Scholar] [CrossRef] [Green Version]
- Koca, S.S.; Ozgen, M.; Dagli, F.; Tuzcu, M.; Ozercan, I.H.; Sahin, K.; Isik, A. Proteasome inhibition prevents development of experimental dermal fibrosis. Inflammation 2012, 35, 810–817. [Google Scholar] [CrossRef]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2019, 38, e101812. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, D.; Porzig, B.B.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Chang, I.; Wang, C.Y. Inhibition of HDAC6 protein enhances bortezomib-induced apoptosis in head and neck squamous cell carcinoma (HNSCC) by reducing autophagy. J. Biol. Chem. 2016, 291, 18199–18209. [Google Scholar] [CrossRef] [Green Version]
- Haggadone, M.D.; Peters-Golden, M. Microenvironmental influences on extracellular vesicle-mediated communication in the lung. Trends Mol. Med. 2018, 24, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haggadone, M.D.; Mancuso, P.; Peters-Golden, M. Oxidative Inactivation of the Proteasome Augments Alveolar Macrophage Secretion of Vesicular SOCS3. Cells 2020, 9, 1589. https://doi.org/10.3390/cells9071589
Haggadone MD, Mancuso P, Peters-Golden M. Oxidative Inactivation of the Proteasome Augments Alveolar Macrophage Secretion of Vesicular SOCS3. Cells. 2020; 9(7):1589. https://doi.org/10.3390/cells9071589
Chicago/Turabian StyleHaggadone, Mikel D., Peter Mancuso, and Marc Peters-Golden. 2020. "Oxidative Inactivation of the Proteasome Augments Alveolar Macrophage Secretion of Vesicular SOCS3" Cells 9, no. 7: 1589. https://doi.org/10.3390/cells9071589