Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease

 , and
, and 
Abstract
:1. Introduction
2. NAFLD
2.1. Epidemiology
2.2. Etiology
2.2.1. Ethnicity
2.2.2. Genetic Factors
2.2.3. Metabolic Factors
2.2.4. Environmental Factors
2.2.5. Gut Microbiota
2.3. Pathophysiology
2.4. Progression and Associated Diseases
3. Current Therapeutic Strategies for NAFLD
3.1. Lifestyle Modification and Bariatric Surgery
3.1.1. Exercise
3.1.2. Dietary Interventions
3.1.3. Bariatric Surgery
3.2. Pharmacotherapy
3.2.1. Pioglitazone
3.2.2. Vitamin E
3.2.3. Other Current and Emerging Medications
3.2.4. Drugs Targeting Nuclear Receptors
4. PPARs as Promising Targets for the Treatment of NAFLD
4.1. Overview of PPARs
4.1.1. Structure, Tissue Expression, and Mode of Action
4.1.2. PPARs in Glucose and Lipid Metabolism
4.1.3. PPARs in Inflammation and HSC Activation
4.1.4. PPARs in NAFLD
4.2. Available PPAR Agonists
4.2.1. PPARα Agonists
4.2.2. PPARβ/δ Agonists
4.2.3. PPARγ Agonists
4.3. Novel PPAR Agonists
4.3.1. Pemafibrate
4.3.2. PPARα and β/δ Dual Agonist Elafibranor
4.3.3. PPARα and γ Dual Agonist Saroglitazar
4.3.4. Pan-PPAR Agonist Lanifibranor
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Zhou, J.; Wang, W.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Zhu, L.; Cai, J.; Li, H. Unexpected Rapid Increase in the Burden of NAFLD in China From 2008 to 2018: A Systematic Review and Meta-Analysis. Hepatology 2019, 70, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; Therneau, T.M.; Larson, J.J.; Coward, A.; Somers, V.K.; Kamath, P.S. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology 2018, 67, 1726–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A guide to non-alcoholic fatty liver disease in childhood and adolescence. Int. J. Mol. Sci. 2016, 17, 947. [Google Scholar] [CrossRef] [PubMed]
- Goldner, D.; Lavine, J.E. Nonalcoholic Fatty Liver Disease in Children: Unique Considerations and Challenges. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Bellentani, S.; Argo, C.K.; Ballestri, S.; Byrne, C.D.; Caldwell, S.H.; Cortez-Pinto, H.; Grieco, A.; Machado, M.V.; Miele, L.; et al. Epidemiological modifiers of non-alcoholic fatty liver disease: Focus on high-risk groups. Dig. Liver Dis. 2015, 47, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in NAFLD: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Vandel, J.; Dubois-Chevalier, J.; Gheeraert, C.; Derudas, B.; Raverdy, V.; Thuillier, D.; Van Gaal, L.; Francque, S.; Pattou, F.; Staels, B.; et al. Hepatic molecular signatures highlight the sexual dimorphism of Non-Alcoholic SteatoHepatitis (NASH). Hepatology 2020. [Google Scholar] [CrossRef]
- Rich, N.E.; Oji, S.; Mufti, A.R.; Browning, J.D.; Parikh, N.D.; Odewole, M.; Mayo, H.; Singal, A.G. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 198–210.e2. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic overexpression of PNPLA3 I148M in mouse liver causes hepatic steatosis. J. Clin. Invest. 2012, 122, 4130–4144. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.A.; Larrieta, E.; Kershenobich, D.; Torre, A. The Expression of PNPLA3 Polymorphism could be the Key for Severe Liver Disease in NAFLD in Hispanic Population. Ann. Hepatol. 2017, 16, 909–915. [Google Scholar] [CrossRef]
- Krawczyk, M.; Liebe, R.; Lammert, F. Toward Genetic Prediction of Nonalcoholic Fatty Liver Disease Trajectories: PNPLA3 and Beyond. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; Mcpherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. ARTICLE TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahali, B.; Liu, Y.L.; Daly, A.K.; Day, C.P.; Anstee, Q.M.; Speliotes, E.K. TM6SF2: Catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology 2015, 148, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Holmen, O.L.; Zhang, H.; Fan, Y.; Hovelson, D.H.; Schmidt, E.M.; Zhou, W.; Guo, Y.; Zhang, J.; Langhammer, A.; Løchen, M.L.; et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat. Genet. 2014, 46, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, N.; Doche, M.E.; Chen, S.; Mao, H.Z.; Walsh, M.T.; Bedoya, C.; Guindi, M.; Xiong, W.; Ignatius Irudayam, J.; Iqbal, J.; et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum. Mol. Genet. 2017, 26, 2719–2731. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [Green Version]
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef] [Green Version]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.; et al. Obesity-linked suppression of membrane-bound o-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 2019, 8, 1–69. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Beer, N.L.; Tribble, N.D.; McCulloch, L.J.; Roos, C.; Johnson, P.R.V.; Orho-Melander, M.; Gloyn, A.L. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum. Mol. Genet. 2009, 18, 4081–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Darlay, R.; Cockell, S.; Meroni, M.; Govaere, O.; Tiniakos, D.; Burt, A.D.; Bedossa, P.; Palmer, J.; Liu, Y.-L.; et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically-characterised cohort. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Männistö, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xue, F.; Meng, J.; Liu, S.S.; Chen, L.Z.; Gao, H.; Geng, N.; Jin, W.W.; Xin, Y.N.; Xuan, S.Y. TRIB1 rs17321515 and rs2954029 gene polymorphisms increase the risk of non-alcoholic fatty liver disease in Chinese Han population. Lipids Health Dis. 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Crudele, A.; Panera, N.; Romito, I.; Meroni, M.; De Stefanis, C.; Palma, A.; Comparcola, D.; Fracanzani, A.L.; Miele, L.; et al. β-Klotho gene variation is associated with liver damage in children with NAFLD. J. Hepatol. 2020, 72, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Kempinska-Podhorodecka, A.; Wunsch, E.; Milkiewicz, P.; Stachowska, E.; Milkiewicz, M. The Association between SOCS1−1656G>A Polymorphism, Insulin Resistance and Obesity in Nonalcoholic Fatty Liver Disease (NAFLD) Patients. J. Clin. Med. 2019, 8, 1912. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Pirola, C.J.; Valenti, L.; Davidson, N.O. Genetic pathways in nonalcoholic fatty liver disease: Insights from systems biology. Hepatology. 2020. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.M.; Ioannou, G.N.; Boyko, E.J.; Utzschneider, K.M. Non-alcoholic fatty liver disease as an independent manifestation of the metabolic syndrome: Results of a US national survey in three ethnic groups. J. Gastroenterol. Hepatol. 2013, 28, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Ye, L.; Liu, A.; Wen, S.W.; Deng, J.; Wu, X.; Lai, Z. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: A meta-analysis. Medicine (USA) 2017, 96, e8179. [Google Scholar] [CrossRef]
- Leite, N.C.; Villela-Nogueira, C.A.; Pannain, V.L.N.; Bottino, A.C.; Rezende, G.F.M.; Cardoso, C.R.L.; Salles, G.F. Histopathological stages of nonalcoholic fatty liver disease in type 2 diabetes: Prevalences and correlated factors. Liver Int. 2011, 31, 700–706. [Google Scholar] [CrossRef]
- Loomba, R.; Abraham, M.; Unalp, A.; Wilson, L.; Lavine, J.; Doo, E.; Bass, N.M. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012, 56, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, D.W.; Yan, H.Y.; Wang, Z.Y.; Zhao, S.H.; Wang, B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: Evidence from a meta-analysis of 21 cohort studies. Obes. Rev. 2016, 17, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Katrina Loomis, A.; Kabadi, S.; Preiss, D.; Hyde, C.; Bonato, V.; Louis, M.S.; Desai, J.; Gill, J.M.R.; Welsh, P.; Waterworth, D.; et al. Body mass index and risk of nonalcoholic fatty liver disease: Two electronic health record prospective studies. J. Clin. Endocrinol. Metab. 2016, 101, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Kim, W.; Kim, D.; Yoon, J.H.; Lee, K.; Kim, J.H.; Cho, E.J.; Lee, J.H.; Kim, H.Y.; Kim, Y.J.; et al. Visceral obesity predicts significant fibrosis in patients with nonalcoholic fatty liver disease. Medicine (USA) 2015, 94. [Google Scholar] [CrossRef] [PubMed]
- van der Poorten, D.; Milner, K.L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Chung, G.E.; Kwak, M.S.; Seo, H.B.; Kang, J.H.; Kim, W.; Kim, Y.J.; Yoon, J.H.; Lee, H.S.; Kim, C.Y. Body Fat Distribution and Risk of Incident and Regressed Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 132–138.e4. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Sun, X.; Yuan, G.; Zhou, X.; Lu, H.; Lin, X.; Yu, X. Lipid phenotypes in patients with nonalcoholic fatty liver disease. Metabolism 2016, 65, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Sugino, I.; Kuboki, K.; Matsumoto, T.; Murakami, E.; Nishimura, C.; Yoshino, G. Influence of fatty liver on plasma small, dense LDL- cholesterol in subjects with and without metabolic syndrome. J. Atheroscler. Thromb. 2011, 18, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Wu, S.J.; Zou, H.; Zhu, G.Q.; Wang, L.R.; Zhang, Q.; Shi, K.Q.; Han, J.B.; Huang, W.J.; Braddock, M.; Chen, Y.P.; et al. Increased levels of systolic blood pressure within the normal range are associated with significantly elevated risks of nonalcoholic fatty liver disease. Medicine (USA) 2015, 94. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Barrera, F.; George, J. The role of diet and nutritional intervention for the management of patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- Berná, G.; Romero-Gomez, M. The role of nutrition in non-alcoholic fatty liver disease: Pathophysiology and management. Liver Int. 2020, 40, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.C.P.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean diet and NAFLD: What we know and questions that still need to be answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marjot, T.; Moolla, A.; Cobbold, J.F.; Hodson, L.; Tomlinson, J.W. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr. Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Invest. 2018, 128, 545–555. [Google Scholar] [CrossRef]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.H.; Guan, B.J.; Gao, H.Y.; Peng, X.E. Omega-3 polyunsaturated fatty acid supplementation and non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Medicine (USA) 2018, 97, e12271. [Google Scholar] [CrossRef]
- Argo, C.K.; Patrie, J.T.; Lackner, C.; Henry, T.D.; De Lange, E.E.; Weltman, A.L.; Shah, N.L.; Al-Osaimi, A.M.; Pramoonjago, P.; Jayakumar, S.; et al. Effects of n-3 fish oil on metabolic and histological parameters in NASH: A double-blind, randomized, placebo-controlled trial. J. Hepatol. 2015, 62, 190–197. [Google Scholar] [CrossRef]
- Martin, P.G.P.; Guillou, H.; Lasserre, F.; Déjean, S.; Lan, A.; Pascussi, J.M.; SanCristobal, M.; Legrand, P.; Besse, P.; Pineau, T. Novel aspects of PPARα-mediated regulation of lipid and xenobiotic metabolism revealed through a nutrigenomic study. Hepatology 2007, 45, 767–777. [Google Scholar] [CrossRef]
- Ducheix, S.; Montagner, A.; Polizzi, A.; Lasserre, F.; Marmugi, A.; Bertrand-Michel, J.; Podechard, N.; Al Saati, T.; Chétiveaux, M.; Baron, S.; et al. Essential fatty acids deficiency promotes lipogenic gene expression and hepatic steatosis through the liver X receptor. J. Hepatol. 2013, 58, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Benhamed, F.; Pégorier, J.P.; Foufelle, F.; Viollet, B.; Vaulont, S.; Girard, J.; Postic, C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Invest. 2005, 115, 2843–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Najima, Y.; Nakakuki, M.; Nagai, R.; Ishibashi, S.; Osuga, J.I.; Yamada, N.; Shimano, H. Polyunsaturated Fatty Acids Ameliorate Hepatic Steatosis in Obese Mice by SREBP-1 Suppression. Hepatology 2003, 38, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Bozzetto, L.; Costabile, G.; Luongo, D.; Naviglio, D.; Cicala, V.; Piantadosi, C.; Patti, L.; Cipriano, P.; Annuzzi, G.; Rivellese, A.A. Reduction in liver fat by dietary MUFA in type 2 diabetes is helped by enhanced hepatic fat oxidation. Diabetologia 2016, 59, 2697–2701. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986.e7. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Ioannou, G.N.; Kuver, R.; Lee, S.P.; Teoh, N.C.; Farrell, G.C. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Caraballo, S.C.; Comhair, T.M.; Verheyen, F.; Gaemers, I.; Schaap, F.G.; Houten, S.M.; Hakvoort, T.B.M.; Dejong, C.H.C.; Lamers, W.H.; Koehler, S.E. Prevention and reversal of hepatic steatosis with a high-protein diet in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia Caraballo, S.C.; Comhair, T.M.; Houten, S.M.; Dejong, C.H.C.; Lamers, W.H.; Koehler, S.E. High-protein diets prevent steatosis and induce hepatic accumulation of monomethyl branched-chain fatty acids. J. Nutr. Biochem. 2014, 25, 1263–1274. [Google Scholar] [CrossRef]
- Schwarz, J.; Tomé, D.; Baars, A.; Hooiveld, G.J.E.J.; Müller, M. Dietary Protein Affects Gene Expression and Prevents Lipid Accumulation in the Liver in Mice. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Charidemou, E.; Ashmore, T.; Li, X.; McNally, B.D.; West, J.A.; Liggi, S.; Harvey, M.; Orford, E.; Griffin, J.L. A randomized 3-way crossover study indicates that high-protein feeding induces de novo lipogenesis in healthy humans. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD + and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.G.; Reily, M.D.; Lehman-Mckeeman, L.D.; Vaillancourt, R.R.; Cherrington, N.J. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids 2015, 47, 603–615. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Grzych, G.; Vonghia, L.; Bout, M.-A.; Weyler, J.; Verrijken, A.; Dirinck, E.; Joncquel, M.; Van Gaal, L.; Paumelle, R.; Francque, S.; et al. Plasma BCAA changes in Patients with NAFLD are Sex Dependent. J. Clin. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, S.; Yan, W.; Xia, Y.; Chen, X.; Wang, W.; Zhang, J.; Gao, C.; Peng, C.; Yan, F.; et al. Branched Chain Amino Acids Cause Liver Injury in Obese/Diabetic Mice by Promoting Adipocyte Lipolysis and Inhibiting Hepatic Autophagy. EBioMedicine 2016, 13, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Y.; Wan, B.; Zhang, H.; Wu, S.; Zhu, Z.; Lin, Y.; Wang, M.; Zhang, N.; Lin, S.; et al. Association between vitamin d status and non-alcoholic fatty liver disease: A population-based study. J. Nutr. Sci. Vitaminol. (Tokyo) 2019, 65, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef]
- Zein, C.O.; Unalp, A.; Colvin, R.; Liu, Y.C.; McCullough, A.J. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 54, 753–759. [Google Scholar] [CrossRef] [Green Version]
- La Merrill, M.A.; Johnson, C.L.; Smith, M.T.; Kandula, N.R.; Macherone, A.; Pennell, K.D.; Kanaya, A.M. Exposure to Persistent Organic Pollutants (POPs) and Their Relationship to Hepatic Fat and Insulin Insensitivity among Asian Indian Immigrants in the United States. Environ. Sci. Technol. 2019, 53, 13906–13918. [Google Scholar] [CrossRef] [PubMed]
- al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Marmugi, A.; Ducheix, S.; Lasserre, F.; Polizzi, A.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.P.; et al. Low doses of bisphenol a induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology 2012, 55, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.E.; Guo, G.L. Understanding Environmental Contaminants’ Direct Effects on Non-alcoholic Fatty Liver Disease Progression. Curr. Environ. Health. Rep. 2019, 6, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Reports 2019, 1, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.e2. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The role of the microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 2005, 115. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals With Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Anghel, S.I.; Wahli, W. Fat poetry: A kingdom for PPARγ. Cell Res. 2007, 17, 486–511. [Google Scholar] [CrossRef] [Green Version]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control oef Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Santoleri, D.; Titchenell, P.M. Resolving the Paradox of Hepatic Insulin Resistance. CMGH 2019, 7, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.I.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [Green Version]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.B.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef] [Green Version]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yuan, B.; Lo, K.A.; Patterson, H.C.; Sun, Y.; Lodish, H.F. Adiponectin regulates expression of hepatic genes critical for glucose and lipid metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 14568–14573. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Farrell, G.C.; Haczeyni, F.; Chitturi, S. Pathogenesis of NASH: How metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. In Advances in Experimental Medicine and Biology; Springer LLC: New York, NY, USA, 2018; Volume 1061, pp. 19–44. [Google Scholar]
- Ioannou, G.N.; Landis, C.S.; Jin, G.; Haigh, W.G.; Farrell, G.C.; Kuver, R.; Lee, S.P.; Savard, C. Cholesterol Crystals in Hepatocyte Lipid Droplets Are Strongly Associated With Human Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 776–791. [Google Scholar] [CrossRef]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Ratziu, V. Journal of Hepatology; Elsevier, B.V.: Amsterdam, The Netherlands, 1 May 2015; pp. 1002–1004. [Google Scholar]
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wai-Sun Wong, V.; Peleg, N.; et al. Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015, 149. [Google Scholar] [CrossRef] [Green Version]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [Green Version]
- Reig, M.; Gambato, M.; Man, N.K.; Roberts, J.P.; Victor, D.; Orci, L.A.; Toso, C. Should Patients with NAFLD/NASH Be Surveyed for HCC? Transplantation 2019, 103, 39–44. [Google Scholar] [CrossRef]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef]
- Sanna, C.; Rosso, C.; Marietti, M.; Bugianesi, E. Non-alcoholic fatty liver disease and extra-hepatic cancers. Int. J. Mol. Sci. 2016, 17, 717. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L. A nutrigenomic approach to non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutoukidis, D.A.; Astbury, N.M.; Tudor, K.E.; Morris, E.; Henry, J.A.; Noreik, M.; Jebb, S.A.; Aveyard, P. Association of Weight Loss Interventions with Changes in Biomarkers of Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2019, 179, 1262–1271.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ge, J.; Zhao, C.; Le, S.; Yang, Y.; Ke, D.; Wu, N.; Tan, X.; Zhang, X.; Du, X.; et al. Effect of aerobic exercise and diet on liver fat in pre-diabetic patients with non-alcoholic-fatty-liver-disease: A randomized controlled trial. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.A.; King, N.; McFarlane, J.R.; Graham, P.L.; Dieberg, G. Effect of exercise training on liver function in adults who are overweight or exhibit fatty liver disease: A systematic review and meta-analysis. Br. J. Sports Med. 2018, 52, 834–843. [Google Scholar] [CrossRef]
- Hallsworth, K.; Thoma, C.; Hollingsworth, K.G.; Cassidy, S.; Anstee, Q.M.; Day, C.P.; Trenell, M.I. Modified high-intensity interval training reduces liver fat and improves cardiac function in non-alcoholic fatty liver disease: A randomized controlled trial. Clin. Sci. 2015, 129, 1097–1105. [Google Scholar] [CrossRef]
- Abdelbasset, W.K.; Tantawy, S.A.; Kamel, D.M.; Alqahtani, B.A.; Soliman, G.S. A randomized controlled trial on the effectiveness of 8-week high-intensity interval exercise on intrahepatic triglycerides, visceral lipids, and health-related quality of life in diabetic obese patients with nonalcoholic fatty liver disease. Medicine (USA) 2019, 98. [Google Scholar] [CrossRef]
- Oh, S.; So, R.; Shida, T.; Matsuo, T.; Kim, B.; Akiyama, K.; Isobe, T.; Okamoto, Y.; Tanaka, K.; Shoda, J. High-intensity aerobic exercise improves both hepatic fat content and stiffness in sedentary obese men with nonalcoholic fatty liver disease. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hallsworth, K.; Fattakhova, G.; Hollingsworth, K.G.; Thoma, C.; Moore, S.; Taylor, R.; Day, C.P.; Trenell, M.I. Resistance exercise reduces liver fat and its mediators in non-alcoholic fatty liver disease independent of weight loss. Gut 2011, 60, 1278–1283. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Buch, A.; Yeshua, H.; Vaisman, N.; Webb, M.; Harari, G.; Kis, O.; Fliss-Isakov, N.; Izkhakov, E.; Halpern, Z.; et al. Effect of resistance training on non-alcoholic fatty-liver disease a randomized-clinical trial. World J. Gastroenterol. 2014, 20, 4382–4392. [Google Scholar] [CrossRef]
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol. Metab. 2019, 30, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef]
- Katsagoni, C.N.; Georgoulis, M.; Papatheodoridis, G.V.; Panagiotakos, D.B.; Kontogianni, M.D. Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: A meta-analysis. Metabolism 2017, 68, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golabi, P.; Locklear, C.T.; Austin, P.; Afdhal, S.; Byrns, M.; Gerber, L.; Younossi, Z.M. Effectiveness of exercise in hepatic fat mobilization in nonalcoholic fatty liver disease: Systematic review. World J. Gastroenterol. 2016, 22, 6318–6327. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-T.; Zheng, J.; Peng, H.-W.; Cai, X.-L.; Pan, X.-T.; Li, H.-Q.; Hong, Q.-Z.; Peng, X.-E. Physical activity intervention for non-diabetic patients with non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. BMC Gastroenterol. 2020, 20, 66. [Google Scholar] [CrossRef]
- Yoshioka, N.; Ishigami, M.; Watanabe, Y.; Sumi, H.; Doisaki, M.; Yamaguchi, T.; Ito, T.; Ishizu, Y.; Kuzuya, T.; Honda, T.; et al. Effect of weight change and lifestyle modifications on the development or remission of nonalcoholic fatty liver disease: Sex-specific analysis. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, S.A.; Hodson, L. Managing NAFLD in Type 2 Diabetes: The Effect of Lifestyle Interventions, a Narrative Review. Adv. Ther. 2020, 37, 1381–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, G.; Day, C.P.; Dufour, J.F.; Canbay, A.; Nobili, V.; Ratziu, V.; Tilg, H.; Roden, M.; Gastaldelli, A.; Yki-Jarvinen, H.; et al. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.C.; Itsiopoulos, C.; Thodis, T.; Ward, G.; Trost, N.; Hofferberth, S.; O’Dea, K.; Desmond, P.V.; Johnson, N.A.; Wilson, A.M. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 138–143. [Google Scholar] [CrossRef]
- Kontogianni, M.D.; Tileli, N.; Margariti, A.; Georgoulis, M.; Deutsch, M.; Tiniakos, D.; Fragopoulou, E.; Zafiropoulou, R.; Manios, Y.; Papatheodoridis, G. Adherence to the Mediterranean diet is associated with the severity of non-alcoholic fatty liver disease. Clin. Nutr. 2014, 33, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Saeed, N.; Nadeau, B.; Shannon, C.; Tincopa, M. Evaluation of dietary approaches for the treatment of non-alcoholic fatty liver disease: A systematic review. Nutrients 2019, 11, 3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosavian, S.P.; Arab, A.; Paknahad, Z. The effect of a Mediterranean diet on metabolic parameters in patients with non-alcoholic fatty liver disease: A systematic review of randomized controlled trials. Clin. Nutr. ESPEN 2020, 35, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 365–367. [Google Scholar] [CrossRef]
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: A pilot study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Dufour, S.; Lyu, K.; Zhang, X.-M.; Hakkarainen, A.; Lehtimäki, T.E.; Cline, G.W.; Petersen, K.F.; Shulman, G.I.; Yki-Järvinen, H. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2020, 117, 7347–7354. [Google Scholar] [CrossRef] [Green Version]
- Vilar-Gomez, E.; Athinarayanan, S.J.; Adams, R.N.; Hallberg, S.J.; Bhanpuri, N.H.; McKenzie, A.L.; Campbell, W.W.; McCarter, J.P.; Phinney, S.D.; Volek, J.S.; et al. Post hoc analyses of surrogate markers of non-alcoholic fatty liver disease (NAFLD) and liver fibrosis in patients with type 2 diabetes in a digitally supported continuous care intervention: An open-label, non-randomised controlled study. BMJ Open 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Bergentall, M.; Wu, H.; Bergh, P.-O.; Hakkarainen, A.; Nielsen, J.; Williams, K.J.; Uhlén, M.; Lee, S.; Snyder, M.; Romeo, S.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e5. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: A comprehensive review of the literature. Obes. Rev. 2020. [Google Scholar] [CrossRef] [Green Version]
- Markova, M.; Pivovarova, O.; Hornemann, S.; Sucher, S.; Frahnow, T.; Wegner, K.; Machann, J.; Petzke, K.J.; Hierholzer, J.; Lichtinghagen, R.; et al. Isocaloric Diets High in Animal or Plant Protein Reduce Liver Fat and Inflammation in Individuals With Type 2 Diabetes. Gastroenterology 2017, 152, 571–585.e8. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.W.; Lin, S.X.; Shen, Z.H.; Luo, W.W.; Wang, X.Y. Systematic review with meta-analysis: The effects of probiotics in nonalcoholic fatty liver disease. Gastroenterol. Res. Pract. 2019, 2019, 1484598. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, S.; Zhang, Q.; Liu, L.; Meng, G.; Wu, H.; Bao, X.; Gu, Y.; Sun, S.; Wang, X.; et al. Insoluble dietary fibre intake is associated with lower prevalence of newly-diagnosed non-alcoholic fatty liver disease in Chinese men: A large population-based cross-sectional study. Nutr. Metab. 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorletti, E.; Afolabi, P.R.; Miles, E.A.; Smith, D.E.; Almehmadi, A.; Alshathry, A.; Childs, C.E.; Del Fabbro, S.; Bilson, J.; Moyses, H.E.; et al. Synbiotics Alter Fecal Microbiomes, But Not Liver Fat or Fibrosis, in a Randomized Trial of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1597–1610.e7. [Google Scholar] [CrossRef] [PubMed]
- George, E.S.; Forsyth, A.; Itsiopoulos, C.; Nicoll, A.J.; Ryan, M.; Sood, S.; Roberts, S.K.; Tierney, A.C. Practical dietary recommendations for the prevention andmanagement of nonalcoholic fatty liver disease in adults. Adv. Nutr. 2018, 9, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A.; Tinsley, G.; Bianco, A.; Moro, T. The influence of meal frequency and timing on health in humans: The role of fasting. Nutrients 2019, 11, 719. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.S.; Wong, G.L.H.; Chan, R.S.M.; Shu, S.S.T.; Cheung, B.H.K.; Li, L.S.; Chim, A.M.L.; Chan, C.K.M.; Leung, J.K.Y.; Chu, W.C.W.; et al. Beneficial effects of lifestyle intervention in non-obese patients with non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 1349–1356. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and genetics in NAFLD: The perfect binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef] [Green Version]
- Taitano, A.A.; Markow, M.; Finan, J.E.; Wheeler, D.E.; Gonzalvo, J.P.; Murr, M.M. Bariatric Surgery Improves Histological Features of Nonalcoholic Fatty Liver Disease and Liver Fibrosis. J. Gastrointest. Surg. 2015, 19, 429–437. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, T.K.; Mhaskar, R.; Schwitalla, T.; Muradova, E.; Gonzalvo, J.P.; Murr, M.M. Bariatric surgery improves nonalcoholic fatty liver disease: A contemporary systematic review and meta-analysis. Surg. Obes. Relat. Dis. 2019, 15, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.M.; Sheka, A.C.; Kizy, S.; Irey, R.; Benner, A.; Sieger, G.; Simon, G.; Ma, S.; Lake, J.; Aliferis, C.; et al. Bariatric Surgery is Associated with Decreased Progression of Nonalcoholic Fatty Liver Disease to Cirrhosis. Ann. Surg. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.L.; Hagemann, C.A.; Wei, C.; Kazankov, K.; Thomsen, K.L.; Knop, F.K.; Grønbæk, H. Bariatric surgery in patients with non-alcoholic fatty liver disease—From pathophysiology to clinical effects. World J. Hepatol. 2019, 11, 138–249. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Hollebecque, A.; Arnalsteen, L.; Buob, D.; Leteurtre, E.; Caiazzo, R.; Pigeyre, M.; Verkindt, H.; Dharancy, S.; Louvet, A.; et al. Prospective Study of the Long-Term Effects of Bariatric Surgery on Liver Injury in Patients Without Advanced Disease. Gastroenterology 2009, 137, 532–540. [Google Scholar] [CrossRef]
- Mantovani, A.; Byrne, C.D.; Scorletti, E.; Mantzoros, C.S.; Targher, G. Efficacy and safety of anti-hyperglycaemic drugs in patients with non-alcoholic fatty liver disease with or without diabetes: An updated systematic review of randomized controlled trials. Diabetes Metab. 2020. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.P.; Sanyal, A.J.; Kowdley, K.V.; Robuck, P.R.; Hoofnagle, J.; Kleiner, D.E.; Ünalp, A.; Tonascia, J. Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 2009, 30, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A Placebo-Controlled Trial of Pioglitazone in Subjects with Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus. Ann. Intern. Med. 2016, 165, 305. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Sanyal, A.J.; Neuschwander-Tetri, B.A. Improvements in Histologic Features and Diagnosis Associated with Improvement in Fibrosis in Nonalcoholic Steatohepatitis: Results From the Nonalcoholic Steatohepatitis Clinical Research Network Treatment Trials. Hepatology 2019, 70, 522–531. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, Placebo-Controlled Trial of Pioglitazone in Nondiabetic Subjects With Nonalcoholic Steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettcher, E.; Csako, G.; Pucino, F.; Wesley, R.; Loomba, R. Meta-analysis: Pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2012, 35, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients With Nonalcoholic Steatohepatitis With vs Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef]
- Kawaguchi-Suzuki, M.; Cusi, K.; Bril, F.; Gong, Y.; Langaee, T.; Frye, R.F. A genetic score associates with pioglitazone response in patients with non-alcoholic steatohepatitis. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Shah, P.; Mudaliar, S. Pioglitazone: Side effect and safety profile. Expert Opin. Drug Saf. 2010, 9, 347–354. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Lomonaco, R.; Barb, D.; Orsak, B.; Bruder, J.M.; Cusi, K. Effect of pioglitazone on bone mineral density in patients with nonalcoholic steatohepatitis: A 36-month clinical trial. J. Diabetes 2019, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y.; Han, J. Pioglitazone and bladder cancer risk: A systematic review and meta-analysis. Cancer Med. 2018, 7, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Ripamonti, E.; Azoulay, L.; Abrahamowicz, M.; Platt, R.W.; Suissa, S. A systematic review of observational studies of the association between pioglitazone use and bladder cancer. Diabet. Med. 2019, 36, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARS in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Czaja, M.J. Pioglitazone: More than just an insulin sensitizer. Hepatology 2009, 49, 1427–1430. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.-Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Metab. 2018, 315. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.J.; Chiou, H.Y.C.; Jiang, H.J.; Lee, M.Y.; Hsieh, T.J.; Kuo, K.K. Pioglitazone Enhances Cytosolic Lipolysis, β-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- de Mendonça, M.; dos Santos, B.d.A.C.; de Sousa, É.; Rodrigues, A.C. Adiponectin is required for pioglitazone-induced improvements in hepatic steatosis in mice fed a high-fat diet. Mol. Cell. Endocrinol. 2019, 493. [Google Scholar] [CrossRef] [PubMed]
- Corey, K.E.; Vuppalanchi, R.; Wilson, L.A.; Cummings, O.W.; Chalasani, N. NASH resolution is associated with improvements in HDL and triglyceride levels but not improvement in LDL or non-HDL-C levels. Aliment. Pharmacol. Ther. 2015, 41, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Corey, K.E.; Wilson, L.A.; Altinbas, A.; Yates, K.P.; Kleiner, D.E.; Chung, R.T.; Krauss, R.M.; Chalasani, N.; Bringman, D.; Dasarathy, S.; et al. Relationship between resolution of non-alcoholic steatohepatitis and changes in lipoprotein sub-fractions: A post-hoc analysis of the PIVENS trial. Aliment. Pharmacol. Ther. 2019, 49, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of Vitamin e or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents the tonic randomized controlled trial. JAMA J. Am. Med. Assoc. 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banini, B.A.; Cazanave, S.C.; Yates, K.P.; Asgharpour, A.; Vincent, R.; Mirshahi, F.; Le, P.; Contos, M.J.; Tonascia, J.; Chalasani, N.P.; et al. Haptoglobin 2 Allele is Associated with Histologic Response to Vitamin E in Subjects with Nonalcoholic Steatohepatitis. J. Clin. Gastroenterol. 2019, 53, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of Vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2019, 42, 1481–1488. [Google Scholar] [CrossRef]
- Tsou, P.; Wu, C.-J. Serum Vitamin E Levels of Adults with Nonalcoholic Fatty Liver Disease: An Inverse Relationship with All-Cause Mortality in Non-Diabetic but Not in Pre-Diabetic or Diabetic Subjects. J. Clin. Med. 2019, 8, 1057. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin E cancer prevention trial (SELECT). JAMA J. Am. Med. Assoc. 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. BMJ 2010, 341, c5702. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abner, E.L.; Schmitt, F.A.; Mendiondo, M.S.; Marcum, J.L.; Kryscio, R.J. Vitamin E and All-Cause Mortality: A Meta-Analysis. Curr. Aging Sci. 2012, 4, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.; Li, A.; John, N.; Sallam, S.; Shah, N.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Xu, Y.; Ren, X.; Xiang, D.; Lei, K.; Zhang, C.; Liu, D. Vitamin E Ameliorates Lipid Metabolism in Mice with Nonalcoholic Fatty Liver Disease via Nrf2/CES1 Signaling Pathway. Dig. Dis. Sci. 2019, 64, 3182–3191. [Google Scholar] [CrossRef]
- Landrier, J.F.; Gouranton, E.; El Yazidi, C.; Malezet, C.; Balaguer, P.; Borel, P.; Amiot, M.J. Adiponectin expression is induced by vitamin E via a peroxisome proliferator-activated receptor γ-dependent mechanism. Endocrinology 2009, 150, 5318–5325. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Polyzos, S.A.; Kang, E.S.; Boutari, C.; Rhee, E.J.; Mantzoros, C.S. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism. 2020. [Google Scholar] [CrossRef]
- Muthiah, M.D.; Sanyal, A.J. Current management of non-alcoholic steatohepatitis. Liver Int. 2020, 40, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Neuschwander-Tetri, B.A. Therapeutic Landscape for NAFLD in 2020. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease11This article is part of a Special Issue entitled: Xenobiotic nuclear receptors: New Tricks for An Old Dog, edited by Dr. Wen Xie. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Gonzalez, F.J.; Huang, M.; Bi, H. Nuclear receptors and non-alcoholic fatty liver disease: An update. Liver Res. 2020. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13. [Google Scholar] [CrossRef] [Green Version]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Invest. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef]
- Takeuchi, S.; Matsuda, T.; Kobayashi, S.; Takahashi, T.; Kojima, H. In vitro screening of 200 pesticides for agonistic activity via mouse peroxisome proliferator-activated receptor (PPAR)α and PPARγ and quantitative analysis of in vivo induction pathway. Toxicol. Appl. Pharmacol. 2006, 217, 235–244. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-Mediated Toxicology and Applied Pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casals-Casas, C.; Desvergne, B. Endocrine Disruptors: From Endocrine to Metabolic Disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the role of PPARβ/δ in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Brunmeir, R.; Xu, F. Functional regulation of PPARs through post-translational modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [Green Version]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J.; Yang, X.; Downes, M.; Yu, R.T.; Bookout, A.L.; et al. Nuclear Receptor Expression Links the Circadian Clock to Metabolism. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Lemberger, T.; Saladin, R.; Vázquez, M.; Assimacopoulos, F.; Staels, B.; Desvergne, B.; Wahli, W.; Auwerx, J. Expression of the peroxisome proliferator-activated receptor α gene is stimulated by stress and follows a diurnal rhythm. J. Biol. Chem. 1996, 271, 1764–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Régnier, M.; Polizzi, A.; Lippi, Y.; Fouché, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARα activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; et al. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 1995, 15, 3012–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, M.; Baugé, E.; Lalloyer, F.; Lefebvre, P.; Staels, B. Ketone body therapy protects from lipotoxicity and acute liver failure upon Ppar α deficiency. Mol. Endocrinol. 2015, 29, 1134–1143. [Google Scholar] [CrossRef] [Green Version]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gälman, C.; Lundåsen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafström, I.; Dahlin, M.; Åmark, P.; Angelin, B.; Rudling, M. The Circulating Metabolic Regulator FGF21 Is Induced by Prolonged Fasting and PPARα Activation in Man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Mangelsdorf, D.J. A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21. Cell Metab. 2019, 29, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Mandard, S.; Tan, N.S.; Escher, P.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 2000, 275, 28488–28493. [Google Scholar] [CrossRef] [Green Version]
- Smati, S.; Régnier, M.; Fougeray, T.; Polizzi, A.; Fougerat, A.; Lasserre, F.; Lukowicz, C.; Tramunt, B.; Guillaume, M.; Burnol, A.F.; et al. Regulation of hepatokine gene expression in response to fasting and feeding: Influence of PPAR-α and insulin-dependent signalling in hepatocytes. Diabetes Metab. 2020, 46, 129–136. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Rajak, S.; Singh, B.K.; Yen, P.M. Hepatic lipid catabolism via PPARα-lysosomal crosstalk. Int. J. Mol. Sci. 2020, 21, 2391. [Google Scholar] [CrossRef] [Green Version]
- Siong Tan, H.W.; Anjum, B.; Shen, H.M.; Ghosh, S.; Yen, P.M.; Sinha, R.A. Lysosomal inhibition attenuates peroxisomal gene transcription via suppression of PPARA and PPARGC1A levels. Autophagy 2019, 15, 1455–1459. [Google Scholar] [CrossRef]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARα. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S. Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Alvarez, A.; Soledad Alvarez, M.; Gonzalez, R.; Cucarella, C.; Muntané, J.; Casado, M. Human SREBP1c expression in liver is directly regulated by Peroxisome Proliferator-activated Receptor α (PPARα). J. Biol. Chem. 2011, 286, 21466–21477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, B.L.; Hebbach, A.; Hauton, D.; Brown, A.M.; Wiggins, D.; Patel, D.D.; Gibbons, G.F. A role for PPARα in the control of SREBP activity and lipid synthesis in the liver. Biochem. J. 2005, 389, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; März, W.; Tafuri, S.; Wahli, W.; et al. PPARalpha governs glycerol metabolism. J. Clin. Invest. 2004, 114, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Mandard, S.; Escher, P.; Gonzalez, F.J.; Tafuri, S.; Desvergne, B.; Wahli, W. The peroxisome proliferator-activated receptor α regulates amino acid metabolism. FASEB J. 2001, 15, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M.; Way, J.M.; Tanner, C.J.; Winegar, D.A.; Kliewer, S.A.; Houmard, J.A.; Kraus, W.E.; Lynis Dohm, G. Peroxisome proliferator-activated receptor-α regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 2002, 51, 901–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, C.; Montastier, E.; Valle, C.; Bezaire, V.; Mazzucotelli, A.; Mairal, A.; Viguerie, N.; Langin, D. Peroxisome proliferator-activated receptor-α control of lipid and glucose metabolism in human white adipocytes. Endocrinology 2010, 151, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G.; Bharadwaj, K.G.; Fong, J.L.; Mitra, R.; Sambandam, N.; Courtois, M.R.; Lavine, K.J.; Goldberg, I.J.; Kelly, D.P. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-α transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-α activators. Circulation 2010, 121, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPAR modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, M.K.; Bayliss, J.; Keenan, S.; Rhost, S.; Ting, S.B.; Watt, M.J. The role of Ap2a2 in PPARα-mediated regulation of lipolysis in adipose tissue. FASEB J. 2019, 33, 13267–13279. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Sanada, K.; Nagai, H.; Li, Y.; Aoki, Y.; Ara, T.; Seno, S.; Matsuda, H.; Yu, R.; Kawada, T.; et al. Over-expression of PPARα in obese mice adipose tissue improves insulin sensitivity. Biochem. Biophys. Res. Commun. 2017, 493, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Rosell, M.; Diaz-Delfin, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16 1296. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barquissau, V.; Beuzelin, D.; Pisani, D.F.; Beranger, G.E.; Mairal, A.; Montagner, A.; Roussel, B.; Tavernier, G.; Marques, M.A.; Moro, C.; et al. White-to-brite conversion in human adipocytes promotes metabolic reprogramming towards fatty acid anabolic and catabolic pathways. Mol. Metab. 2016, 5, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Defour, M.; Dijk, W.; Ruppert, P.; Nascimento, E.B.M.; Schrauwen, P.; Kersten, S. The Peroxisome Proliferator-Activated Receptor α is dispensable for cold-induced adipose tissue browning in mice. Mol. Metab. 2018, 10, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Lasar, D.; Rosenwald, M.; Kiehlmann, E.; Balaz, M.; Tall, B.; Opitz, L.; Lidell, M.E.; Zamboni, N.; Krznar, P.; Sun, W.; et al. Peroxisome Proliferator Activated Receptor Gamma Controls Mature Brown Adipocyte Inducibility through Glycerol Kinase. Cell Rep. 2018, 22, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Su, Y.; Silva, F.J.; Weller, A.H.; Sostre-Colón, J.; Titchenell, P.M.; Steger, D.J.; Seale, P.; Soccio, R.E. Shared PPARα/γ Target Genes Regulate Brown Adipocyte Thermogenic Function. Cell Rep. 2020, 30, 3079–3091.e5. [Google Scholar] [CrossRef] [Green Version]
- Sommars, M.A.; Ramachandran, K.; Senagolage, M.D.; Futtner, C.R.; Germain, D.M.; Allred, A.L.; Omura, Y.; Bederman, I.R.; Barish, G.D. Dynamic repression by BCL6 controls the genome-wide liver response to fasting and steatosis. Elife 2019, 8. [Google Scholar] [CrossRef]
- Li, G.; Brocker, C.N.; Yan, T.; Xie, C.; Krausz, K.W.; Xiang, R.; Gonzalez, F.J. Metabolic adaptation to intermittent fasting is independent of peroxisome proliferator-activated receptor alpha. Mol. Metab. 2018, 7, 80–89. [Google Scholar] [CrossRef]
- Leuenberger, N.; Pradervand, S.; Wahli, W. Sumoylated PPARalpha mediates sex specific gene repression and protects the liver from estrogen induced toxicity in mice. J. Clin. Invest. 2009, 119, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor β/δ in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Koh, J.H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.H. PPARβ Is Essential for Maintaining Normal Levels of PGC-1α and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Holloszy, J.O.; Kim, K.; Koh, J.H. Exercise training-induced PPARβ increases PGC-1α protein stability and improves insulin-induced glucose uptake in rodent muscles. Nutrients 2020, 12, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1α expression is controlled in skeletal muscles by PPARβ, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARδ regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor δ/β in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Xie, X.; Fan, Y.; Tian, J.; Guan, Y.; Wang, X.; Zhu, Y.; Wang, N. Peroxisome proliferator-activated receptor-δ induces insulin-induced gene-1 and suppresses hepatic lipogenesis in obese diabetic mice. Hepatology 2008, 48, 432–441. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol. Genomics 2010, 41, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Risérus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Bedu, E.; Wahli, W.; Desvergne, B. Peroxisome proliferator-activated receptor β/δ as a therapeutic target for metabolic diseases. Expert Opin. Ther. Targets 2005, 9, 861–873. [Google Scholar] [CrossRef]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic regulation of VLDL receptor by PPARβ/δ and FGF21 modulates non-alcoholic fatty liver disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, M.; Zwarts, I.; Van Zutphen, T.; Van DIjk, T.H.; Bloks, V.W.; Harkema, L.; De Bruin, A.; Downes, M.; Evans, R.M.; Verkade, H.J.; et al. Intestinal PPARδ protects against diet-induced obesity, insulin resistance and dyslipidemia. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Wagner, K.-D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor γ is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Mullican, S.E.; DiSpirito, J.R.; Peed, L.C.; Lazar, M.A. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc. Natl. Acad. Sci. USA 2013, 110, 18656–18661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.Q.; Phillips, C.T.; et al. A PPARγ-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.G.; Moreira, R.J.; Castro, É.; Caron, A.; Côté, M.; Andrade, M.L.; Oliveira, T.E.; Ortiz-Silva, M.; Peixoto, A.S.; Dias, F.A.; et al. PPARγ is a major regulator of branched-chain amino acid blood levels and catabolism in white and brown adipose tissues. Metabolism 2018, 89, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.-H.; Gavrilova, O.; Ward, J.M.; Brewer, B.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003, 111, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Kusakabe, T.; Noguchi, T.; Takiguchi, S.; Suzuki, T.; Yamano, S.; Gonzalez, F.J. Hepatic Steatosis in Leptin-Deficient Mice Is Promoted by the PPARγ Target Gene Fsp27. Cell Metab. 2008, 7, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xu, M.J.; Cai, Y.; Zhou, Z.; Cao, H.; Mukhopadhyay, P.; Pacher, P.; Zheng, S.; Gonzalez, F.J.; Gao, B. Inflammation is independent of steatosis in a murine model of steatohepatitis. Hepatology 2017, 66, 108–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasenfuss, S.C.; Bakiri, L.; Thomsen, M.K.; Williams, E.G.; Auwerx, J.; Wagner, E.F. Regulation of steatohepatitis and PPARγ signaling by distinct AP-1 dimers. Cell Metab. 2014, 19, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Park, J.E.; Lee, M.; Hardwick, J.P. Hepatic lipid homeostasis by peroxisome proliferator-activated receptor gamma 2. Liver Res. 2018, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARγ contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Stienstra, R.; Mandard, S.; Tan, N.S.; Wahli, W.; Trautwein, C.; Richardson, T.A.; Lichtenauer-Kaligis, E.; Kersten, S.; Müller, M. The Interleukin-1 receptor antagonist is a direct target gene of PPARα in liver. J. Hepatol. 2007, 46, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Baugé, E.; Bourguet, W.; De Bosscher, K.; Lalloyer, F.; Tailleux, A.; Lebherz, C.; Lefebvre, P.; Staels, B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 2014, 60, 1593–1606. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARα-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Jordan, S.; Tung, N.; Casanova-Acebes, M.; Chang, C.; Cantoni, C.; Zhang, D.; Wirtz, T.H.; Naik, S.; Rose, S.A.; Brocker, C.N.; et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell 2019, 178, 1102–1114.e17. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.N.; Yue, J.; Kim, D.; Qu, A.; Bonzo, J.A.; Gonzalez, F.J. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G283–G299. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.; Hall, P.; Robertson, G.; Leclercq, I. Administration of the Potent PPARα Agonist, Wy-14,643, Reverses Nutritional Fibrosis and Steatohepatitis in Mice. Hepatology 2004, 39, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, L.; Chen, J.; Li, L.; Zheng, Z.; Ren, J.; Qiu, Y. Oleoylethanolamide, an endogenous PPAR-α ligand, attenuates liver fibrosis targeting hepatic stellate cells. Oncotarget 2015, 6, 42530–42540. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 Activation of Kupffer Cells by PPARδ Ameliorates Obesity-Induced Insulin Resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhuang, Y.; Sng, M.K.; Tan, N.S.; Wahli, W. The potential of the FSP1cre-Pparb/d−/− mouse model for studying Juvenile NAFLD. Int. J. Mol. Sci. 2019, 20, 5115. [Google Scholar] [CrossRef] [Green Version]
- Kostadinova, R.; Montagner, A.; Gouranton, E.; Fleury, S.; Guillou, H.; Dombrowicz, D.; Desreumaux, P.; Wahli, W. GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Hellemans, K.; Michalik, L.; Dittie, A.; Knorr, A.; Rombouts, K.; De Jong, J.; Heirman, C.; Quartier, E.; Schuit, F.; Wahli, W.; et al. Peroxisome proliferator-activated receptor-β signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology 2003, 124, 184–201. [Google Scholar] [CrossRef]
- Iwaisako, K.; Haimerl, M.; Paik, Y.H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-inflammatory Properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue T reg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, J.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazra, S.; Xiong, S.; Wang, J.; Rippe, R.A.; Chatterjee, V.K.K.; Tsukamoto, H. Peroxisome Proliferator-activated Receptor γ Induces a Phenotypic Switch from Activated to Quiescent Hepatic Stellate Cells. J. Biol. Chem. 2004, 279, 11392–11401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, A.; Crabb, D.W.; Ceni, E.; Salzano, R.; Mello, T.; Svegliati-Baroni, G.; Ridolfi, F.; Trozzi, L.; Surrenti, C.; Casini, A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002, 122, 1924–1940. [Google Scholar] [CrossRef] [PubMed]
- Morán-Salvador, E.; Titos, E.; Rius, B.; González-Périz, A.; García-Alonso, V.; López-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Clària, J. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. 2004, 61, 393–416. [Google Scholar] [CrossRef]
- Kersten, S.; Rakhshandehroo, M.; Knoch, B.; Müller, M. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Costet, P.; Legendre, C.; Moré, J.; Edgar, A.; Galtier, P.; Pineau, T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J. Biol. Chem. 1998, 273, 29577–29585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stienstra, R.; Mandard, S.; Patsouris, D.; Maass, C.; Kersten, S.; Müller, M. Peroxisome proliferator-activated receptor α protects against obesity-induced hepatic inflammation. Endocrinology 2007, 148, 2753–2763. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Yoo, S.-H.; Henderson, L.E.; Gonzalez, F.J.; Woodcroft, K.J.; Song, B.-J. PPARα Expression Protects Male Mice from High Fat–Induced Nonalcoholic Fatty Liver. J. Nutr. 2011, 141, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARα-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tanaka, N.; Guo, R.; Lu, Y.; Nakajima, T.; Gonzalez, F.J.; Aoyama, T. PPARα protects against trans-fatty-acid-containing diet-induced steatohepatitis. J. Nutr. Biochem. 2017, 39, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C.; et al. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Liang, N.; Damdimopoulos, A.; Goñi, S.; Huang, Z.; Vedin, L.L.; Jakobsson, T.; Giudici, M.; Ahmed, O.; Pedrelli, M.; Barilla, S.; et al. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARα. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Shan, W.; Nicol, C.J.; Ito, S.; Bility, M.T.; Kennett, M.J.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-β/δ protects against chemically induced liver toxicity in mice. Hepatology 2008, 47, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Nadra, K.; Anghel, S.I.; Joye, E.; Tan, N.S.; Basu-Modak, S.; Trono, D.; Wahli, W.; Desvergne, B. Differentiation of Trophoblast Giant Cells and Their Metabolic Functions Are Dependent on Peroxisome Proliferator-Activated Receptor β/δ. Mol. Cell. Biol. 2006, 26, 3266–3281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilardi, F.; Winkler, C.; Quignodon, L.; Diserens, J.G.; Toffoli, B.; Schiffrin, M.; Sardella, C.; Preitner, F.; Desvergne, B. Systemic PPARγ deletion in mice provokes lipoatrophy, organomegaly, severe type 2 diabetes and metabolic inflexibility. Metabolism 2019, 95, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán-Salvador, E.; López-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; López-Vicario, C.; Barak, Y.; Arroyo, V.; Clària, J. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, A.W.; Majumdar, N.; Yang, P.; Subbaiah, P.V.; Kineman, R.D.; Cordoba-Chacon, J. Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol. 2017, 232, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Patitucci, C.; Couchy, G.; Bagattin, A.; Cañeque, T.; De Reyniès, A.; Scoazec, J.Y.; Rodriguez, R.; Pontoglio, M.; Zucman-Rossi, J.; Pende, M.; et al. Hepatocyte nuclear factor 1α suppresses steatosisassociated liver cancer by inhibiting PPARγ transcription. J. Clin. Invest. 2017, 127, 1873–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duszka, K.; Ellero-Simatos, S.; Ow, G.S.; Defernez, M.; Paramalingam, E.; Tett, A.; Ying, S.; König, J.; Narbad, A.; Kuznetsov, V.A.; et al. Complementary intestinal mucosa and microbiota responses to caloric restriction. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Korecka, A.; Polizzi, A.; Lippi, Y.; Blum, Y.; Canlet, C.; Tremblay-Franco, M.; Gautier-Stein, A.; Burcelin, R.; Yen, Y.-C.C.; et al. Hepatic circadian clock oscillators and nuclear receptors integrate microbiome-derived signals. Sci. Rep. 2016, 6, 20127. [Google Scholar] [CrossRef] [PubMed]
- Ul Hasan, A.; Rahman, A.; Kobori, H. Interactions between host PPARs and gut microbiota in health and disease. Int. J. Mol. Sci. 2019, 20, 387. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Okopień, B.; Bułdak, Ł.; Bołdys, A. Benefits and risks of the treatment with fibrates––a comprehensive summary. Expert Rev. Clin. Pharmacol. 2018, 11, 1099–1112. [Google Scholar] [CrossRef]
- Larter, C.Z.; Yeh, M.M.; van Rooyen, D.M.; Brooling, J.; Ghatora, K.; Farrell, G.C. Peroxisome proliferator-activated receptor-α agonist, Wy 14643, improves metabolic indices, steatosis and ballooning in diabetic mice with non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 341–350. [Google Scholar] [CrossRef]
- Toyama, T.; Nakamura, H.; Harano, Y.; Yamauchi, N.; Morita, A.; Kirishima, T.; Minami, M.; Itoh, Y.; Okanoue, T. PPARα ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 2004, 324, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; García-Pagán, J.C. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Nikam, A.; Patankar, J.V.; Somlapura, M.; Lahiri, P.; Sachdev, V.; Kratky, D.; Denk, H.; Zatloukal, K.; Abuja, P.M. The PPARα Agonist Fenofibrate Prevents Formation of Protein Aggregates (Mallory-Denk bodies) in a Murine Model of Steatohepatitis-like Hepatotoxicity. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of Fenofibrate and Niacin on Intrahepatic Triglyceride Content, Very Low-Density Lipoprotein Kinetics, and Insulin Action in Obese Subjects with Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- El-Haggar, S.M.; Mostafa, T.M. Comparative clinical study between the effect of fenofibrate alone and its combination with pentoxifylline on biochemical parameters and liver stiffness in patients with non-alcoholic fatty liver disease. Hepatol. Int. 2015, 9, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Oscarsson, J.; Önnerhag, K.; Risérus, U.; Sundén, M.; Johansson, L.; Jansson, P.A.; Moris, L.; Nilsson, P.M.; Eriksson, J.W.; Lind, L. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: A double-blind, randomized, placebo-controlled study. J. Clin. Lipidol. 2018, 12, 1390–1403.e4. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Pahan, K. Gemfibrozil, stretching arms beyond lipid lowering. Immunopharmacol. Immunotoxicol. 2009, 31, 339–351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, S.; Hu, L.; Wang, J.; Liu, Y.; Shi, P. Gemfibrozil reduces lipid accumulation in smmc-7721 cells via the involvement of pparα and srebp1. Exp. Ther. Med. 2019, 17, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Yaghoubi, M.; Jafari, S.; Sajedi, B.; Gohari, S.; Akbarieh, S.; Heydari, A.H.; Jameshoorani, M. Comparison of fenofibrate and pioglitazone effects on patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2017, 29, 1385–1388. [Google Scholar] [CrossRef]
- Garbacz, W.G.; Huang, J.T.J.; Higgins, L.G.; Wahli, W.; Palmer, C.N.A. PPAR Is Required for PPAR δ Action in Regulation of Body Weight and Hepatic Steatosis in Mice. PPAR Res. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Barroso, E.; Palomer, X.; Escolà-Gil, J.C.; Cedó, L.; Wahli, W.; Vázquez-Carrera, M. Pharmacological PPARβ/δ activation upregulates VLDLR in hepatocytes. Clin. e Investig. en Arterioscler. 2019, 31, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Bojic, L.A.; Telford, D.E.; Fullerton, M.D.; Ford, R.J.; Sutherland, B.G.; Edwards, J.Y.; Sawyez, C.G.; Gros, R.; Kemp, B.E.; Steinberg, G.R.; et al. PPAR—Activation attenuates hepatic steatosis in Ldlr Mice by enhanced fat oxidation, reduced lipogenesis, and improved insulin sensitivity. J. Lipid Res. 2014, 55, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.H.; Lee, M.Y.; Choi, R.; Kim, H.M.; Cho, E.J.; Kim, B.H.; Choi, Y.S.; Naowaboot, J.; Lee, E.Y.; Yang, Y.C.; et al. Peroxisome proliferator-activated receptor δ agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp. Mol. Med. 2012, 44, 578–585. [Google Scholar] [CrossRef]
- Silva-Veiga, F.M.; Rachid, T.L.; de Oliveira, L.; Graus-Nunes, F.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. GW0742 (PPAR-beta agonist) attenuates hepatic endoplasmic reticulum stress by improving hepatic energy metabolism in high-fat diet fed mice. Mol. Cell. Endocrinol. 2018, 474, 227–237. [Google Scholar] [CrossRef]
- Shan, W.; Palkar, P.S.; Murray, I.A.; Mcdevitt, E.I.; Kennett, M.J.; Kang, B.H.; Isom, H.C.; Perdew, G.H.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator—Activated receptor β/δ (PPARβ/δ) attenuates carbon tetrachloride hepatotoxicity by downregulating proinflammatory gene expression. Toxicol. Sci. 2008, 105, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.-J.; McWherter, C.A.; Teoh, N.C.-H.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, S.J.; Leeming, D.J.; Detlefsen, S.; Bruun, M.F.; Hjuler, S.T.; Henriksen, K.; Hein, P.; Karsdal, M.A.; Brockbank, S.; Cruwys, S. Biochemical and histological characterisation of an experimental rodent model of non-alcoholic steatohepatitis – Effects of a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist and a glucagon-like peptide-1 analogue. Biomed. Pharmacother. 2019, 111, 926–933. [Google Scholar] [CrossRef]
- Ahn, H.Y.; Kim, H.H.; Hwang, J.Y.; Park, C.; Cho, B.Y.; Park, Y.J. Effects of pioglitazone on nonalcoholic fatty liver disease in the absence of constitutive androstane receptor expression. PPAR Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, C.; Huan, Y.; Liu, S.; Hou, S.; Sun, S.; Li, C.; Liu, Q.; Jiang, Q.; Wang, Y.; Shen, Z. Effect of chronic pioglitazone treatment on hepatic gene expression profile in obese C57BL/6J mice. Int. J. Mol. Sci. 2015, 16, 12213–12229. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: A meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
- Wu, H.M.; Ni, X.X.; Xu, Q.Y.; Wang, Q.; Li, X.Y.; Hua, J. Regulation of lipid-induced macrophage polarization through modulating peroxisome proliferator-activated receptor-gamma activity affects hepatic lipid metabolism via a Toll-like receptor 4/NF-κB signaling pathway. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Nan, Y.M.; Fu, N.; Wu, W.J.; Liang, B.L.; Wang, R.Q.; Zhao, S.X.; Zhao, J.M.; Yu, J. Rosiglitazone prevents nutritional fibrosis and steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 358–365. [Google Scholar] [CrossRef]
- Wei, Z.; Zhao, D.; Zhang, Y.; Chen, Y.; Zhang, S.; Li, Q.; Zeng, P.; Li, X.; Zhang, W.; Duan, Y.; et al. Rosiglitazone ameliorates bile duct ligation-induced liver fibrosis by down-regulating NF-κB-TNF-α signaling pathway in a PPARγ-dependent manner. Biochem. Biophys. Res. Commun. 2019, 519, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.A.; Zhang, F.; Jiang, L.; Ye, R.; An, Y.; Shao, M.; Tao, C.; Gupta, R.K.; Scherer, P.E. Peroxisome Proliferator-Activated Receptor γ and Its Role in Adipocyte Homeostasis and Thiazolidinedione-Mediated Insulin Sensitization. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-γ ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for Nonalcoholic Steatohepatitis: One-Year Results of the Randomized Placebo-Controlled Fatty Liver Improvement With Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.; Serfaty, L.; Cervera, P.; Capeau, J.; Ratziu, V. Hepatic molecular effects of rosiglitazone in human non-alcoholic steatohepatitis suggest long-term pro-inflammatory damage. Hepatol. Res. 2014, 44, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P.; Barter, P.J.; Ceska, R.; Corsini, A.; Després, J.P.; et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα) paradigm: Conceptual framework and therapeutic potential. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Kambe, A.; Yamamoto, Y.; Arulmozhiraja, S.; Ito, S.; Nakagawa, Y.; Tokiwa, H.; Nakano, S.; Shimano, H. Elucidation of molecular mechanism of a selective PPARα modulator, pemafibrate, through combinational approaches of x-ray crystallography, thermodynamic analysis, and first-principle calculations. Int. J. Mol. Sci. 2020, 21, 361. [Google Scholar] [CrossRef] [Green Version]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome analysis of K-877 (A novel selective PPARα modulator (SPPARMα))-regulated genes in primary human hepatocytes and the mouse liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Raza-Iqbal, S.; Tanaka, T.; Murakami, K.; Anai, M.; Osawa, T.; Matsumura, Y.; Sakai, J.; Kodama, T. Gene expression profiles induced by a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMA) pemafibrate. Int. J. Mol. Sci. 2019, 20, 5682. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Nakagawa, Y.; Wang, Y.; Han, S.-I.; Satoh, A.; Sekiya, M.; Matsuzaka, T.; Shimano, H. Effects of K-877, a novel selective PPARα modulator, on small intestine contribute to the amelioration of hyperlipidemia in low-density lipoprotein receptor knockout mice. J. Pharmacol. Sci. 2017, 133, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Hennuyer, N.; Duplan, I.; Paquet, C.; Vanhoutte, J.; Woitrain, E.; Touche, V.; Colin, S.; Vallez, E.; Lestavel, S.; Lefebvre, P.; et al. The novel selective PPARα modulator (SPPARMα) pemafibrate improves dyslipidemia, enhances reverse cholesterol transport and decreases inflammation and atherosclerosis. Atherosclerosis 2016, 249, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Sairyo, M.; Kobayashi, T.; Masuda, D.; Kanno, K.; Zhu, Y.; Okada, T.; Koseki, M.; Ohama, T.; Nishida, M.; Sakata, Y.; et al. A novel selective PPARαmodulator (SPPARMα), K-877 (pemafibrate), attenuates postprandial hypertriglyceridemia in mice. J. Atheroscler. Thromb. 2018, 25, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Nakagawa, Y.; Oishi, A.; Han, S.I.; Wang, Y.; Kumagai, K.; Ohno, H.; Mizunoe, Y.; Iwasaki, H.; Sekiya, M.; et al. The peroxisome proliferator-activated receptor α (PPARα) agonist pemafibrate protects against diet-induced obesity in mice. Int. J. Mol. Sci. 2018, 19, 2148. [Google Scholar] [CrossRef] [Green Version]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator, improves the pathogenesis in a rodent model of nonalcoholic steatohepatitis. Sci. Rep. 2017, 7, 42477. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.C.; et al. Pemafibrate, a selective PPARα modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Yamashita, S.; Arai, H.; Yokote, K.; Araki, E.; Matsushita, M.; Nojima, T.; Suganami, H.; Ishibashi, S. Efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα): Pooled analysis of phase 2 and 3 studies in dyslipidemic patients with or without statin combination. Int. J. Mol. Sci. 2019, 20, 5537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and safety of pemafibrate (K-877), a selective peroxisome proliferator-activated receptor α modulator, in patients with dyslipidemia: Results from a 24-week, randomized, double blind, active-controlled, phase 3 trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S. Efficacy and safety of pemafibrate versus fenofibrate in patients with high triglyceride and low HDL cholesterol levels: A multicenter, placebo-controlled, double-blind, randomized trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of pemafibrate, a novel selective PPARa modulator, on lipid and glucose metabolism in patients with type 2 diabetes and hypertriglyceridemia: A randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.D.; Paynter, N.P.; Everett, B.M.; Glynn, R.J.; Amarenco, P.; Elam, M.; Ginsberg, H.; Hiatt, W.R.; Ishibashi, S.; Koenig, W.; et al. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am. Heart J. 2018, 206, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E.T.T.L. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58. [Google Scholar] [CrossRef] [PubMed]
- Tølbøl, K.S.; Kristiansen, M.N.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Roth, J.D.; Veidal, S.S.; Fensholdt, L.K.D.; Rigbolt, K.T.G.; Papazyan, R.; Nielsen, J.C.; Feigh, M.; Vrang, N.; Young, M.; Jelsing, J.; et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Briand, F.; Heymes, C.; Bonada, L.; Angles, T.; Charpentier, J.; Branchereau, M.; Brousseau, E.; Quinsat, M.; Fazilleau, N.; Burcelin, R.; et al. A 3-week nonalcoholic steatohepatitis mouse model shows elafibranor benefits on hepatic inflammation and cell death. Clin. Transl. Sci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, T.H.; Yang, Y.Y.; Huang, C.C.; Liu, C.W.; Tsai, H.C.; Lin, M.W.; Tsai, C.Y.; Huang, S.F.; Wang, Y.W.; Lee, T.Y.; et al. Elafibranor interrupts adipose dysfunction-mediated gut and liver injury in mice with alcoholic steatohepatitis. Clin. Sci. 2019, 133, 531–544. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator- activated receptor α/δ agonist gft505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, R. The First Approved Agent in the Glitazar’s Class: Saroglitazar. Curr. Drug Targets 2014, 15, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.R.; Giri, S.R.; Trivedi, C.; Bhoi, B.; Rath, A.; Vanage, G.; Vyas, P.; Ranvir, R.; Patel, P.R. Saroglitazar, a novel PPARα/γ agonist with predominant PPARα activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol. Res. Perspect. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Goand, U.K.; Gupta, S.; Shankar, K.; Varshney, S.; Rajan, S.; Srivastava, A.; Gupta, A.; Vishwakarma, A.L.; Srivastava, A.K.; et al. Saroglitazar reduces obesity and associated inflammatory consequences in murine adipose tissue. Eur. J. Pharmacol. 2018, 822, 32–42. [Google Scholar] [CrossRef]
- Hassan, N.F.; Nada, S.A.; Hassan, A.; El-Ansary, M.R.; Al-Shorbagy, M.Y.; Abdelsalam, R.M. Saroglitazar Deactivates the Hepatic LPS/TLR4 Signaling Pathway and Ameliorates Adipocyte Dysfunction in Rats with High-Fat Emulsion/LPS Model-Induced Non-alcoholic Steatohepatitis. Inflammation 2019, 42, 1056–1070. [Google Scholar] [CrossRef]
- Makled, M.N.; Sharawy, M.H.; El-Awady, M.S. The dual PPAR-α/γ agonist saroglitazar ameliorates thioacetamide-induced liver fibrosis in rats through regulating leptin. Naunyn. Schmiedebergs. Arch. Pharmacol. 2019, 392, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Pai, V.; Paneerselvam, A.; Mukhopadhyay, S.; Bhansali, A.; Kamath, D.; Shankar, V.; Gambhire, D.; Jani, R.H.; Joshi, S.; Patel, P. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of saroglitazar 2 and 4 mg compared to pioglitazone 45 mg in diabetic dyslipidemia (PRESS V). J. Diabetes Sci. Technol. 2014, 8, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Bhansali, S.; Kurpad, A.V.; Hawkins, M.; Sharma, A.; Kaur, S.; Rastogi, A.; Bhansali, A. Effect of a Dual PPAR α/γ agonist on Insulin Sensitivity in Patients of Type 2 Diabetes with Hypertriglyceridemia- Randomized double-blind placebo-controlled trial. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist—Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]
- Wettstein, G.; Luccarini, J.-M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrêne, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ruzehaji, N.; Frantz, C.; Ponsoye, M.; Avouac, J.; Pezet, S.; Guilbert, T.; Luccarini, J.M.; Broqua, P.; Junien, J.L.; Allanore, Y. Pan PPAR agonist IVA337 is effective in prevention and treatment of experimental skin fibrosis. Ann. Rheum. Dis. 2016, 75, 2175–2183. [Google Scholar] [CrossRef] [Green Version]
- Avouac, J.; Konstantinova, I.; Guignabert, C.; Pezet, S.; Sadoine, J.; Guilbert, T.; Cauvet, A.; Tu, L.; Luccarini, J.M.; Junien, J.L.; et al. Pan-PPAR agonist IVA337 is effective in experimental lung fibrosis and pulmonary hypertension. Ann. Rheum. Dis. 2017, 76, 1931–1940. [Google Scholar] [CrossRef]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Z.; Fan, R. PPARα and NCOR/SMRT corepressor network in liver metabolic regulation. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kalliora, C.; Kyriazis, I.D.; Oka, S.I.; Lieu, M.J.; Yue, Y.; Area-Gomez, E.; Pol, C.J.; Tian, Y.; Mizushima, W.; Chin, A.; et al. Dual PPARα/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, B.; Du, Y.; Lin, Y.; Liu, J.; Huang, S.; Zhang, A.; Jia, Z.; Zhang, Y. Targeting PPARα for the treatment and understanding of cardiovascular diseases. Cell. Physiol. Biochem. 2019, 51, 2760–2775. [Google Scholar] [CrossRef]
- Krishnappa, M.; Patil, K.; Parmar, K.; Trivedi, P.; Mody, N.; Shah, C.; Faldu, K.; Maroo, S.; Parmar, D. Effect of saroglitazar 2 mg and 4 mg on glycemic control, lipid profile and cardiovascular disease risk in patients with type 2 diabetes mellitus: A 56-week, randomized, double blind, phase 3 study (PRESS XII study). Cardiovasc. Diabetol. 2020, 19, 93. [Google Scholar] [CrossRef] [PubMed]
- Afroze, S.H.; Munshi, M.K.; Martínez, A.K.; Uddin, M.; Gergely, M.; Szynkarski, C.; Guerrier, M.; Nizamutdinov, D.; Dostal, D.; Glaser, S. Activation of the renin-angiotensin system stimulates biliary hyperplasia during cholestasis induced by extrahepatic bile duct ligation. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G691–G701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saran, A.R.; Dave, S.; Zarrinpar, A. Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease. Gastroenterology 2020, 158. [Google Scholar] [CrossRef]


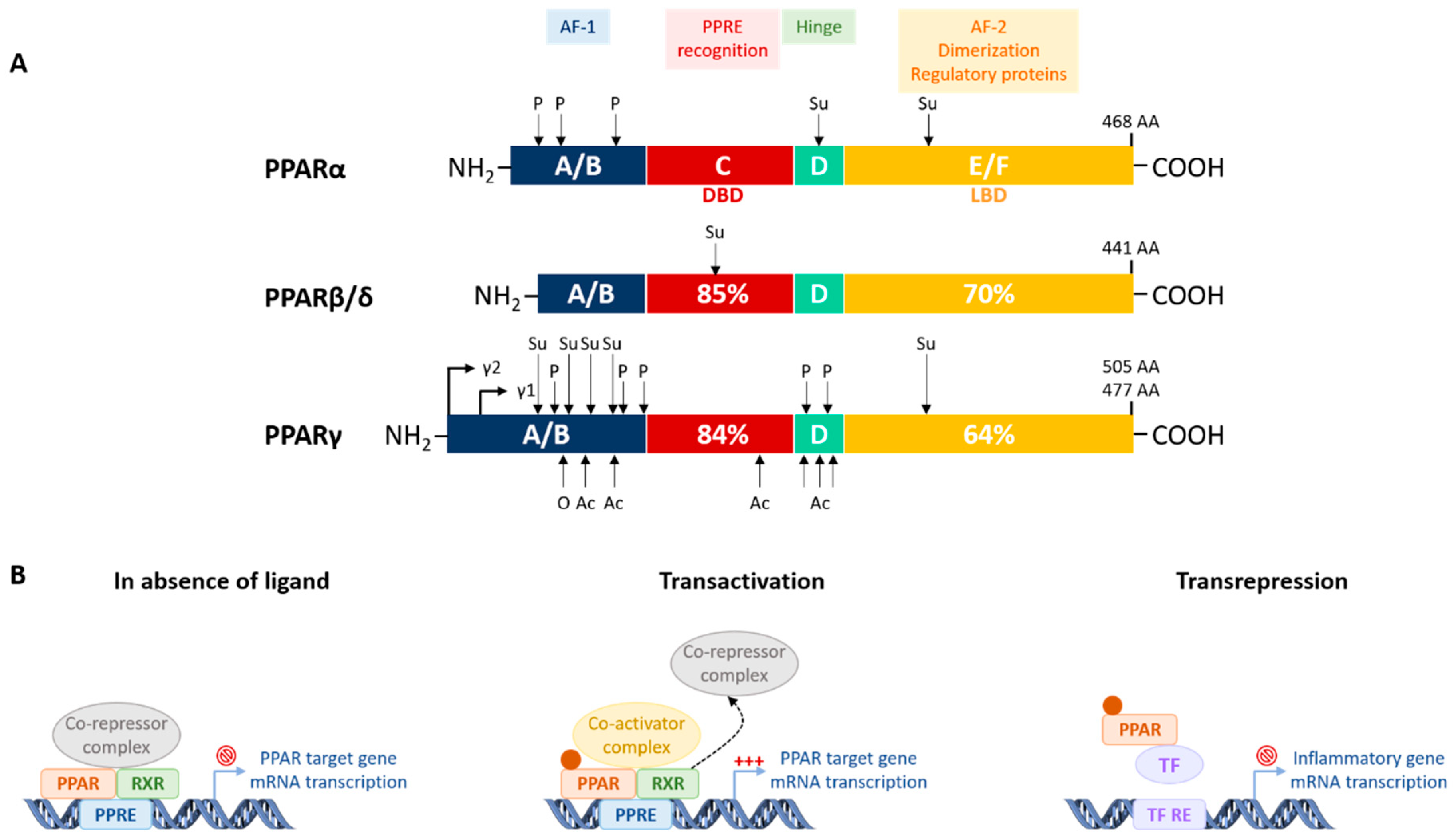



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isotypes | PPARα | PPARβ/δ | PPARγ |
|---|---|---|---|
| Main tissue expression | Liver | Skeletal & cardiac muscles | WAT |
| Skeletal muscles | BAT | ||
| Heart | Liver | Macrophages | |
| Kidney | WAT | ||
| BAT | BAT | ||
| Intestine | Macrophages | ||
| Main natural ligands | FA | FA | FA |
| Eicosanoids | VLDL components | Arachidonic acid metabolites | |
| Phospholipids | |||
| Main synthetic single agonists | Fenofibrate | GW501516 | Pioglitazone |
| Wy14643 | GW0742 | Rosiglitazone | |
| Gemfibrazil | Seladelpar | ||
| Pemafibrate | |||
| Biological functions related to NAFLD | TG hydrolysis | Muscle FA storage | Adipogenesis |
| FA catabolism | FA catabolism | Adipose FA storage | |
| Ketogenesis | Lipoprotein metabolism | Adipokine secretion | |
| FGF21 production | Glucose utilization | Anti-inflammatory | |
| Glycerol metabolism | Anti-inflammatory | Anti-fibrotic | |
| Anti-Inflammatory | |||
| Potential therapeutic target for | Hypertriglyceridemia | Atherogenic dyslipidemia | Insulin resistance |
| Atherogenic dyslipidemia | Insulin resistance | Obesity | |
| NAFLD | Obesity | T2DM | |
| T2DM | NAFLD | ||
| NAFLD |
| Compounds | Chemical Structure | Description |
|---|---|---|
| Pemafibrate |  | Selective and potent PPARα modulator |
| Clinical application for dyslipidemia | ||
| Safety profile in clinical studies | ||
| Currently in phase 3: effect on CV events in T2DM | ||
| Elafibranor |  | High activity on PPARα, moderate activity on PPARβ/δ |
| Safety profile in clinical studies | ||
| Currently in phase 3: effect on fibrosis in NASH | ||
| Saroglitazar |  | High activity on PPARα, moderate activity on PPARγ |
| Clinical application for dyslipidemia | ||
| Safety profile in clinical studies | ||
| Currently in phase 2: effect on NAFLD/NASH, phase 3: effect on fibrosis compared to pioglitazone and vitamin E | ||
| Lanifibranor |  | Moderate and balanced activity on PPARα, PPARβ/δ, PPARγ |
| Currently in phase 2: effect on NAFLD/NASH |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. https://doi.org/10.3390/cells9071638
Fougerat A, Montagner A, Loiseau N, Guillou H, Wahli W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells. 2020; 9(7):1638. https://doi.org/10.3390/cells9071638
Chicago/Turabian StyleFougerat, Anne, Alexandra Montagner, Nicolas Loiseau, Hervé Guillou, and Walter Wahli. 2020. "Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease" Cells 9, no. 7: 1638. https://doi.org/10.3390/cells9071638